

Abstract
The first in vivo measurements of a protein diffusion coefficient versus cytoplasmic biopolymer volume fraction are presented. Fluorescence recovery after photobleaching yields the effective diffusion coefficient on a 1-μm-length scale of green fluorescent protein within the cytoplasm of Escherichia coli grown in rich medium. Resuspension into hyperosmotic buffer lacking K+ and nutrients extracts cytoplasmic water, systematically increasing mean biopolymer volume fraction, <φ>, and thus the severity of possible crowding, binding, and confinement effects. For resuspension in isosmotic buffer (osmotic upshift, or Δ, of 0), the mean diffusion coefficient, <D>, in cytoplasm (6.1 ± 2.4 μm2 s−1) is only 0.07 of the in vitro value (87 μm2 s−1); the relative dispersion among cells, σD/<D> (standard deviation, σD, relative to the mean), is 0.39. Both <D> and σD/<D> remain remarkably constant over the range of Δ values of 0 to 0.28 osmolal. For a Δ value of ≥0.28 osmolal, formation of visible plasmolysis spaces (VPSs) coincides with the onset of a rapid decrease in <D> by a factor of 380 over the range of Δ values of 0.28 to 0.70 osmolal and a substantial increase in σD/<D>. Individual values of D vary by a factor of 9 × 104 but correlate well with fVPS, the fractional change in cytoplasmic volume on VPS formation. The analysis reveals two levels of dispersion in D among cells: moderate dispersion at low Δ values for cells lacking a VPS, perhaps related to variation in φ or biopolymer organization during the cell cycle, and stronger dispersion at high Δ values related to variation in fVPS. Crowding effects alone cannot explain the data, nor do these data alone distinguish crowding from possible binding or confinement effects within a cytoplasmic meshwork.
Most biochemical and biophysical studies of the reaction mechanisms, kinetics, and thermodynamics of proteins and nucleic acids are carried out in vitro using dilute aqueous solutions of purified biopolymer constituents. However, the cytoplasm of both prokaryotic and eukaryotic cells contains a very high total concentration of proteins, nucleic acids, lipids, and supramolecular assemblies of these constituents. In Escherichia coli grown at moderate osmolality, the typical total mass density of protein and nucleic acid within the cytoplasm is ∼220 mg/ml (5), distributed among the nucleoid, ribosomes, and a diverse collection of smaller proteins, tRNA, and mRNA (3). Taken together, these macromolecules occupy some 20 to 30% of the total cytoplasmic volume.
In a crowded fluid, each molecule is excluded from much of the total volume by the presence of other biopolymers (11, 37). In thermodynamic terms, excluded volume decreases the translational entropy of each species, increases free energy, and, thus, increases the thermodynamic driving force to react or bind. These effects could be very large; the thermodynamic activity (the “effective concentration”) of a typical globular protein could be >100 times higher in the E. coli cytoplasm than at the same concentration in an uncrowded solution (9). Crowding also dramatically affects diffusion (26), which is critical for normal cell function and growth (28). Excluded volume slows diffusion by making it less likely that a probe particle can find space in which to move without simultaneous, cooperative motion of several or many background particles (26).
While the bacterial cytoplasm is often assumed to be a crowded aqueous solution (37), the physical state of the cytoplasm is uncertain. Particularly for the low water content induced by hyperosmotic stress, the cytoplasm might become a biopolymer meshwork comprising the nucleoid, associated proteins, nascent mRNA, ribosomes, polypeptide chains, and strongly associated water (13, 38). Such conditions are reminiscent of a polymeric hydrogel (1). Confinement within the pores of the meshwork would enhance protein binding equilibria and slow protein diffusion in a manner qualitatively similar to crowding.
Strong effects of crowding and confinement in vitro have been observed for the tracer diffusion of globular proteins (26) and for the diffusion of tracer proteins in concentrated solutions of hydrophilic polymers (2, 8) and in hydrogels (1). The apparent diffusion coefficient decreases roughly exponentially with the macromolecular volume fraction φ, in agreement with a parametrized model called scaled particle theory (SPT) (14, 26). Additional studies in vitro have shown crowding effects on protein folding (36) and association (24), on the thermodynamics of the protein-nucleic acid interactions critical to replication (27), on enzyme kinetics (25), and on the stability of protein oligomers such as F-actin (16, 20) and of fibrils such as β-amyloid (15).
We know of no experimental studies of crowding/confinement effects on protein diffusion or binding in a live cell. Verkman and coworkers (18) recently reported a sixfold decrease in the diffusion coefficient of the small fluorescent probe BCECF in the cytoplasm of Swiss 3T3 fibroblasts over a range of hyperosmotic upshifts that increased φ by a factor of three. Here we present a quantitative study of the effects of crowding/confinement on diffusion of green fluorescent protein (GFP) in the cytoplasm of E. coli grown in rich medium at 0.24 osmolal. Over the range of osmotic upshift (Δ) values of 0 to 0.7 osmolal, the mean diffusion coefficient (<D>) decreases by a factor of 430. The relative dispersion σD/<D> (standard deviation, σD, relative to the mean) among cells at fixed value of Δ is already 0.39 for unstressed cells (Δ of 0) and increases by a factor of 2 to 4 at larger upshifts for which formation of visible plasmolysis spaces (VPSs) is observed. We compare the data qualitatively with different models of crowding and confinement and suggest possible underlying causes of cell-to-cell heterogeneity.
MATERIALS AND METHODS
Bacterial strain and sample preparation.
We studied a B strain of E. coli, specifically the Tuner strain BL21(DE3) (Novagen, Madison, WI). We transfected the strain with the plasmid pQBI63 expressing Superglow GFP and ampicillin resistance (Qbiogene, Carlsbad, CA) and also the plasmid pLacI expressing the lac repressor and chloramphenicol resistance (a derivative of pACYC184 from Novagen). The Tuner strain of E. coli BL21(DE3) lacks lacIq but expresses T7 RNA polymerase from the lacUV5 promoter. pQBI63 expresses GFP from a T7 RNA polymerase promoter containing lac operators. Thus, repression of GFP expressed from pQBI63 and of T7 RNA polymerase expressed by BL21(DE3) requires lac repressor expressed from pLacI. The pLacI plasmid is compatible with pQBI63; i.e., both can replicate in the same cell.
Our intention was to induce GFP production using isopropyl-β-d-thiogalactopyranoside (IPTG). We verified that expression of GFP occurs after the addition of IPTG and that the level of induction of GFP increased as the concentration of IPTG increased. However, even at very low IPTG concentrations, the level of induction of GFP was very high. In the absence of IPTG, no GFP formed during the initial exponential growth phase following inoculation; the cell doubling time was 51 min. However, GFP was expressed during stationary phase by an unknown mechanism, perhaps because expression of lacI is σ-70 dependent. To reduce the level of GFP, the cytoplasmic GFP expressed in stationary phase cultures was diluted by subculturing overnight in fresh medium in the absence of IPTG. We subsequently made two or three subcultures, each time harvesting cells in exponential growth and resuspending them in fresh medium. The doubling time in such exponential growth phases remained 51 min, suggesting that the expression level of GFP does not greatly affect cell physiology. IPTG was not used.
To test for sensitivity to the E. coli strain, a small number of diffusion measurements were carried out using a K-12 strain, specifically MG1655. In this case, we inserted plasmids expressing GFPmut2 from the lac promoter (pGFP; Clontech) and lac repressor constitutively (pLacI; Novagen). This strain required IPTG to induce expression of GFP.
GFP is a 27-kDa β-barrel prone to dimerization due to a hydrophobic patch on its surface (dimerization constant of ∼104 M−1 in buffer [30]). From direct absorption measurements, we estimated the mean GFP concentration in the cytoplasm to be ∼300 μM. It is likely we studied the diffusion of GFP dimers. GFP has no concentrated surface patches of positive or negative charge. We know of no evidence that GFP forms higher oligomers under any conditions.
Cells were grown to stationary phase in the medium Sigma EZMix Lennox L broth (LB), diluted 1:100 in growth medium, grown to mid-log phase at 0.24 osmolal growth osmolality, harvested, resuspended in a phosphate-buffered medium in which K+ is replaced by Na+ and nutrients are omitted, and studied immediately for a period of at most 45 min to ensure minimal change in physiology. LB is a rich medium containing yeast extract, which includes amino acids and nucleic acids. Bacteria cultures were grown overnight at 30°C with constant shaking in LB containing 34 μg/ml chloramphenicol and 100 μg/ml ampicillin. The resuspension step was repeated to thoroughly remove growth medium. Cells were harvested by centrifugation of 300 μl of culture at 4,000 × g for 1 min and resuspended in a phosphate-buffered medium (64.4 mM Na2HPO4, 14.0 mM NaH2PO4, 6.9 mM NaCl, 15.1 mM NH4Cl, 81 μM CaCl2, 0.81 mM MgSO4 for isosmotic conditions). Variations in the resuspension buffer osmolality were made by adding NaCl. The osmolality of all solutions was measured with a commercial vapor pressure osmometer (Wescor). Omission of K+ and of energy sources from the resuspension buffer minimized the ability of E. coli to recover from osmotic stress. Under such conditions, the cytoplasmic biopolymer and osmolyte content were fixed at the values achieved during growth (5). This study emphasizes osmotic upshifts in the range of 0 to 0.7 osmolal (total osmolality range, 0.24 to 0.94 osmolal). Over this range, the cytoplasm loses more and more water so that the mean cytoplasmic biopolymer fraction (<φ>) increases (4).
For fluorescence recovery after photobleaching (FRAP) measurements, approximately 7 μl of bacterial suspension was placed on a glass slide precoated with poly-l-lysine to improve adhesion. A coverslip was added and sealed to prevent water evaporation. All measurements were taken at room temperature (25 ± 2°C). Diffusion measurements for cells resuspended in isosmotic buffer were taken from every sample to check for consistency over time. We obtained the same baseline distribution of GFP diffusion coefficients with or without poly-l-lysine on the slide. The cells selected for study were well isolated, adhered cells that lay flat on the glass surface. Typical cell lengths were 2 to 4 μm. In addition, we required the presence of a continuous segment of cytoplasm at least 0.9 μm in length with no intervening VPSs (see images below). Cells forming septa were not studied.
Fluorescence microscopy.
The effective diffusion coefficient of GFP within each cell was measured by FRAP (21). Elowitz et al. first applied this method to GFP diffusion in unstressed E. coli (10). A commercial inverted microscope (Nikon Eclipse TE-300) equipped with a 100× oil immersion objective with a numerical aperture of 1.25 was modified to admit two laser beams in epi-illumination, both derived from the same 488-nm laser using beam splitters (Fig. 1a). The weak, expanded probe beam (full width at half maximum [FWHM], 18 μm at the sample; peak intensity, 100 W/cm2) was centered on the single cell under study. The probe beam was mechanically chopped in synchrony with the camera frame cycle to provide a continuous sequence of “snapshots” of the entire cell. The camera and the probe beam shutter were controlled by Metamorph Software (version 4.6r5). The more tightly focused bleach beam (full width at half maximum, 0.9 μm; peak intensity, 32 kW/cm2) fired once at the beginning of each diffusion measurement to photobleach GFP in a selected subregion of the bacterium (Fig. 1b). A 20-nm wide bandpass filter centered at 520 nm isolated GFP fluorescence for imaging on the CoolSNAP HQ charge-coupled-device camera (6.45 μm by 6.45 μm square pixels, corresponding to 64.5 nm by 64.5 nm in real space). Magnification was checked by imaging the grid of an improved Neubauer hemacytometer using white light. We acquired one prebleach image, fired a single 100- to 300-ms bleach pulse, and then acquired a stream of images during the fluorescence recovery. The exposure time was fixed at 26 ms/frame. For fast recoveries, we acquired a continuous stream of 26-ms frames with no dead time between. For slower recoveries, we used a time-lapse mode to acquire a sequence of 26-ms frames with an adjustable delay time between frames.
FIG. 1.

(a) Optical layout showing laser paths, beam splitters (a), mirrors (e), electronic shutters (c and d), collimating telescope (f), and lenses (b and g). (b) Schematic of camera region of interest, including one cell, bleach beam area, and probe beam area.
Data analysis.
For the B strain grown in 0.24 osmolal LB, four representative examples of the FRAP for different Δ values are shown in Fig. 2. Typically, more than half of the total cell fluorescence was initially bleached. For clarity, we renormalized each false-color scale to the maximum value in the entire cell so that the recovered image looked similar to the prebleach image. Cell autofluorescence and irreversible photobleaching caused by the probe beam were insignificant. See the supplemental material for two movies of fluorescence recovery, one at a Δ of 0 and one at a Δ of 0.44 osmolal.
FIG. 2.

Sequences of FRAP images for B-strain cells grown in 0.24 osmolal (Osm) LB and upshifted as shown. The intensity scale is renormalized to 100 for each image. The prebleach (Pre) image and subsequent times are indicated. Bar, 2 μm. The range of x of 0 to L over which the diffusion analysis was carried out is shown in the first image of each sequence. Rel, relative.
Following Elowitz et al. (10), we converted the recovery of the GFP spatial distribution into a one-dimensional diffusion problem. Cell images were rotated relative to pixel space so that the cell axis was parallel to the new pixel grid. Intensity data were converted to one-dimensional intensity profiles I(x, t) (the intensity I at position x at time t) by averaging the intensity within each column of pixels perpendicular to the cell axis and subtracting a baseline intensity obtained far away from the cell image. Figure 3a shows the intensity profile versus time for the unstressed cell (Δ of 0) of Fig. 2. In the prebleach image, the intensity falls off discontinuously at either end due to tapering of the cell end caps. The intensity profile is truncated to remove the end-cap regions. The remaining approximately cylindrical segment defines x at 0 to L (where L is length), the range of the diffusion analysis. We assume that C(x, t), the concentration of unbleached GFP at position x and time t, is proportional to I(x, t).
FIG. 3.

(a) Mean intensity in each pixel column (each value of x) versus distance along the cell axis for the Δ = 0 recovery sequence shown in Fig. 1 at different times as shown; the upper trace is the prebleach image. (b) Least-squares single exponential fits of equation 4 to the decay of n = 1 cosine mode intensity for cells shown in Fig. 1 at the osmotic upshifts shown.
The one-dimensional diffusion equation governing the recovery of intensity in a cylindrical region is as follows:
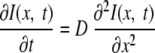 |
(1) |
Here D is an effective GFP diffusion coefficient relevant to the ∼1-μm-length scale under study. The solution to a symmetrized model problem defined on [−L, L], when I(−x) = I(x) and with perfectly reflecting boundary planes (no end caps) when x = −L and L, is as follows:
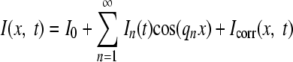 |
(2) |
where the wave number qn ☰ nπ/L and n = 1, 2, 3,… , the mode number. For the model problem, the Fourier amplitude of each cosine mode at each time t is calculated from the intensity data as follows:
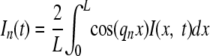 |
(3) |
In the model problem, the cosine mode amplitudes In(t), n = 1, 2,… decay as simple exponentials in time:
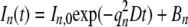 |
(4) |
The constant term Bn allows for the possibility of nonuniform intensity at t = 0 that persists after recovery. The diffusion coefficient (D) would be calculated from the decay rate (k1) of the amplitude of mode 1 as k1L2/π2.
The real bacteria under study have one or two hemispherical end caps that perturb the diffusion relative to this model problem. In practice, we found that the experimental amplitude of the n = 1 mode defined on the interval 0 to L is remarkably well fit by equation 4 in all cases studied, in spite of the end cap(s). Examples of single-exponent fits are shown in Fig. 3b. The quality of the exponential fits is high for both long and short cells with either one or two end caps. Due to the initial bleach profile, the intensity is dominated by the constant I0 and the first cosine mode of amplitude I1(t) at all times. The time-independent asymmetry is generally small (B1≪I1,0) and can be well estimated by scaling the asymmetry prior to the bleach pulse.
To correct for end-cap effects, we numerically modeled diffusion as a three-dimensional random walk in two geometries: a cylinder of length L and radius R plus two hemispherical end caps, and a cylinder of length L and radius R plus one hemispherical end cap. For initial conditions similar to our bleach profile and for the relevant range of the ratio L/R of 1.7 to 5.0, each geometry exhibits single-exponent decay of the n = 1 mode when analyzed as a one-dimensional problem in the same way as the experimental intensity data. However, in order to obtain the correct value of D from the measured decay rate k1, we found it necessary to redefine the wave vector (q1) of mode 1 as follows: q1 ≡ π/Leff, where Leff is an effective length for the problem having one or two end caps. The numerical modeling indicates that each end cap adds approximately 0.67R to the effective length. In practice, we measure L for each cell as the length between sharp breaks in the slope of the one-dimensional intensity profile (Fig. 3a). We obtain R by deconvolving the point spread function of the microscope from the intensity profile perpendicular to the cylinder axis; see details in the discussion of volume measurements below. L is then corrected using the equation Leff = L + 0.67mR + (2 − m)δ. Here, an m value of 1 or 2 is the number of end caps and δ is an additional correction for cells having one flat end due to the broadening of the image by the point spread function of the microscope. We measured δ at the half-height intensity on the flat edge of the image; it is typically 3 pixels, or 0.19 μm. The diffusion coefficient is finally obtained as follows: D ≡ k1L2eff/π2, where k1 is the exponential decay rate of the mode 1 amplitude in time. The correction in D due to Leff is typically a factor of 1.5 to 2.0 relative to the value that would have been obtained using the uncorrected L.
We have sufficient signal to carry out four or five bleach/recovery cycles on the same cell. Multiple measurements on one cell typically yield exponential decay rates (k1) that are constant within ±10% of the mean for that cell, which defines the precision of the measurements. However, we estimate that the absolute accuracy of D is limited to ±30% by uncertainties in the correction involving Leff.
RESULTS
GFP diffusion coefficient versus osmotic upshift.
The top of Fig. 4 shows images of two cells prior to photobleaching, one at a Δ of 0 and the other at a Δ of 0.44 osmolal. The cytoplasm of the unstressed cell looks ellipsoidal and axially symmetric about its long axis, with smooth edges and well-rounded end caps. The distribution of GFP along the length of the cell is quite uniform. In contrast, the cytoplasm of the cell at a Δ of 0.44 osmolal exhibits two VPSs, marked by arrows. One is a lateral invagination, and the other is a flattening of the pole (end cap) on the right. Osmotic upshift values of ≥0.28 osmolal cause such changes in the morphology (volume and shape) of the cytoplasmic space. VPSs arise when the cytoplasm loses a sufficient volume of water and the surface/volume ratio becomes so high as to necessitate local detachment of the cytoplasmic membrane from the cell wall (19, 39). In such images, we can discern the outline of the cell from the faint but smooth ellipsoidal fluorescent halo surrounding the plasmolyzed cytoplasm (deep purple in the false-color images). In this work, VPSs consistently appear at a Δ of 0.28 osmolal, with roughly half the cells exhibiting a single polar VPS. At Δ values of >0.28 osmolal, essentially all cells exhibit VPSs, and multiple VPSs become common. In all such cases, we studied diffusion only in “straight” cell segments with L values of at least 0.6 μm, bleaching one end of this segment as before.
FIG. 4.

(Top) Images of B-strain cells grown in 0.24 osmolal (Osm) LB in isosmotic resuspension buffer (a) or with an upshift of 0.44 osmolal (b). Two VPSs are marked by arrows. Bar, 1 μm. (Bottom) Distribution of D and log D for single cells grown in 0.24 osmolal LB and upshifted as shown. Table 1 contains additional quantitative details.
The sequences of recovery images for cells at different osmotic upshifts (Fig. 2) show clearly that the time scale of recovery lengthens dramatically with increasing Δ. The unstressed cells recover on a ∼0.2-s time scale. As Δ increases to 0.28 osmolal, 0.44 osmolal, and 0.70 osmolal, the recovery time increases to ∼3 s, ∼60 s, and ∼20 min, respectively, although there is great cell-to-cell dispersion. For cells with no VPS, we obtained the same diffusion coefficient on bleaching of either end. In several cells exhibiting a lateral VPS, we were able to analyze diffusion within two segments on opposite sides of the invagination. The two diffusion coefficients match within experimental error. Diffusion across a deep lateral VPS is very slow, occurring on time scales of at least 3 min regardless of the Δ value.
We obtained diffusion coefficients describing the recovery of 125 cells grown in 0.24 osmolal LB and studied over the range of osmotic upshift values of 0 to 0.7 osmolal. The quantitative results are summarized in Table 1. The 39 cells studied at a Δ of 0 exhibit a broad distribution of diffusion coefficients, as shown by the histograms of D and of log D in Fig. 4. The <D> of 6.1 ± 2.4 μm2/s (± σD, one standard deviation of single measurements) is 14 times smaller than the diffusion coefficient of GFP in aqueous buffer, where D0 is 87 μm2 s−1 (34). Individual values of D vary by a factor of 6.3 across the 39 cells. The dispersion in D from cell to cell, as measured by a σD/<D> value of 0.39, far exceeds the ±10% measurement precision for a single cell. The data show no significant correlation of measured D with length of cell over the range 2 to 4 μm or with initial cell brightness over the observed range of a factor of five in total GFP fluorescence intensity.
TABLE 1.
Quantitative details of GFP diffusion coefficient versus osmotic upshift for B-strain cells
| Δ (milliosmolal)a | Cells lacking VPSs
|
Cells having VPS(s)
|
||
|---|---|---|---|---|
| <D> ± σDb (μm2 s−1) | Range of D (μm2 s−1) | <D> ± σDb (μm2 s−1) | Range of D (μm2 s−1) | |
| 0 ± 5 | 6.1 ± 2.4 (n = 39)c | 1.7-10.7 | ||
| 162 ± 5 | 6.8 ± 1.0 (n = 3) | 5.8-7.7 | ||
| 283 ± 5 | 5.3 ± 1.9 (n = 14) | 0.52-7.7 | 1.3 ± 1.5 (n = 16) | 0.15-5.6 |
| 392 ± 5 | 0.94 ± 0.55 (n = 17) | 0.38-2.5 | ||
| 440 ± 5 | 0.32 ± 0.50 (n = 26) | 0.0042-2.0 | ||
| 532 ± 5 | 0.12 ± 0.10 (n = 4) | 0.023-0.26 | ||
| 701 ± 5 | 0.014 ± 0.021 (n = 4) | 1.2 × 10−4-0.045 | ||
Uncertainty of ±5 milliosmolal due to variations in solution preparation; measurement uncertainty of ±3 milliosmolal.
Standard deviation of single measurements.
The number of individual cells measured is given in parentheses.
For Δ values of 0.28, 0.39, and 0.44 osmolal, the distributions of D and of log D were compared with those for unstressed cells in Fig. 4. The histograms of log D provide the best visual estimate of σD/<D>, the relative dispersion in D. The blue bars refer to cells that lack VPSs, while the red bars refer to cells exhibiting VPSs. For Δ values of 0 and 0.16 osmolal, essentially no cells have VPSs. At a Δ of 0.28 osmolal, a smaller diffusion coefficient clearly correlates with the presence of a VPS, although the dispersion is broad for both morphologies. For cells with a single polar VPS, the mean diffusion coefficient, <D>, is 1.3 μm2 s−1, four times smaller than for those with no VPS (<D> = 5.3 μm2 s−1). The two distributions of D overlap only slightly (P < 6 × 10−7 that the two distributions have the same mean; Student t test).
In Fig. 5, we plot log D versus osmotic upshift, with each horizontal dash representing a single measurement and the symbols showing log <D>, the logarithm of the arithmetic mean at each Δ. As Δ increases from 0 to 0.28 osmolal, <D> changes little for cells lacking VPSs. Over the range of Δ values of 0.28 to 0.70, <D> for cells exhibiting VPSs decreases by a factor of 93, and individual cells vary in diffusion coefficient by a factor of 9 × 104. The onset of VPS formation coincides with an abrupt change in slope of log <D> versus Δ. The relative dispersion in D evidently increases with Δ, as judged primarily by the distribution of log D for a Δ of 0.44 osmolal. At still higher Δ values, it became difficult to obtain large data sets because cells with sufficiently long unplasmolyzed segments to photobleach become rare. As shown by Fig. 4 and 5, essentially all the cells exhibiting VPSs have smaller D values than all the cells lacking VPSs. Among cells with VPSs, the distributions of D for different values of Δ overlap substantially. We show below that the primary determinant of D for these cells is not the upshift but, rather, the fractional change in cytoplasmic volume induced by VPS formation.
FIG. 5.

Semilog plot of <D> versus Δ for cells grown in 0.24 osmolal LB. Horizontal bars are individual measurements. Dashed lines are merely guides to the eye. The GFP diffusion coefficient in buffer is indicated. mOsm, milliosmolal.
Finally, we spot-checked the behavior of a different strain of E. coli, the K-12 strain, under otherwise identical conditions of growth and resuspension (Table 2). The data for the K-12 strain closely mimic the data for the B strain in all respects, including the “plateau” in <D> at Δ values of ≤0.28 osmolal, the correlation of D with the two different cell morphologies observed at a Δ of 0.28 osmolal, and the exponential fall-off of <D> versus Δ at values above 0.28 osmolal.
TABLE 2.
Quantitative details of GFP diffusion coefficient versus osmotic upshift for K-12 cells
| Δ (milliosmolal)a | Cells lacking VPSsb
|
Cells having VPS(s)b
|
||
|---|---|---|---|---|
| <D> ± σDc (μm2 s−1) | Range of D (μm2 s−1) | <D> ± σDc (μm2 s−1) | Range of D (μm2 s−1) | |
| 0 ± 5 | 4.8 ± 1.4 (n = 15)c | 2.9-7.2 | ||
| 279 ± 5 | 6.0 (n = 1) | 0.84 ± 0.2 (n = 4) | 0.58-1.07 | |
| 439 ± 5 | 0.10 ± 0.06 (n = 3) | 0.033-0.16 | ||
Uncertainty of ±5 milliosmolal due to variations in solution preparation; measurement uncertainty of ±3 milliosmolal.
The number of individual cells measured is given in parentheses.
Standard deviation of single measurements.
Plasmolysis spaces and the dispersion of diffusion coefficients.
At a Δ of 0.28 osmolal, we observe two distinct cytoplasmic morphologies (without and with VPSs) and two very different corresponding distributions of D. Formation of a VPS reduces <D> from 5.3 ± 1.9 μm2 s−1 to 1.3 ± 1.5 μm2 s−1 (± σD) and also increases the relative dispersion σD/<D> by a factor of three (Table 1). This surprising effect was reproducible across different days and occurred for both the B strain and the K-12 strain.
The observations imply an underlying distribution of threshold ΔVPS values, the upshift at which plasmolysis would first occur in a particular cell if we were to study single cells as Δ gradually increased. This might arise from cell-to-cell variation in turgor pressure (19) related to different membrane-derived oligosaccharide concentrations in the periplasm and thus different cytoplasmic osmolalities (4), from variation in cytoplasmic content at different phases of the cell cycle (32), or from variation in the mechanical strength of the noncovalent binding between the end caps of the cytoplasmic membrane and the peptidoglycan layer (19, 22). The fact that half the cells exhibit VPSs shows that the distribution's median value is ∼0.28 osmolal.
At the three Δ values of 0.28, 0.39, and 0.44 osmolal, we used the prebleach images to roughly estimate the fractional change in volume V induced by VPS formation: fVPS = −ΔVVPS/ Vpre-VPS. This fraction lies between 0 and +1 because ΔVVPS is negative. The corresponding ratio of biopolymer volume fractions “after” and “before” VPS formation is given by φ/φ0 = (1 − fVPS)−1, which is ≥1. The denominator Vpre-VPS refers to the volume of a hypothetical cytoplasm that has been shrunken by perhaps 20% due to the external osmotic upshift but has not yet formed a VPS. We use the faint, ellipsoidal “halo” of the plasmolyzed cytoplasm (the 0.15 contour on a scale in which white is 1.00) (Fig. 2) to estimate Vpre-VPS, assuming the cell is a cylinder with two hemispherical end caps before plasmolysis.
The radius of the cylinder is obtained as follows. We mathematically “squashed” the intensity of a three-dimensional cylinder with a variable radius in the relevant range of R of 0.4 to 0.6 μm into a plane and convolved the resulting intensity distribution with the two-dimensional point spread function of the microscope (approximated as a Gaussian with an FWHM of λ/2, or 0.26 μm). This yields a model image of each cylinder. We fit the intensity profile in the direction transverse to the cylinder axis to a Gaussian whose FWHM increases with R. This provides a model mapping from the FWHM of an experimental transverse intensity profile to the true, underlying cylinder radius, R. In practice, we averaged the values of R obtained from five transverse line scans across the cylindrical part of each image to obtain the mean cylinder radius used in the volume calculation. Each contribution to ΔVVPS is estimated as the volume of a spherical cap.
The resulting estimates of fVPS values are deemed accurate to ±20% at all values of Δ; i.e., the uncertainty in ΔVVPS increases as plasmolysis becomes more extensive. For the cells exhibiting one or more VPSs, the <fVPS> (mean fractional change in volume induced by VPS formation) shifts upward from 0.16 ± 0.05 to 0.17 ± 0.05 to 0.22 ± 0.08 (± 1σ of single measurements) as Δ increases from 0.28 to 0.39 to 0.44 osmolal. The mean total volume <Vpre-VPS> and the distribution of Vpre-VPS do not change significantly with Δ.
Among all cells with VPS(s), the diffusion coefficient correlates remarkably well with fVPS, as shown in Fig. 6. The combined data from the three upshifts span almost three decades in D and intersperse substantially, but when plotted versus fVPS they fall within ±0.5 log units of a smooth curve (Fig. 6, hand-drawn gray swath). Extrapolation of that curve to an fVPS of 0 roughly coincides with the mean diffusion coefficient observed for the cells lacking VPSs at Δ values of 0 to 0.28 osmolal. The residual deviations of the data from the smooth curve along the log D axis are comparable to the dispersion observed in cells lacking VPSs. The residual deviations along the fVPS axis can be explained by the estimated ±20% uncertainty in fVPS.
FIG. 6.

Correlation of log D versus fVPS = − ΔVVPS/Vpre-VPS for cells exhibiting VPSs at Δ values of 0.28 osmolal (Osm), 0.39 osmolal, and 0.44 osmolal. Horizontal error bars indicate ±20% uncertainty in fVPS. Gray swath, smooth curve drawn by hand through the data; blue horizontal lines, log D for 14 cells at a Δ of 0.28 osmolal exhibiting no VPSs; black horizontal lines, log D for 39 cells at a Δ of 0.
Cell viability and recovery of diffusion.
We briefly explored the viability of cells that had been subjected to different osmotic upshifts and then plated on agar plates made either from the standard 0.24 osmolal LB growth medium or from buffer isosmotic to the upshifted condition. In each case, three samples of the culture were diluted by 105, and the plates were incubated for 24 h at 37°C before counting. The benchmark viability (100%) was taken as the mean over three plates of the number of colonies observed for cells grown at 0.24 osmolal LB and plated directly onto agar of 0.24 osmolal LB (i.e., not subjected to upshift). For cells initially upshifted by 0.16, 0.28, 0.39, and 0.44 osmolal and then plated on isosmotic agar, mean viabilities were 55%, 30%, 5%, and 2%, respectively. Under such isosmotic plating, the cells should not “reinflate” (import cytoplasmic water) until the cell machinery adjusts. Interestingly, the viability fraction is roughly proportional to the fraction of cells that do not exhibit VPSs after the initial upshift (100%, 47%, 0%, and 0%, respectively). The data are consistent with the suggestion that VPS formation inhibits recovery in the presence of K+ and nutrients.
For Δ values of 0.28 and 0.39 osmolal (total upshifted osmolality of 0.52 and 0.63 osmolal), plating at the original growth value of 0.24 osmolal substantially enhanced viability to 108% and 53%, respectively (compared with 30% and 5% for isosmotic plating). To test the effects of a similar upshift/downshift cycle on GFP diffusion, we upshifted cells by 0.39 osmolal for 15 min (which gives 100% VPS formation), downshifted them back to the original growth condition of 0.24 osmolal in the absence of K+ or nutrients, and measured the diffusion coefficient as usual. A sample of n = 8 cells subjected to such upshift/downshift recovered 70% of the mean diffusion coefficient of a control sample of n = 5 cells that had never been upshifted. The reinflated cells looked normal; there was no evidence of VPSs. The differential osmotic pressure that exists following the downshift evidently reinflates the cells independent of any active cell mechanism. Taken together, the data suggest that such reinflation enhances the viability of cells that exhibit VPS formation immediately following the upshift.
DISCUSSION
A long-term goal of this work is a quantitative model of binding, crowding, and confinement effects on protein diffusion in the cytoplasm. The key observations from this initial study of GFP diffusion in the cytoplasm of E. coli grown in rich medium are as follows: the 14-fold decrease in the GFP diffusion coefficient from buffer to cytoplasm at a Δ of 0; the plateau in <D> versus Δ at small Δ values; the abrupt change in slope of <D> versus Δ above 0.28 osmolal, correlating with the onset of VPS formation; and the wide dispersion in D among cells with identical history at each value of Δ.
The data reveal at least two sources of dispersion in D among cells. First, for cells lacking VPSs (Δ of ≤0.28 osmolal), the relatively mild dispersion (σD/<D> = 0.39) might arise from differences in cytoplasmic composition or internal structure at different phases of the cell cycle (17), which is not controlled in these experiments. A single cell studied throughout a cell cycle might or might not exhibit the same wide range of diffusion coefficients. Second, there is two to four times greater relative dispersion in D among cells having VPSs (Δ values of ≥0.28 osmolal). Most of this second type of dispersion is closely related to variability in the fractional cytoplasmic volume change accompanying VPS formation (Fig. 6). Clearly, some cells plasmolyze much more extensively than others at the same value of Δ; a corresponding variability in the biopolymer volume fraction φ would directly impact the GFP diffusion coefficient. This type of heterogeneity could be related to differences in cytoplasmic content or periplasmic content that affect turgor pressure and cytoplasmic crowding. It could also arise from variability in mechanical properties of the cell, e.g., in the strength of adhesion of the cytoplasmic membrane to the peptidoglycan layer. It will be fascinating to test whether or not these cell-to-cell differences are phased to the cell cycle.
This first glimpse of protein diffusion data versus osmotic stress somewhat constrains the set of possibly relevant physical models of the cytoplasm. Verkman (37) argues for three independent, multiplicative factors that slow diffusion in the cytoplasm versus buffer: increased viscosity of the fluid, binding of the probe to larger intracellular components, and molecular crowding effects. To this we would add the possibility of confinement effects within a cytoplasmic meshwork. Crowding and confinement are not mutually exclusive; the cytoplasm is heterogeneous, at least in part due to the presence of the nucleoid (29, 35).
We can state with confidence that our observations cannot be understood by crowding theory alone. The most practical model of diffusion in crowded fluids is SPT (14, 26). Direct quantitative comparison of our B-strain data with SPT would require knowledge of the mean cytoplasmic biopolymer volume fraction (<φ>) versus osmotic upshift. We presently lack such data for E. coli grown in LB or any other rich medium; we know only that <φ> increases monotonically with Δ. Nevertheless, we have extensively explored both the Muramatsu-Minton (26) and the Han-Herzfeld (14) parametrizations of SPT. These can be tuned to give essentially identical results for D/D0 versus φ. Either version of SPT can explain the roughly exponential decrease in <D> versus Δ at high Δ values. However, no combination of SPT parameters can explain the 14-fold decrease in D from buffer to the unstressed cells, followed by a plateau in <D> at low Δ values, followed by a strong, roughly exponential decrease at high Δ. In other words, if crowding effects were primarily responsible for D0/D = 14 at a Δ of 0, the diffusion coefficient would already be sensitive to removal of small amounts of cytoplasmic water by osmotic upshift. The experiments show clearly that this is not the case.
For E. coli grown in 0.1 osmolal tryptone broth, Woldringh (39) showed that as Δ increases and turgor pressure is relieved, the mean relative decrease in total cell volume prior to the onset of VPS formation is ∼18%. The cytoplasmic volume presumably shrinks more than 18%, because the periplasmic volume is less sensitive to osmotic upshift (4). In our results, this implies substantial cytoplasmic shrinkage over Δ values of 0 to 0.28 osmolal, the range in which <D> and σD do not change for cells lacking VPSs. The data of Fig. 4 and 5 can be qualitatively understood by postulating a combination of crowding plus reversible binding of GFP to less mobile entities such as the nucleoid, ribosomes, or the cytoplasmic membrane. GFP might bind to other cytoplasmic entities via its hydrophobic patch. In such a binding-crowding model, the binding must be sufficiently strong to explain D0/D = 14 for unstressed cells. For example, if GFP were bound to an immobile particle 93% of the time and diffused freely in a low-viscosity fluid 7% of the time, D would decrease 14-fold from buffer to cytoplasm. Alternatively, 86% binding combined with a factor-of-2 decrease in microviscosity would also be consistent with the data. In this view, crowding must be a relatively minor effect up to a Δ of ∼0.28 osmolal, at which upshift a crossover occurs to a regime in which crowding effects dominate the GFP mobility and thus explain the exponential decay of <D> at high Δ values.
However, the roughly exponential decrease in <D> versus Δ for Δ values of ≥0.28 osmolal conforms equally well to theories of confinement in a porous meshwork (23). Such theories require detailed information about the morphology of the meshwork (pore shape and size distributions and connectivity). In a confinement picture, lower microviscosity combined with the tortuous geometry of the cytoplasmic fluid enveloping the nucleoid might explain the 14-fold decrease in <D> from buffer to unstressed cytoplasm. As water is extracted, GFP diffusion might initially be unaffected because diffusion pathways or channels remain sufficiently large to accommodate the size of GFP. In this view, the abrupt change in slope of <D> versus Δ at a Δ of 0.28 osmolal indicates that formation of a VPS concentrates the cytoplasmic content sufficiently to change key determinants of diffusion.
Perhaps the cytoplasmic medium remains fluid at all Δ values. Formation of VPS(s) compresses the nucleoid sufficiently to render more and more diffusion pathways comparable to, and eventually smaller than, the size of GFP. Diffusion then depends not only on the detailed geometry of the confining meshwork but also on the ability of the nucleoid to remodel locally (7) and enable GFP translational motion. Alternatively, perhaps a cytoplasmic phase transition occurs to a type of gel state (31). Here, we are imagining a condensed “supermolecule” comprising the nucleoid, RNA polymerase, newly synthesized RNA, ribosomes, and nascent proteins, all bound together by noncovalent interactions (38). Cells growing well in rich broth have especially large cytoplasmic mole fractions of DNA, RNA polymerase, and ribosomes (3). The high charge density of these constituents is reminiscent of synthetic polymer hydrogels, with noncovalent interactions playing the role of the polymeric cross-links. In such a hydrogel, much of the water would be bound to biopolymer surfaces, consistent with measurements of water content versus Δ for bacteria grown in minimal medium (4).
A priori it appears impossible to choose among binding, crowding, and confinement mechanisms based on diffusion measurements alone, and indeed all three effects could be important. In future work, measurement of the GFP rotational correlation time might distinguish binding from confinement effects at low Δ. Measurement of the frequency dependence of the cytoplasmic viscoelasticity (6) versus Δ would test the possibility of gel formation.
Finally, we briefly compared the present results with earlier diffusion studies in both E. coli and eukaryotic cells. The mean diffusion coefficient <D> of 6.1 μm2 s−1 for GFP in B-strain cells grown in LB at 0.24 osmolal is 14 times slower than a D0 of 87 μm2 s−1 (34), the GFP diffusion coefficient in solution phase. The only other measurements of GFP translational diffusion in E. coli are from Elowitz et al. (10), who found a similar <D> value of 7.7 ± 2.5 μm2 s−1 (D0/<D> = 11) in the DH5α strain grown in Luria broth, also a rich medium. Our preliminary data on the K-12 strain grown in minimal morpholinepropanesulfonic acid-buffered medium at 0.28 osmolal found a significantly larger <D> value, ∼14 μm2 s−1.
In contrast, the diffusion coefficient of GFP in the cytoplasm of CHO-K1 cells is only three times smaller than in buffer (33). Rotational correlation times of GFP measured by time-dependent fluorescence anisotropy are only 1.5 times longer in the cytoplasm of CHO cells compared with buffer solution (12). The prevailing view is that the cytoplasm of eukaryotic cells is a moderately crowded solution with microviscosity not much larger than that of pure water. The comparison of D0/<D> values between CHO cells and E. coli is consistent with the suggestion that the eukaryotic cytoplasm is less crowded than that of E. coli, in spite of presumably similar biopolymer fractions (9). Accordingly, all excluded-volume theories would predict that the concentration of biopolymer mass into large “particles” such as organelles and cytoskeletal elements minimizes crowding in the cytoplasmic fluid.
Measurement of the effects of osmotic upshift on protein diffusion in a eukaryotic cell are not yet available. Verkman and coworkers (18) studied osmotic effects on diffusion of the small-molecule fluorescent probe BCECF in Swiss 3T3 fibroblasts. For cells resuspended in isosmotic buffer, BCECF showed a D0/<D> of 3. In cells whose volume was shrunk by a factor of 3 in hypertonic buffer, <D> decreased by only a factor of 6. These effects are mild in comparison with the presently observed 430-fold decrease in <D> for GFP at a Δ of 0.70 osmolal in E. coli. It is again tempting to suggest that crowding/confinement effects are less important in eukaryotic cells, but the comparison is complicated at present by the very different sizes of the fluorescent probes.
Conclusion.
In future work, much will be learned from single-cell measurements of cytoplasmic volume, the GFP diffusion coefficient, and GFP rotational correlation time versus both upshift and phase of the cell cycle. By optimizing the data acquisition, we will be able to measure the diffusion coefficient of a single cell as many as 10 different times. On the theoretical side, it remains important to develop models of crowding effects on diffusion that can accurately predict D/D0 from a given distribution of biopolymer sizes, shapes, and concentrations.
Supplementary Material
Acknowledgments
J.C.W. thanks the NSF (grants CHE-0071458 and CHE-0452375) and the University of Wisconsin-Madison Graduate School and Department of Chemistry for support of this research. M.T.R. thanks the NIH (grant GM-47022) for research support.
We acknowledge enlightening discussions with Ian Booth and C. L. Woldringh and assistance in data analysis from John Larsen.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Amsden, B. 1998. Solute diffusion within hydrogels. Mechanisms and models. Macromolecules 31:8382-8395. [Google Scholar]
- 2.Banks, D. S., and C. Fradin. 2005. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 89:2960-2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bremer, H., and P. P. Dennis. 1996. Modulation of the chemical composition and other parameters of the cell by growth rate, p. 1553-1569. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology, vol. 2. ASM Press, Washington, D.C. [Google Scholar]
- 4.Cayley, D. S., H. J. Guttman, and M. T. Record, Jr. 2000. Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress. Biophys. J. 78:1748-1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cayley, S., and M. T. Record, Jr. 2003. Roles of cytoplasmic osmolytes, water, and crowding in the response of Escherichia coli to osmotic stress: biophysical basis of osmoprotection by glycine betaine. Biochemistry 42:12596-12609. [DOI] [PubMed] [Google Scholar]
- 6.Crocker, J. C., M. T. Valentine, E. R. Weeks, T. Gisler, P. D. Kaplan, A. G. Yodh, and D. A. Weitz. 2000. Two-point microrheology of inhomogeneous soft materials. Phys. Rev. Lett. 85:888-891. [DOI] [PubMed] [Google Scholar]
- 7.Cunha, S., C. L. Woldringh, and T. Odijk. 2005. Restricted diffusion of DNA segments within the isolated Escherichia coli nucleoid. J. Struct. Biol. 150:226-232. [DOI] [PubMed] [Google Scholar]
- 8.Dauty, E., and A. S. Verkman. 2004. Molecular crowding reduces to a similar extent the diffusion of small solutes and macromolecules: measurement by fluorescence correlation spectroscopy. J. Mol. Recognit. 17:441-447. [DOI] [PubMed] [Google Scholar]
- 9.Ellis, R. J. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597-604. [DOI] [PubMed] [Google Scholar]
- 10.Elowitz, M. B., M. G. Surette, P. E. Wolf, J. B. Stock, and S. Leibler. 1999. Protein mobility in the cytoplasm of Escherichia coli. J. Bacteriol. 181:197-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garner, M. M., and M. B. Burg. 1994. Macromolecular crowding and confinement in cells exposed to hypertonicity. Am. J. Physiol. 266:C877-C892. [DOI] [PubMed] [Google Scholar]
- 12.Gautier, I., M. Tramier, C. Durieux, J. Coppey, R. B. Pansu, J. C. Nicolas, K. Kemnitz, and M. Coppey-Moisan. 2001. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys. J. 80:3000-3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gitai, Z. 2005. The new bacterial cell biology: moving parts and subcellular architecture. Cell 120:577-586. [DOI] [PubMed] [Google Scholar]
- 14.Han, J., and J. Herzfeld. 1993. Macromolecular diffusion in crowded solutions. Biophys. J. 65:1155-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatters, D. M., A. P. Minton, and G. J. Howlett. 2002. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 277:7824-7830. [DOI] [PubMed] [Google Scholar]
- 16.Herzfeld, J. 2004. Crowding-induced organization in cells: spontaneous alignment and sorting of filaments with physiological control points. J. Mol. Recognit. 17:376-381. [DOI] [PubMed] [Google Scholar]
- 17.Jensen, R. B., S. C. Wang, and L. Shapiro. 2002. Dynamic localization of proteins and DNA during a bacterial cell cycle. Nat. Rev. Mol. Cell Biol. 3:167-176. [DOI] [PubMed] [Google Scholar]
- 18.Kao, H. P., J. R. Abney, and A. S. Verkman. 1993. Determinants of the translational mobility of a small solute in cell cytoplasm. J. Cell Biol. 120:175-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koch, A. L. 1998. The biophysics of the gram-negative periplasmic space. Crit. Rev. Microbiol. 24:23-59. [DOI] [PubMed] [Google Scholar]
- 20.Kulp, D. T., and J. Herzfeld. 1995. Crowding-induced organization of cytoskeletal elements. III. Spontaneous bundling and sorting of self-assembled filaments with different flexibilities. Biophys. Chem. 57:93-102. [DOI] [PubMed] [Google Scholar]
- 21.Lakowicz, J. R. 1999. Principles of fluorescence spectroscopy, 2nd ed. Kluwer Academic, New York, N.Y.
- 22.Leduc, M., K. Ishidate, N. Shakibai, and L. Rothfield. 1992. Interactions of Escherichia coli membrane lipoproteins with the murine sacculus. J. Bacteriol. 174:7982-7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minton, A. P. 1992. Confinement as a determinant of macromolecular structure and reactivity. Biophys. J. 63:1090-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minton, A. P. 2005. Influence of macromolecular crowding upon the stability and state of association of proteins: predictions and observations. J. Pharm. Sci. 94:1668-1675. [DOI] [PubMed] [Google Scholar]
- 25.Minton, A. P., and J. Wilf. 1981. Effect of macromolecular crowding upon the structure and function of an enzyme: glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 20:4821-4826. [DOI] [PubMed] [Google Scholar]
- 26.Muramatsu, N., and A. P. Minton. 1988. Tracer diffusion of globular proteins in concentrated protein solutions. Proc. Natl. Acad. Sci. USA 85:2984-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy, L. D., and S. B. Zimmerman. 1994. Macromolecular crowding effects on the interaction of DNA with Escherichia coli DNA-binding proteins: a model for bacterial nucleoid stabilization. Biochim. Biophys. Acta 1219:277-284. [DOI] [PubMed] [Google Scholar]
- 28.Neidhardt, F. C., R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.). 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 29.Odijk, T. 1998. Osmotic compaction of supercoiled DNA into a bacterial nucleoid. Biophys. Chem. 73:23-29. [DOI] [PubMed] [Google Scholar]
- 30.Phillips, G. N., Jr. 1997. Structure and dynamics of green fluorescent protein. Curr. Opin. Struct. Biol. 7:821-827. [DOI] [PubMed] [Google Scholar]
- 31.Pollack, G. H. 2001. Cells, gels and the engines of life: a new, unifying approach to cell function. Ebner and Sons, Seattle, WA.
- 32.Shapiro, L., D. Kaiser, and R. Losick. 1993. Development and behavior in bacteria. Cell 73:835-836. [DOI] [PubMed] [Google Scholar]
- 33.Swaminathan, R., C. P. Hoang, and A. S. Verkman. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72:1900-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terry, B. R., E. K. Matthews, and J. Haseloff. 1995. Molecular characterisation of recombinant green fluorescent protein by fluorescence correlation microscopy. Biochem. Biophys. Res. Commun. 217:21-27. [DOI] [PubMed] [Google Scholar]
- 35.Thanbichler, M., P. H. Viollier, and L. Shapiro. 2005. The structure and function of the bacterial chromosome. Curr. Opin. Genet. Dev. 15:153-162. [DOI] [PubMed] [Google Scholar]
- 36.Tokuriki, N., M. Kinjo, S. Negi, M. Hoshino, Y. Goto, I. Urabe, and T. Yomo. 2004. Protein folding by the effects of macromolecular crowding. Protein Sci. 13:125-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verkman, A. S. 2002. Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27:27-33. [DOI] [PubMed] [Google Scholar]
- 38.Woldringh, C. L. 2002. The role of cotranscriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol. Microbiol. 45:17-29. [DOI] [PubMed] [Google Scholar]
- 39.Woldringh, C. L. 1994. Significance of plasmolysis spaces as markers for periseptal annuli and adhesion sites. Mol. Microbiol. 14:597-607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.