

Abstract
Many gram-negative bacteria harbor a copper/zinc-containing superoxide dismutase (CuZnSOD) in their periplasms. In pathogenic bacteria, one role of this enzyme may be to protect periplasmic biomolecules from superoxide that is released by host phagocytic cells. However, the enzyme is also present in many nonpathogens and/or free-living bacteria, including Escherichia coli. In this study we were able to detect superoxide being released into the medium from growing cultures of E. coli. Exponential-phase cells do not normally synthesize CuZnSOD, which is specifically induced in stationary phase. However, the engineered expression of CuZnSOD in growing cells eliminated superoxide release, confirming that this superoxide was formed within the periplasm. The rate of periplasmic superoxide production was surprisingly high and approximated the estimated rate of cytoplasmic superoxide formation when both were normalized to the volume of the compartment. The rate increased in proportion to oxygen concentration, suggesting that the superoxide is generated by the adventitious oxidation of an electron carrier. Mutations that eliminated menaquinone synthesis eradicated the superoxide formation, while mutations in genes encoding respiratory complexes affected it only insofar as they are likely to affect the redox state of menaquinone. We infer that the adventitious autoxidation of dihydromenaquinone in the cytoplasmic membrane releases a steady flux of superoxide into the periplasm of E. coli. This endogenous superoxide may create oxidative stress in that compartment and be a primary substrate of CuZnSOD.
In 1969, McCord and Fridovich reported the isolation from bovine erythrocytes of a protein that scavenged superoxide (29) by the following chemical reaction: O2− + O2− + 2H+ → H2O2 + O2. They subsequently demonstrated that this activity, which they designated superoxide dismutase (SOD), is ubiquitous among aerobic organisms. Eukaryotes contain a copper- and zinc-cofactored isozyme (CuZnSOD) in their cytosol and a manganese-cofactored isozyme in their mitochondria. Bacteria employ in their cytoplasms either manganese- or iron-cofactored enzymes that are structurally homologous to one another and to the eukaryotic mitochondrial manganoenzyme.
The discovery of SOD implied both that superoxide is formed as a by-product of aerobic metabolism and that, if it is not scavenged, it can harm cells. Subsequent studies have revealed that molecular oxygen adventitiously steals electrons from the reduced cofactors of redox enzymes, thereby forming superoxide in all aerobic organisms (19, 28). Further, superoxide can directly oxidize the iron-sulfur clusters of a family of dehydratases, inactivating the enzymes and blocking the pathways to which they belong (10, 12, 13). By rapidly scavenging superoxide, SOD protects these enzymes and permits metabolism to occur in environments that contain oxygen (19, 28).
In 1974 a CuZnSOD was isolated for the first time from a bacterium, Photobacterium leiognathi, a symbiont of the ponyfish (35). The structure of that enzyme closely resembled that of the eukaryotic CuZnSOD, and initially it was suspected that the gene had been laterally transferred from host to symbiont. However, CuZnSODs have since been discovered in all clusters of the Proteobacteria and in sequenced members of several other bacterial phyla. It now seems clear that eukaryotes inherited their CuZnSODs from the bacterial ancestor of the mitochondrion.
In gram-negative bacteria, CuZnSODs are invariably located in the periplasm. Thus, there must be conditions under which this compartment is exposed to superoxide. The enzymes that are known to release superoxide are all cytosolic, and O2− cannot cross membranes at physiological pH (22, 27); therefore, bacterial CuZnSOD must exist to scavenge O2− that is either generated inside the periplasm or that diffuses into it from outside the cell. Some studies have implicated periplasmic SODs in bacterial virulence, raising the prospect that these enzymes scavenge superoxide that is released by the NADPH oxidase of phagocytes (7, 9, 14, 23, 36, 43, 46). However, several observations suggest that this cannot be the sole role of the enzyme. First, periplasmic SODs are found in Caulobacter crescentus, a free-living organism (41), and in nonpathogenic strains of Escherichia coli (2). Second, Salmonella species synthesize both chromosomal and phage-encoded isozymes, and only the latter are required for pathogenesis (23, 46). Implicitly, the chromosomally encoded CuZnSOD, which is a close homologue of the E. coli enzyme, has a role unrelated to pathogenesis. Finally, Salmonella and E. coli mutants that lack periplasmic SODs exhibit a mild sensitivity to H2O2 in vitro (15). Although the basis of that sensitivity is not understood, its existence implies that superoxide must stress the periplasm even when the bacteria are grown in pure culture. This observation raises the prospect that periplasmic superoxide might be formed by the bacterium itself.
This process has been observed in one specialized situation. Huycke and colleagues found that the gram-positive bacterium Enterococcus faecalis, a natural heme auxotroph, releases superoxide into the medium when heme is not provided (17, 18). In the absence of this cofactor, cytochrome d oxidase cannot be activated, and respiration cannot proceed. The superoxide is apparently generated on the outer aspect of the cytoplasmic membrane as a vent for electrons that have entered the respiratory chain. Supplementation of hematin restores oxidase function and eliminates superoxide excretion.
In contrast, gram-negative bacteria that express periplasmic SOD can synthesize their own heme and do not face this dilemma. Nevertheless, we were interested in determining whether superoxide might be formed within the periplasm during aerobic growth. We report here that substantial superoxide is indeed released into this compartment as an incidental by-product of respiration, apparently due to the adventitious oxidation of menaquinone.
MATERIALS AND METHODS
Chemicals.
Cytochrome c (horse heart, type IV), superoxide dismutase (bovine erythrocytes), horseradish peroxidase, thiamine, Casamino Acids, ampicillin, chloramphenicol, kanamycin, potassium ferricyanide, EDTA, isopropylthiogalactoside, 30% hydrogen peroxide, NADH, plumbagin, lactose, and fumarate were from Sigma. Tryptone, yeast extract, and Bacto agar were purchased from Becton Dickinson. Potassium phosphate salts, ammonium sulfate, sodium citrate, sodium chloride, glucose, potassium cyanide, calcium chloride, magnesium sulfate, and Tris base were from Fisher. Amplex red and PicoGreen reagent were obtained from Molecular Probes, and Coomassie reagent and ovalbumin were from Pierce.
Strains.
Bacterial strains used in this study are listed in Table 1. Plasmids were transformed by electroporation. Mutations were introduced by P1 transductions (32) and selection on antibiotic-containing plates. The presence of mutant alleles was subsequently confirmed using screens for the appropriate phenotypic properties. Inheritance of the menA allele was verified by the inability of the mutant to grow on anaerobic glycerol-fumarate plates within 4 days. The ubiCA mutants were unable to grow on aerobic succinate plates, and they also exhibited a diminished rate of growth on aerobic LB plates. The menA ubiCA double mutants were unable to grow on aerobic LB plates at all. Enzymatic assay of NADH dehydrogenase II activity was used to confirm the presence of ndh mutations. Overproduction of SodC1 was verified by enzymatic assay. In combination with some mutations that were used in this study, the cyo and cyd mutations do not exhibit an unambiguous phenotype. The cyo cyd double mutants, however, can neither grow nor respire in aerobic media. Therefore, the individual mutations were validated by a subsequent transduction into the complementary single mutant to generate the double mutant and recreate this phenotype.
TABLE 1.
Strains and plasmids used
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Escherichia coli strains | ||
| AN387 | F−rpsL gal | 47 |
| SSK1 | AN387 plus menA::cam | P1(LC131) × AN387 |
| SSK2 | AN387 plus ΔubiCA::kan | P1(RKP4152) × AN387 |
| SSK3 | AN387 plus menA::cam ΔubiCA::kan | P1(RKP4152) × SSK1 |
| SSK4 | AN387 plus cydAB::cam | P1(KM38) × AN387 |
| SSK6 | AN387 plus (ahpC-ahpF′)Δkan::′ahpF katE12::Tn10 Δ(katG17::Tn10)1 menA::cam | P1(SSK1) × JI377 |
| SSK7 | AN387 plus Δ(cyoABCDE)456::kan | P1(KM39) × AN387 |
| SSK9 | AN387 plus sodC::spc menA::cam | P1(SSK1) × AS454 |
| SSK10 | AN387 plus ΔubiCA::kan ndh::cam | P1(MW03) × SSK1 |
| SSK12 | AN387 plus ΔcydAB::cam Δ(cyoABCDE)456::kan | P1(SSK4) × SSK7 |
| SSK13 | AN387 plus ΔcydAB::cam | P1(GO105) × AN387 |
| SSK14 | AN387 plus menA::cam ΔcydAB::cam | P1(SSK1) × SSK13 |
| SSK15 | RKP4152 plus malE52::Tn10 | P1(TST1) × RKP4152 |
| SSK16 | AN387 plus ΔcydAB::cam malE52::Tn10 ΔubiCA::kan | P1(SSK15) × SSK4 |
| SSK18 | AN387 plus sodC::spc nuo zej-223::Tn10 | P1(JI301) × AS454 |
| SSK19 | AN387 plus sodC::spc nuo zej-223::Tn10 ndh::cam | P1(MW03) × SSK18 |
| SSK20 | AN387 plus sodC::spc ndh::cam | P1(MW03) × AS454 |
| SSK21 | AN387 plus sodC::spc ΔubiCA::kan malE52::Tn10 | P1(SSK15) × AS454 |
| SSK22 | AN387 plus sodC::spc ΔcydAB::cam | P1(SSK4) × AS454 |
| SSK23 | AN387 plus sodC::spc Δ(cyoABCDE)456::kan | P1(SSK7) × AS454 |
| SSK24 | AN387 plus sodC::spc Δ(cyoABCDE)456::kan ΔcydAB::cam | P1(SSK4) × SSK23 |
| ALN21 | AN387 plus nuo zej-223::Tn10 | Lab collection |
| AS454 | AN387 plus sodC::spc | 15 |
| JI301 | AN387 plus nuo zej-223::Tn10 | 38 |
| JI377 | AN387 plus (ahpC-ahpF′) Δkan::′ahpF katE12::Tn10 Δ(katG17::Tn10)1 | 37 |
| SLC22 | AN387 plus ndh::cam | 51 |
| GO105 | Δ(cydAB′)455 zbg-2200::kan cyo-123 recA srlC300::Tn10 pRG110 | Bob Gennis |
| KM38 | UM1 plus cydAB::cam | 37 |
| KM39 | katG14 katE1 Δ(cyoABCDE)456::kan | 37 |
| LC132 | lacI rrnB ΔlacZ hsdK ΔaraBAD ΔrhaBAD menA::cam | 38 |
| MG1655 | Wild-type K-12 strain | 3 |
| MW03 | recA56 srlC300::Tn10 thr-1 leuB6 proA2 his-4 thi-1 argE2 lacY1 galK2 rpsL2 supE44 ara-14 xyl-15 mtl-1 tsx-33 ndh::cam pMW01 | 38 |
| RKP4152 | ΔubiCA::kan | 40 |
| TST1 | araD139 Δ(argF-lac)169 flhD5301 Δ(fruK-yeiR) relA1 rpsL150 rbsR22 malE52::Tn10 Δ(fimB-fimE)632 deoC1 | E. coli Genetic Stock Center |
| UM1 | katE1 katG14 lacY rpsL thi-1 | 26 |
| Enterococcus faecalis strain | ||
| OG1RF | Wild type | 18 |
| Plasmids | ||
| pBR322 | Ampr | 4 |
| pBR328 | Ampr | 4 |
| pRG110 | pBR322 plus 5.8-kb cyoABCD insertion | 1 |
| pMW01 | pBR322 plus EcoRI-SalI fragment containing ndh | 5 |
| psodCI | pWKS30 sodCI | 23 |
| pWKS30 | Ampr | 49 |
| pKK1 | pBR328 plus sodB insert | 21 |
Buffers, media, and growth conditions.
LB medium (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl; pH 7.0) (32) was routinely supplemented with 0.2% glucose (or 0.2% lactose plus 0.7 mM isopropyl-β-d-thiogalactopyranoside [IPTG] if specified). Most bacterial growth in defined medium employed minimal A salts (7.5 mM ammonium sulfate, 2 mM sodium citrate, 33 mM potassium dihydrophosphate, 60 mM potassium hydrophosphate, 1 mM magnesium sulfate, 5 mg/liter thiamine; pH 7.3) (32) that were supplemented with 0.2% Casamino Acids, 0.5 mM tryptophan, and 0.2% glucose. Where indicated below, Casamino Acids were replaced with a 0.5 mM concentration of specified amino acids. Menaquinone mutants were screened in medium containing minimal A salts, 0.6% glycerol, and 30 mM fumarate, and aerobic respiratory proficiency was tested in minimal A salts containing 30 mM succinate. Ampicillin (0.1 mg/ml) was added to cultures of plasmid-bearing strains. Washing buffer (WB) contained 7.5 mM ammonium sulfate, 6 mM sodium chloride, 33 mM potassium dihydrophosphate, 60 mM potassium hydrophosphate, and 1 mM magnesium, adjusted to pH 7.3. Phosphate buffer contained 50 mM potassium phosphate at pH 7.8.
Aerobic cultures were grown in flasks with vigorous shaking at 37°C. Anaerobic cultures were grown in an anaerobic chamber (90% nitrogen, 5% hydrogen, 5% carbon dioxide; Coy Laboratory Products). To remove oxygen, anaerobic buffers and media were moved into the chamber immediately after being autoclaved, and they were allowed to equilibrate with the anaerobic atmosphere for at least 24 h prior to use.
Detection and measurement of extracellular superoxide.
The production of extracellular superoxide was monitored using the ability of superoxide to reduce ferricytochrome c that was added to the cell suspension. This approach was used previously to detect the superoxide that is abundantly released by Enterococcus faecalis (18). The method was modified here to permit the quantitation of the lesser amount that is generated by E. coli and to distinguish superoxide that is generated directly by the bacteria from that which is formed by the autoxidation of excreted metabolites.
To measure total extracellular superoxide production, cells either from exponential-phase (optical density at 600 nm [OD600] of 0.25) or stationary-phase (OD600 of 2.4) cultures were centrifuged, washed with WB, and resuspended to an OD600 of 0.2 in two 50-ml flasks, each containing 10 ml of prewarmed WB supplemented with 0.2% glucose and 20 μM cytochrome c. (Complex medium cannot be used, because medium components directly reduce cytochrome c.) Superoxide dismutase (30 U/ml) was added to one of the two flasks, and they were incubated at 37°C in a shaking water bath. At intervals, 1.5 ml of cell suspension was withdrawn from each flask and filtered using syringe filters. The filtrates were kept on ice, and the amount of reduced cytochrome c was determined as soon as possible (within 2 to 3 min) by the method described below.
To quantify superoxide formation by excreted metabolites, cell suspensions were incubated without the addition of cytochrome c. At various time points, cells were removed by filtration, cytochrome c was added to the filtrate, each aliquot was split, and SOD was added to one of each of the paired samples. The samples were then incubated at 37°C for 15 min, which is sufficient to essentially complete the autoxidation of excreted metabolites.
To quantify the amount of reduced cytochrome c in each sample, the absorbance spectrum was recorded between 570 and 530 nm. Then 0.2 mM of potassium ferricyanide was added to the filtrates to oxidize cytochrome c, and the spectra were again recorded. The amount of reduced cytochrome c was calculated using the ferricyanide-induced absorbance change at 550 nm (Δɛ550 upon reduction of cytochrome c, 0.21 mM−1 cm−1) relative to the 556.5-nm isobestic point. These reduced-minus-oxidized spectra provided the precision that was needed to obtain quantitatively reproducible measurements. The fraction of cytochrome c reduction that had been mediated by superoxide was then determined by comparing the degrees of reduction in the paired samples with and without SOD (Fig. 1).
FIG. 1.

Detection of extracellular superoxide released by exponentially growing cultures of the wild-type strain AN387. (A) Total reduction of extracellular cytochrome c in the absence (line 1) and presence (line 2) of exogenous superoxide dismutase. Line 3 indicates the calculated release of superoxide. The superoxide formed by the excreted metabolite of panel B has been subtracted from the overall rate of superoxide-mediated cytochrome c reduction. (B) Superoxide formation by an excreted metabolite. At time zero, the wild-type strain AN387 was suspended in fresh medium.
The value produced in this way revealed the total amount of extracellular superoxide that had been produced in cell cultures, the amount that had been generated by the autoxidation of metabolites, and, by subtraction, the amount produced directly by the cells. We anticipate that essentially all of the superoxide that is produced in the periplasm will be detected by this assay. While superoxide can spontaneously dismute, reaction with 20 μM cytochrome c (k = 2.5 × 105 M−1 s−1) (11) is overwhelmingly favored. Similarly, it is unlikely that significant superoxide will dismute before it leaves the periplasm. Using the measured rate of superoxide formation and the dismutation rate constant, we calculate that the half-life of superoxide should exceed 0.6 s—which, given the high diffusion coefficient of small molecules (∼10−5 cm2/s), implies a mean single-dimension diffusion distance of 35 μm. The distance between inner and outer membranes is only about 0.1 μm, so most superoxide molecules should persist for long enough to encounter a porin and efflux from the cell. Periplasmic superoxide formation would be underreported only if a periplasmic molecule reacted catalytically with superoxide at a high rate. No such molecule (other than SOD) is known.
Statistical analysis.
All experimental results were determined multiple times with discrete independent cultures. Where rates of superoxide formation differed significantly from those of wild-type cells, the rates were measured four to five times, and error bars represent the standard deviations of those data.
Measurement of hydrogen peroxide formation.
Total H2O2 formation was measured using the fluorescent dye Amplex red (37). This method is subject to some interference by medium components and cannot be performed at all in complex media; therefore, measurements were performed in minimal glucose medium that was supplemented with His, Pro, Leu, Arg, Thr, Phe, Tyr, and Trp. These amino acids are sufficient for good growth of strains that cannot scavenge H2O2. Cells (strains JI377 and SSK6) were grown for at least five generations in this medium to an OD600 of 0.25. They were then pelleted at room temperature and washed once with WB. The cells were resuspended to an OD600 of 0.1 in 10 ml of the same prewarmed medium in a 37°C shaking water bath. Every 3 min, 1.5 ml of cell suspension was withdrawn and filtered using a syringe filter. To 0.5 ml of the cell filtrate were added 1 ml of 0.2 mM solution of Amplex red in 50 mM phosphate buffer (pH 7.8) and then 0.5 ml of horseradish peroxidase (0.02 mg/ml). Fluorescence was measured after 30 seconds in a Shimadzu RF-Mini 150 fluorometer. Fluorescence values were converted to H2O2 concentration using a standard curve obtained from additions of known amounts of H2O2 to growth medium.
Respiration.
Oxygen consumption by growing cells was measured using a Clark oxygen electrode. Cells were centrifuged, washed, and resuspended in WB plus 0.2% glucose at an OD600 of 0.1. Measurements were carried out at 37°C until 50% of the oxygen was consumed.
Enzyme assays.
NADH dehydrogenase II activity was monitored spectrophotometrically by the oxidation of NADH by membrane vesicles. The vesicles were obtained from cells collected at mid-exponential phase and were washed twice with cold 50 mM phosphate buffer (pH 7.8). The concentrated cells were then lysed with a French press and centrifuged for 20 min at 17,000 × g at 4°C to remove cell debris. The supernatant was decanted and centrifuged at 4°C for 1.5 h at 100,000 × g. Vesicles were resuspended in 5 ml of cold phosphate buffer and were kept on ice for about 16 h to deactivate NADH dehydrogenase I (30). NADH oxidation was monitored by the decrease in absorbance of NADH at 340 nm in the presence of 3 mM KCN and 0.1 mM plumbagin. SodCI activity was assayed according to the xanthine oxidase/cytochrome c method (29); in parallel samples, 2 mM KCN, an inhibitor of CuZnSOD, was added to distinguish CuZnSOD activity from that of the cytosolic manganese- and iron-containing SODs. Total protein was determined using Coomassie reagent.
RESULTS
Detection of extracellular superoxide.
When cytochrome c was added to a culture of growing cells, it was continuously reduced, as determined by an increase in absorbance at 550 nm (Fig. 1A, line 1). Reduction occurred at a rate proportionate to cell density, approximately 6 pmol per 0.1 OD600 unit of cells. The addition of superoxide dismutase to the culture diminished the rate by about 30% (line 2), indicating that approximately one-third of the cytochrome c reduction was mediated by superoxide and that the remainder occurred when either the growing cells or an excreted metabolite directly transferred an electron to the cytochrome c.
We focused upon the extracellular superoxide. None was produced by sterile medium. Therefore, in principle it could arise from two sources: through its direct release by the cells and through the autoxidation of an excreted metabolite. When cells were removed by filtration and cytochrome c was added to the filtrate, a small amount of SOD-inhibitable cytochrome c reduction still occurred (Fig. 1B). Because superoxide itself spontaneously dismutes within seconds of its formation, this persistent reduction indicated that a low-molecular-weight metabolite was excreted by the cells and gradually reacted with oxygen in the medium, producing superoxide. Kinetic analysis (see Materials and Methods) indicated that this metabolite was responsible for only a minor fraction of the extracellular superoxide that was released by the cultures. The identity of the excreted compound was not determined. We inferred that the remaining superoxide (Fig. 1A, line 3) was either released directly by the bacteria or produced by autoxidation of a metabolite with such a short half-life that it did not survive the filtration process.
Superoxide is formed within the periplasm.
In subsequent analyses, the superoxide that was formed by the filterable metabolite was measured and subtracted from total superoxide formation. If the residual superoxide was released directly by cells, then its release might be blocked by the presence of SOD within the periplasm. Because the periplasmic SOD of E. coli is synthesized only in stationary phase, the Salmonella enterica serovar Typhimurium sodC1 gene, placed behind a lac promoter, was transformed on a plasmid into E. coli. The consequent periplasmic SOD activity was measured in a sodA sodB sodC strain that lacks interfering SOD isozymes and was found to be about 10 U/mg. For the sake of comparison, E. coli and Salmonella laboratory strains typically contain about 3 U/mg and 15 U/mg of periplasmic SOD, respectively, when they are in stationary phase.
The SodC-expressing plasmid eliminated the release of superoxide from wild-type E. coli (Fig. 2A), indicating that the superoxide is released into the medium from the periplasm. In contrast, sevenfold overproduction of cytosolic FeSOD did not diminish superoxide release, and mutations that eliminated cytosolic FeSOD and MnSOD activities caused at most a marginal (<10%) increase in superoxide release (data not shown). Thus, the superoxide that is released by the cell must be formed within the periplasm.
FIG. 2.

Time courses of superoxide release. (A) Expression of periplasmic superoxide dismutase blocks the release of superoxide. Open squares, AN387 pCKR101 (empty vector). Filled squares, AN387 psodCI (overexpressing sodC). (B) Effect of oxygen concentration upon periplasmic superoxide production by wild-type (AN387) and menA (SSK1) E. coli strains and by a wild-type (OG1RF) E. faecalis strain. Gray bars reflect superoxide formation in air-saturated medium, and black bars reflect superoxide formation when 100% oxygen was bubbled through the medium.
The plasmid had no effect upon superoxide formation by the filterable metabolite (data not shown). This result is consistent with our previous observation that periplasmic SOD cannot scavenge superoxide that is formed in the extracellular medium before it reacts with extracellular cytochrome c (22).
Periplasmic superoxide formation is accelerated by high oxygen concentration.
The previous experiments were conducted with cells suspended in air-saturated growth medium. When pure oxygen was bubbled into the cultures, increasing the dissolved oxygen concentration by four- to fivefold, the rate of periplasmic superoxide production increased proportionately (Fig. 2B). This result suggests that the superoxide is formed by the adventitious oxidation of a reduced molecule.
Prior reports demonstrated that heme-starved E. faecalis generates extracellular superoxide at a rate which is far above what we observed with E. coli (17, 18). To ensure that the discrepancy in rates did not arise from differences in methodology, we measured superoxide production by E. faecalis under the same conditions that we used for E. coli. Indeed, the rate for E. faecalis was approximately 25-fold faster than that for E. coli. In contrast to the case with E. coli, superoxide formation by E. faecalis cultures was not significantly accelerated by hyperoxygenation. It is assumed that heme-starved E. faecalis generates superoxide at some site on its incomplete electron transport chain. The unresponsiveness to oxygen concentration indicates either that electron transfer to oxygen is not the rate-limiting step in superoxide formation or that the autoxidizing component has a saturable oxygen-binding site. Therefore, it seems likely that superoxide production within the periplasm of E. coli and on the surface of E. faecalis occurs by distinct mechanisms.
Superoxide is released by the respiratory chain.
One candidate for the site of superoxide formation in E. coli was the pyroquinoline quinone (PQQ) moiety of periplasmic glucose dehydrogenase. E. coli does not synthesize PQQ, but E. coli scavenges it from laboratory media, including the LB medium in which cells had been precultured prior to the measurement of superoxide production. PQQ autoxidizes rapidly, which led to the misidentification of purified glucose dehydrogenase as a glucose oxidase.
However, superoxide formation by intact cells was not diminished when the cells were precultured in defined, PQQ-free media (data not shown). Further, supplementation with PQQ or the use of lactose instead of glucose media did not alter the rates of superoxide production. Thus, glucose dehydrogenase is evidently not the primary superoxide source.
The respiratory chain lies within the cytoplasmic membrane, which raised the possibility that superoxide might be formed by autoxidation of a component that faces the periplasm. To test this possibility, we examined mutant strains that were severely defective in respiration by virtue of mutations in both quinones (menA ubiCA mutant) or both NADH dehydrogenase enzymes (nuo ndh mutant). The former strain grows poorly in air, so to permit comparisons with a wild-type strain, all three strains were precultured anaerobically and then shifted to aerobic medium for the period of superoxide measurement. The wild-type strain generated superoxide at a rate 30% higher than if it had been precultured aerobically. The nuo ndh mutant produced superoxide at a far lower rate (Fig. 3). Virtually no superoxide was released by a menA ubiCA mutant which possesses both NADH dehydrogenases but lacks ubiquinone and menaquinone, the electron carriers that deliver electrons from respiratory dehydrogenases to the cytochrome oxidases.
FIG. 3.

Superoxide is not released from cells that lack respiratory NADH dehydrogenases (strain ALN21, the nuo ndh mutant) or quinones (strain SSK3, the menA ubiCA mutant). See the text for growth conditions.
In aerobic E. coli, ubiquinone is the predominant quinone, while menaquinone (and its derivative demethylmenaquinone) comprises about 20% of the quinone pool (45, 47, 50). A ubiCA single mutant exhibited a 10-fold decrease in respiration and a diminished growth rate relative to those of wild-type cells, but the formation of periplasmic superoxide was elevated significantly (Fig. 4A). This result suggested that when ubiquinone is absent, the electron flow is diverted to a more autoxidizable component of the respiratory chain. Indeed, mutants lacking menaquinone (menA mutants) exhibited a four- to fivefold diminution in superoxide formation, despite robust growth and a wild-type respiratory rate (Fig. 4A). These results indicated that reduced menaquinone either transferred electrons to periplasmic oxygen directly or delivered them to an enzyme complex that did.
FIG. 4.

Effects of the deletion and overexpression of different components of the respiratory chain on superoxide release. (A) AN387 (wild type), SSK1 (menA), and SSK2 (ubiCA) were precultured aerobically and then assayed for superoxide release. (B) The congenic strains AN387 (wild type), JI301 (nuo), SLC22 (ndh), and ALN21 (nuo ndh) were precultured aerobically and then assayed for superoxide release. (C) The rates of superoxide formation were compared among the wild-type (AN387), menA (SSK1), and ubiCA (SSK2) strains that contained only a vector (pBR322 [striped bars]) or a plasmid overexpressing ndh (pMW01 [black bars]). The stippled bar shows results for the ubiCA ndh double mutant without a plasmid. (D) The rates of growth (gray bars) and of superoxide formation (black bars) were compared among cyd (SSK4) and cyo cyd (SSK12) mutants and normalized to that of the wild-type (Wt) strain. Strains contained either an empty vector or plasmid pRG110 overexpressing the cyoABCD operon. Data are shown from a single typical experiment, so that rates of growth and of superoxide formation are from a common culture; error bars reflect the range of variation from multiple time points.
Localizing the site of superoxide formation.
Menaquinone has a lower reduction potential (−0.074 V) than ubiquinone (+0.113 V) and thus is a stronger electron donor. It is the obligatory carrier in anaerobic respiratory processes that employ low-potential terminal acceptors, such as fumarate. The value of its synthesis in aerobic cells is unclear. During growth in aerobic glucose media, both α-glycerolphosphate dehydrogenase and NADH dehydrogenase activities are relatively abundant and are capable of reducing menaquinone. The nuo ndh mutants exhibited a low level of residual H2O2 formation. In this medium NADH dehydrogenase II is much more active than NADH dehydrogenase I; accordingly, we found that ndh mutants were twofold diminished in periplasmic superoxide formation but that nuo mutants were unaffected (Fig. 4B). These results may simply reflect the role of these dehydrogenases in delivering electrons to menaquinone, or they may reveal that menaquinone autoxidizes while complexed to the dehydrogenase active sites. The latter possibility parallels the behavior of ubiquinone while bound in the semiquinone form to the Qo and Qi sites of the mammalian bc1 complex (16, 44). However, 10-fold overproduction of NdhII, which might be expected to increase the concentration of NdhII-complexed menaquinone, did not increase superoxide formation (Fig. 4C).
Elimination of fumarate reductase had no impact upon superoxide production (data not shown). The same was true of cytochrome o oxidase, indicating that neither of these two enzymes was the site of formation (Fig. 4D). Mutations in cyd, encoding cytochrome d oxidase, caused lower rates of respiration and growth and a twofold acceleration of superoxide formation. The effect was amplified when both cyo and cyd were eliminated. A plasmid that overproduced cytochrome o oxidase restored wild-type respiration and growth rates and suppressed superoxide formation.
Collectively, these data are consistent with the notion that mutations that alter the redox state of menaquinone—either by affecting electron delivery to it or transfer of electrons away from it—have congruent impacts upon superoxide formation. Further, we have been unable to identify any respiratory complex that is essential for superoxide production. The simplest explanation is that uncomplexed dihydromenaquinone itself is the predominant superoxide source.
Menaquinone is not the primary source of endogenous hydrogen peroxide.
The rate at which E. coli generates hydrogen peroxide can be measured using an ahp katG katE strain which lacks the three primary enzymes that scavenge H2O2 (38). Under the conditions of these experiments, this strain generated H2O2 at a rate of 25 pmol per min per 0.1 OD unit of cell suspension. At the same time, extracellular superoxide was formed at a rate of 5 pmol per min per 0.1 OD unit. Upon dismutation, the latter superoxide would account for only 2.5 pmol of H2O2, or about 10% of the total. Further, while the addition of a menA mutation eliminated superoxide formation, H2O2 production was essentially unaffected (a <20% change, the limit of the experimental precision [data not shown]). Thus, while the primary process of endogenous H2O2 formation remains unidentified, it is clearly distinct from the process of periplasmic O2− formation.
Periplasmic superoxide formation by stationary-phase cells.
Periplasmic superoxide dismutase is regulated by RpoS and is therefore not synthesized in LB medium until E. coli enters stationary phase (15). When these stationary-phase cells were resuspended in medium lacking a fresh carbon source, no superoxide production was detected. This was true even when sodC was eliminated. Thus, at least under this stationary-phase condition, E. coli does not produce significant periplasmic superoxide.
When these cells were resuspended in medium containing glucose, however, respiration immediately resumed, and superoxide was exuded from the sodC mutants (Fig. 5). In this situation, wild-type (SodC1+) cells did not release superoxide, apparently because it was scavenged by the periplasmic SOD. Surprisingly, the refed stationary-phase cells initially generated superoxide even when menA was deleted, suggesting that the mechanism of formation differed from that of exponentially growing cells. The nuo ndh and cyo cyd mutants did not release superoxide, however, indicating that even under this circumstance, the superoxide depended upon flux through the respiratory chain.
FIG. 5.

Superoxide release by stationary-phase cells depends upon respiration but does not require menaquinone. Black bars, superoxide release by exponential-phase cells; gray bars, superoxide release by stationary-phase cells immediately upon resuspension in glucose medium. The sodC mutant AS454 and the respiratory mutants SSK3, SSK9, SSK18, SSK19, SSK20, SSK21, SSK22, SSK23, and SSK24 were used. Note that no superoxide was detected in the medium of sodC+ derivatives of any of these stationary-phase cells.
DISCUSSION
A role for periplasmic superoxide dismutase.
Early investigations into bacterial periplasmic superoxide dismutases focused on the possibility that they might defend pathogens against superoxide that they encounter as part of the host immune response. However, that idea was inadequate to rationalize the presence of the enzyme in nonpathogenic strains of E. coli and in free-living bacteria, such as Caulobacter (42). The discovery that aerobic E. coli releases superoxide into its periplasm provides an explanation for its synthesis of periplasmic superoxide dismutase.
The rate of periplasmic superoxide formation is quite high: about 3 μM/s, when normalized to the estimated periplasmic volume. That value is comparable to the 5 μM/s that has been estimated for superoxide formation in the cytosol (20). The latter compartment requires the protection of MnSOD and/or FeSOD, as mutants that lack these SODs are defective in aerobic metabolism and biosynthesis, due to the inactivation of key enzymes (6). In contrast, we have not observed any growth defect in sodC mutants (15). In part, this may be because the efflux of periplasmic superoxide through the outer membrane pores is rapid and may keep periplasmic superoxide levels moderate even in sodC mutants. However, the larger puzzle is the identity of the periplasmic or cell surface biomolecules that periplasmic SOD serves to protect.
Most of the enzymes that are damaged by cytosolic superoxide belong to the family of dehydratases that have exposed [4Fe-4S] clusters. Superoxide directly oxidizes these exposed clusters, thereby destabilizing them and leading to inactivation of the enzymes and the pathways to which they belong (10, 12, 13, 24, 25). However, the periplasm is not known to contain enzymes that belong to this family. There are respiratory complexes embedded in the cytoplasmic membrane that utilize Fe-S clusters, but these clusters are typically buried within polypeptides and are not affected by superoxide. Evidently, a second class of superoxide-sensitive enzymes or structural molecules is present in the periplasm. The identification of this molecule(s) is a priority, both to complete our understanding of endogenous superoxide stress in the periplasm and to gain insight into the mechanism by which phagocytic superoxide might incapacitate pathogenic bacteria.
Mechanism of superoxide formation.
Superoxide is a charged species at neutral pH; therefore, it should not efficiently cross membranes (22, 27). This possibility has nevertheless been raised as a possible explanation for the role that Saccharomyces cerevisiae cytosolic SOD plays in protecting mitochondrial enzymes (48). Our own calculations suggest that the release of superoxide from the E. coli cytosol into the periplasm should be vanishingly rare (<1 in 106). This prediction was supported by the fact that genetic alternations that either raised or lowered the amount of cytosolic SOD had no detectable impact upon the rate of superoxide release into the external medium. Therefore, most or all periplasmic superoxide must be formed within the periplasm.
Molecular oxygen is a relatively poor univalent electron acceptor (midpoint potential [Em] = −0.16 V), so only good univalent electron donors can transfer an electron to it. Several cytosolic enzymes that can generate superoxide have been identified, and in all cases a reduced flavin (univalent Em ≈ −0.22 V) is the direct electron donor (31). There are no flavoenzymes in the periplasm of E. coli. However, the chemical principle was sustained by our finding that in exponentially growing cells, the donor is menaquinone, another good univalent reductant (Em = −0.074 V). In contrast, ubiquinone (Em = +0.113 V) seemed uninvolved, despite its greater abundance than menaquinone.
There are three mechanisms by which menaquinone might deliver electrons to oxygen, and at this point we cannot exclude any of them. The first is that dihydromenaquinone might transfer an electron directly, generating superoxide and a menasemiquinone radical that would subsequently transfer an electron to a second molecule of oxygen. Electron spin restrictions may impede the transfer of both electrons to the same molecule of oxygen (with H2O2 as a product): the orbital structure of oxygen requires that it accept electrons only in univalent steps, and the superoxide-menasemiquinone collision complex may not survive long enough to permit the second transfer reaction. Thus, dihydromenaquinone oxidation may predominantly generate two distinct molecules of superoxide.
A second mechanism would be driven by reaction between reduced and oxidized menaquinones, generating two molecules of menasemiquinone (i.e., comproportionation), as follows:
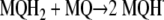 |
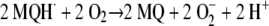 |
Oxygen, a radical species itself, reacts much more rapidly with semiquinones than with dihydroquinones (34). This fact may argue against this mechanism, as this model might predict that comproportionation comprises the rate-limiting step. In contrast, we observed that oxygen was a limiting reactant.
The third mechanism also emphasizes the autoxidizability of semiquinone species. Semiquinones are formed as momentary intermediate species when quinones are divalently reduced by dehydrogenases, and they are also transiently formed when dihydroquinones pass their electrons to metal centers in terminal oxidases. Much of the superoxide that is formed by mammalian mitochondria has been ascribed to the autoxidation of the ubisemiquinone radicals that are formed during the catalytic cycle of the bc1 complex (16, 44). The dependence of superoxide formation upon oxygen concentration would not be an objection to this model, since the radical intermediates are so fleeting that their ability to be oxidized depends upon the concentration of the oxidant. However, we found that each of the major respiratory complexes could individually be removed without diminishing periplasmic superoxide production. We tentatively conclude that unbound dihydromenaquinone is likely to be the autoxidizing species.
Interestingly, Huycke and colleagues observed that E. faecalis generates superoxide when its respiratory chain lacks a functional cytochrome d oxidase and that this activity requires demethylmenaquinone (18). The parallel to our observations is striking, but some differences suggest that the mechanisms may be distinct. First, E. faecalis forms superoxide 25 times more rapidly than does E. coli. E. coli strains did not approach that rate even when cytochrome oxidases were deleted to force electron accumulation on menaquinone. Second, superoxide formation in E. coli occurred in proportion to oxygen concentration, which is what one would expect if the rate-limiting step were the adventitious oxidation of a reduced electron carrier. However, in E. faecalis, superoxide formation was not increased when oxygen levels were elevated. These observations suggest the possibility that E. faecalis, a natural heme auxotroph that must frequently find itself in heme-deficient environments, might have a respiratory enzyme that catalyzes quinone oxidation as a way to discharge an incomplete electron transport chain. Siegele et al. suggested that in E. coli cydCD mutants, the heme-less cytochrome d oxidase might do this directly (39). However, we did not observe any increase in periplasmic superoxide formation in these mutants (data not shown).
Finally, we observed that when stationary-phase cells were refed glucose, superoxide was initially formed at substantial rates even in strains lacking menaquinone. Thus, menaquinone oxidation is not the sole source of periplasmic superoxide in E. coli. This superoxide formation in refed cells was eliminated by combinations of mutations that either blocked electron entry into the respiratory chain (nuo ndh mutant) or that blocked electron flow out of it (cyo cyd mutant). This observation suggests that the autoxidizing species may be a partially reduced enzyme, as it fits the expectation that enzymatic semiquinones are most abundant when enzymes are actively turning over. The structure of the respiratory chain in stationary-phase cells is not well studied, and we have not analyzed this situation further. However, it is notable that even in exponentially growing cells, the absence of menaquinone did not completely eliminate superoxide formation. Therefore, the superoxide source in the refed cells could be the same residual source that is detected in the menaquinoneless mutants.
Why is periplasmic SOD induced only in stationary phase?
Like many oxidative defenses, periplasmic SOD is positively regulated by the RpoS system and is strongly induced in stationary phase. However, in our experiments, the highest yield of superoxide was obtained when cells were growing exponentially; stationary-phase cells evolved periplasmic superoxide only after they were diluted into fresh medium. Thus, the timing of SOD synthesis appeared not to correlate with superoxide formation. This apparent contradiction may turn on the reason that cells enter stationary phase. While in our experiments growth stopped because carbon sources were exhausted, in natural habitats cells may also enter stationary phase for lack of a phosphorus, nitrogen, or sulfur source. If a carbon source is present, superoxide formation might still occur. Indeed, some recent work by Moreau indicates that oxidative stress might be especially severe under these conditions (33).
We tested this idea by culturing sodC mutants in a defined glucose medium containing limiting phosphate. When these cells stopped growing, they were diluted into phosphate-free medium, and superoxide release was measured. Periplasmic superoxide formation occurred at a rate that was 50 to 60% of that of exponentially growing cells (data not shown).
Still, it is not obvious why periplasmic SOD would not be made in exponentially growing cells. We speculate that cells may be particularly vulnerable to oxidative damage in stationary phase; injuries may be most consequential when cells lack the energy and material to repair or replace the damaged molecules. In fact, several other antioxidant enzymes are strongly induced in stationary phase as part of the RpoS regulon, including catalase, exonuclease III, and the iron-sequestering protein Dps (8).
Acknowledgments
This work was supported by Public Health Service grant GM49640 from the National Institutes of Health.
We are grateful to Mark Huycke, Bob Gennis, Jim Slauch, Peter Loewen, and Robert Poole for providing strains that were used in this study.
REFERENCES
- 1.Au, D. C.-T., and R. B. Gennis. 1987. Cloning of the cyo locus encoding the cytochrome o terminal oxidase complex of Escherichia coli. J. Bacteriol. 169:3237-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benov, L. T., and I. Fridovich. 1994. Escherichia coli expresses a copper- and zinc-containing superoxide dismutase. J. Biol. Chem. 269:25310-25314. [PubMed] [Google Scholar]
- 3.Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453-1474. [DOI] [PubMed] [Google Scholar]
- 4.Bolivar, F., R. L. Rodriguez, P. J. Green, M. C. Betlach, H. L. Heyneker, H. W. Boyer, J. H. Crosa, and S. Falkow. 1977. Construction and characterization of new cloning vehicles. I. Ampicillin-resistant derivatives of plasmid pMB9. Gene 2:95-113. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun, M. W., and R. B. Gennis. 1993. Demonstration of separate genetic loci encoding distinct membrane-bound respiratory NADH dehydrogenases in Escherichia coli. J. Bacteriol. 175:3013-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carlioz, A., and D. Touati. 1986. Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? EMBO J. 5:623-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Groote, M. A., D. Granger, Y. Xu, G. Campbell, R. Prince, and F. C. Fang. 1997. Periplasmic superoxide dismutase protects Salmonella from products of phagocyte NADPH-oxidase and nitric oxide synthase. Proc. Natl. Acad. Sci. USA 94:13997-14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenstark, A., M. J. Calcutt, M. Becker-Hapak, and A. Ivanova. 1996. Role of Escherichia coli rpoS and associated genes in defense against oxidative damage. Free Radic. Biol. Med. 21:975-993. [DOI] [PubMed] [Google Scholar]
- 9.Farrant, J. L., A. Sansone, J. R. Canvin, M. J. Pallen, P. R. Langford, T. S. Wallis, G. Dougan, and J. S. Kroll. 1997. Bacterial copper- and zinc-cofactored superoxide dismutase contributes to the pathogenesis of systemic salmonellosis. Mol. Microbiol. 25:785-796. [DOI] [PubMed] [Google Scholar]
- 10.Flint, D. H., J. F. Tuminello, and M. H. Emptage. 1993. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268:22369-22376. [PubMed] [Google Scholar]
- 11.Fridovich, I. 1985. Xanthine oxidase, p. 51-53. In R. A. Greenwald (ed.), Handbook of methods for oxygen radical research. CRC Press, Boca Raton, Fla.
- 12.Gardner, P. R., and I. Fridovich. 1991. Superoxide sensitivity of the Escherichia coli 6-phosphogluconate dehydratase. J. Biol. Chem. 266:1478-1483. [PubMed] [Google Scholar]
- 13.Gardner, P. R., and I. Fridovich. 1991. Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 266:19328-19333. [PubMed] [Google Scholar]
- 14.Gee, J. M., M. W. Valderas, M. E. Kovach, V. K. Grippe, G. T. Robertson, W.-L. Ng, J. M. Richardson, M. E. Winkler, and R. M. Roop II. 2005. The Brucella abortus Cu,Zn superoxide dismutase is required for optimal resistance to oxidative killing by murine macrophages and wild-type virulence in experimentally infected mice. Infect. Immun. 73:2873-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gort, A. S., D. M. Ferber, and J. A. Imlay. 1999. The regulation and role of the periplasmic copper,zinc superoxide dismutase of Escherichia coli. Mol. Microbiol. 32:179-191. [DOI] [PubMed] [Google Scholar]
- 16.Han, D., E. Williams, and E. Cadenas. 2001. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 353:411-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huycke, M. M., W. Joyce, and M. F. Wack. 1996. Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J. Infect. Dis. 173:743-746. [DOI] [PubMed] [Google Scholar]
- 18.Huycke, M. M., D. Moore, W. Joyce, P. Wise, L. Shepard, Y. Kotake, and M. S. Gilmore. 2001. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol. Microbiol. 42:729-740. [DOI] [PubMed] [Google Scholar]
- 19.Imlay, J. A. 2003. Pathways of oxidative damage. Annu. Rev. Microbiol. 57:395-418. [DOI] [PubMed] [Google Scholar]
- 20.Imlay, J. A., and I. Fridovich. 1991. Assay of metabolic superoxide production in Escherichia coli. J. Biol. Chem. 266:6957-6965. [PubMed] [Google Scholar]
- 21.Keyer, K., A. S. Gort, and J. A. Imlay. 1995. Superoxide and the production of oxidative DNA damage. J. Bacteriol. 177:6782-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korshunov, S. S., and J. A. Imlay. 2002. A potential role for periplasmic superoxide dismutase in blocking the penetration of external superoxide into the cytosol of phagocytosed bacteria. Mol. Microbiol. 43:95-106. [DOI] [PubMed] [Google Scholar]
- 23.Krishnakumar, R., M. Craig, J. A. Imlay, and J. M. Slauch. 2004. Differences in enzymatic properties allow SodCI but not SodCII to contribute to virulence in Salmonella enterica serovar Typhimurium strain 14028. J. Bacteriol. 186:5230-5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo, C. F., T. Mashino, and I. Fridovich. 1987. α,β-Dihydroxyisovalerate dehydratase: a superoxide-sensitive enzyme. J. Biol. Chem. 262:4724-4727. [PubMed] [Google Scholar]
- 25.Liochev, S. I., and I. Fridovich. 1992. Fumarase C, the stable fumarase of Escherichia coli, is controlled by the soxRS regulon. Proc. Natl. Acad. Sci. USA 89:5892-5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loewen, P. C. 1984. Isolation of catalase-deficient Escherichia coli mutants and genetic mapping of katE, a locus that affects catalase activity. J. Bacteriol. 157:622-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch, R. E., and I. Fridovich. 1978. Permeation of the erythrocyte stroma by superoxide radical. J. Biol. Chem. 253:4697-4699. [PubMed] [Google Scholar]
- 28.Massey, V., S. Strickland, S. G. Mayhew, L. G. Howell, P. C. Engel, R. G. Matthews, M. Schuman, and P. A. Sullivan. 1969. The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem. Biophys. Res. Commun. 36:891-897. [DOI] [PubMed] [Google Scholar]
- 29.McCord, J. M., and I. Fridovich. 1969. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244:6049-6055. [PubMed] [Google Scholar]
- 30.Messner, K. R., and J. A. Imlay. 1999. The identification of primary sites of superoxide and hydrogen peroxide formation in the aerobic respiratory chain and sulfite reductase complex of Escherichia coli. J. Biol. Chem. 274:10119-10128. [DOI] [PubMed] [Google Scholar]
- 31.Messner, K. R., and J. A. Imlay. 2002. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 277:42563-42571. [DOI] [PubMed] [Google Scholar]
- 32.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 33.Moreau, P. L. 2004. Diversion of the metabolic flux from pyruvate dehydrogenase to pyruvate oxidase decreases oxidative stress during glucose metabolism in nongrowing Escherichia coli cells incubated under aerobic, phosphate starvation conditions. J. Bacteriol. 186:7364-7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller, F. 1987. Flavin radicals: chemistry and biochemistry. Free Radic. Biol. Med. 3:215-230. [DOI] [PubMed] [Google Scholar]
- 35.Puget, K., and A. M. Michelson. 1974. Isolation of a new copper-containing superoxide dismutase bacteriocuprein. Biochem. Biophys. Res. Commun. 58:830-838. [DOI] [PubMed] [Google Scholar]
- 36.San-Mateo, L. R., and M. M. H. H. Kawula. 1998. Periplasmic copper-zinc superoxide dismutase protects Haemophilus ducreyi from exogenous superoxide. Mol. Microbiol. 27:391-404. [DOI] [PubMed] [Google Scholar]
- 37.Seaver, L. C., and J. A. Imlay. 2001. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J. Bacteriol. 183:7173-7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seaver, L. C., and J. A. Imlay. 2004. Are respiratory enzymes the primary sources of intracellular hydrogen peroxide? J. Biol. Chem. 279:48742-48750. [DOI] [PubMed] [Google Scholar]
- 39.Siegele, D. A., K. R. C. Imlay, and J. A. Imlay. 1996. The stationary-phase-exit defect of cydC (surB) mutants is due to the lack of a functional terminal cytochrome oxidase. J. Bacteriol. 178:6091-6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soballe, B., and R. K. Poole. 1998. Requirement for ubiquinone downstream of cytochrome(s) b in the oxygen-terminated respiratory chains of Escherichia coli K-12 revealed using a null mutant allele of ubiCA. Microbiology 144:361-373. [DOI] [PubMed] [Google Scholar]
- 41.Steinman, H. M. 1982. Copper-zinc superoxide dismutase from Caulobacter crescentus CB15. A novel bacteriocuprein form of the enzyme. J. Biol. Chem. 257:10283-10293. [PubMed] [Google Scholar]
- 42.Steinman, H. M. 1993. Function of periplasmic copper-zinc superoxide dismutase in Caulobacter crescentus. J. Bacteriol. 175:1198-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tatum, F. M., P. G. Detilleux, J. M. Sacks, and S. M. Halling. 1992. Construction of Cu-Zn superoxide dismutase deletion mutants of Brucella abortus: analysis of survival in vitro in epithelial and phagocytic cells and in vivo in mice. Infect. Immun. 60:2863-2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turrens, J. F., A. Alexandre, and A. L. Lehninger. 1985. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 237:408-414. [DOI] [PubMed] [Google Scholar]
- 45.Unden, G., and J. Bongaerts. 1997. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320:217-234. [DOI] [PubMed] [Google Scholar]
- 46.Uzzau, S., L. Bossi, and N. Figueroa-Bossi. 2002. Differential accumulation of Salmonella [Cu, Zn] superoxide dismutases SodCI and SodCII in intracellular bacteria: correlation with their relative contribution to pathogenicity. Mol. Microbiol. 46:147-156. [DOI] [PubMed] [Google Scholar]
- 47.Wallace, B. J., and I. G. Young. 1977. Role of quinones in electron transport to oxygen and nitrate in Escherichia coli. Studies with a ubiA menA double quinone mutant. Biochim. Biophys. Acta 461:84-100. [DOI] [PubMed] [Google Scholar]
- 48.Wallace, M. A., L.-L. Liou, J. Martins, M. H. S. Clement, S. Bailey, V. D. Longo, J. S. Valentine, and E. B. Gralla. 2004. Superoxide inhibits 4Fe-4S cluster enzymes involved in amino acid biosynthesis: cross-compartment protection by CuZnSOD. J. Biol. Chem. 279:32055-32062. [DOI] [PubMed] [Google Scholar]
- 49.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195-199. [PubMed] [Google Scholar]
- 50.Wissenbach, U., D. Ternes, and G. Unden. 1992. An Escherichia coli mutant containing only demethylmenaquinone, but no menaquinone: effects on fumarate, dimethylsulfoxide, trimethylamine N-oxide and nitrate respiration. Arch. Microbiol. 158:68-73. [DOI] [PubMed] [Google Scholar]
- 51.Woodmansee, A. N., and J. A. Imlay. 2002. Reduced flavins promote oxidative DNA damage in nonrespiring Escherichia coli by delivering electrons to intracellular free iron. J. Biol. Chem. 277:34055-34066. [DOI] [PubMed] [Google Scholar]