

Abstract
Rieske oxygenase (RO) systems are two- and three-component enzyme systems that catalyze the formation of cis-dihydrodiols from aromatic substrates. Degradation of pollutants in contaminated soil and generation of chiral synthons have been the major foci of RO research. Substrate specificity and product regio- and stereoselectivity have been shown to vary between individual ROs. While directed evolution methods for altering RO function have been successful in the past, rational engineering of these enzymes still poses a challenge due to the lack of structural understanding. Here we examine the structural changes induced by mutation of Phe-352 in naphthalene 1,2-dioxygenase from Pseudomonas sp. strain NCIB 9816-4 (NDO-O9816-4). Structures of the Phe-352-Val mutant in native form and in complex with phenanthrene and anthracene, along with those of wild-type NDO-O9816-4 in complex with phenanthrene, anthracene, and 3-nitrotoluene, are presented. Phenanthrene was shown to bind in a different orientation in the Phe-352-Val mutant active site from that in the wild type, while anthracene was found to bind in similar positions in both enzymes. Two orientations of 3-nitrotoluene were observed, i.e., a productive and a nonproductive orientation. These orientations help explain why NDO-O9816-4 forms different products from 3-nitrotoluene than those made from nitrobenzene dioxygenase. Comparison of these structures among themselves and with other known ROs bound to substrates reveals that the orientation of substrate binding at the active site is the primary determinant of product regio- and stereoselectivity.
Rieske oxygenase (RO) systems have been shown to catalyze the first step in the metabolism of aromatic compounds by various prokaryotic organisms (7). ROs produce cis-dihydrodiols from a large variety of substrates and molecular oxygen (dioxygen). In addition, this enzyme system is also able to perform oxidation with a high degree of stereo-, regio-, and enantiospecificity (35). These factors have made ROs a desirable platform for use in biosynthesis of compounds and in bioremediation (reviewed in references 3, 30, and 40).
RO systems use electrons from NAD(P)H to activate molecular oxygen, which is then used to oxidize the substrate. RO systems are composed of two or three components, including a reductase, a ferredoxin (not found in all systems), and an oxygenase (Fig. 1). The reductase component liberates electrons from NAD(P)H and transfers the electrons to the ferredoxin. The ferredoxin shuttles the electrons to the oxygenase, where they are used in catalysis. In systems where the ferredoxin is absent, the reductase transfers electrons directly to the oxygenase. The oxygenase component of these systems is responsible for catalysis. This component consists of an α subunit, which contains both a Rieske binding domain and a catalytic domain. In some cases, a beta subunit is present, which is believed to primarily function as a stabilizer for the alpha subunits. Structural studies have been performed on a number of ROs; all have been shown to have either an α3 or α3β3 quaternary structure (9). This arrangement positions a Rieske domain of one subunit within ∼12 Å of a catalytic mononuclear iron of another, thus allowing electron transfer to take place.
FIG. 1.

Schematic depiction of the components of a Rieske oxygenase system. Electrons from NAD(P)H are extracted by the reductase and then transferred to the ferredoxin. The ferredoxin transfers these electrons to the Rieske cluster of the oxygenase. In two-component systems, the reductase directly transfers electrons to the oxygenase. The oxygenase binds molecular oxygen and an aromatic substrate in the active site of the mononuclear domain. Electrons are transferred from the oxygenase Rieske cluster to the mononuclear iron, and an oxidized product is formed. The image was made using biphenyl ferredoxin reductase (Protein Data Bank no. 1F3P), biphenyl reductase ferredoxin (Protein Data Bank no. 1FQT), and naphthalene dioxygenase (Protein Data Bank no. 1NDO).
The production of cis-diols occurs at the catalytic mononuclear iron and is coordinated by two histidines and a single aspartic acid. The 2-his-1-carboxylate facial triad motif is found in many enzymes (34). This motif is conserved in all known Rieske oxygenases. While the mononuclear iron provides the platform for catalysis, it alone is not able to control substrate specificity and product regio- and stereoselectivity for the enzyme. Naphthalene 1,2-dioxygenase from Pseudomonas sp. strain NCIB 9816-4 (NDO-O9816-4) has been studied as a model system for the dioxygenation of small molecules containing one to three aromatic rings (35). Previous structural studies have determined the molecular structure of the terminal oxygenase (5, 19, 20), including the positions of both molecular oxygen and the aromatic substrate bound to the enzyme. These studies have demonstrated that the mononuclear iron binds to molecular oxygen in a side-on fashion, while the aromatic substrate binds in the active site above the iron-oxygen complex. Structural studies of other ROs have demonstrated that the substrate binds in the active site with the atoms that would be oxidized closest to the mononuclear iron (10, 12, 13, 26). This led to the hypothesis that interactions between substrate and enzyme control substrate orientation, which determines both substrate selectivity and product regio- and stereoselectivity.
The Phe-352-Val mutation in NDO-O9816-4 was chosen as the focus of this set of structural investigations. Previous studies have shown that specific mutations in the active site of NDO-O9816-4 can affect the regio- and stereoselectivity of product formation (31, 32, 41). These studies demonstrated that mutations of Phe-352 change the product regioselectivities for naphthalene, phenanthrene, and biphenyl (Fig. 2). Wild-type NDO-O9816-4 catalyzes the stereospecific oxidation of naphthalene to (1R,2S)-1,2-dihydronaphthalene-1,2-diol. The Phe-352-Val mutant, in particular, had the largest effect on the stereochemistry of the naphthalene product of any single point mutation studied (32). Approximately 8% of the substrate was converted to (1S,2R)-1,2-dihydronaphthalene-1,2-diol, a product not formed by wild-type NDO-O9816-4. This mutation also allowed a new product to be formed from phenanthrene, (1S,2R)-1,2-dihydrophenanthrene-1,2-diol. Mutation of analogous residues in nitrobenzene 1,2-dioxygenase from Comamonas sp. strain JS765 (18) and 2-nitrotoluene 2,3-dioxygenase from Acidovorax sp. strain JS42 (24) also altered product regioselectivity.
FIG. 2.

Products formed by NDO-O9816-4 and NBDO-OJS765 from anthracene (1, 16, 31), phenanthrene (31, 32), and 3-nitrotoluene (25, 38). The percentage of each anthracene and phenanthrene dihydrodiol product formed by wild-type NDO-O9816-4 is shown below the product name. The percentage of each anthracene and phenanthrene product formed by Phe-352-Val NDO-O9816-4 is shown in brackets. N.R., products not reported to be formed.
Here we compare the structures of Phe-352-Val NDO-O9816-4 and wild-type NDO-O9816-4, in both the substrate-bound and unbound forms. The Phe-352-Val NDO-O9816-4 structure shows no significant changes in other residues that contribute to the active-site topology. The structures of wild-type NDO-O9816-4 and the Phe-352-Val mutant bound to phenanthrene demonstrate that enzyme-ligand interactions lead to the differences in product regioselectivity observed in biotransformation assays. We also show through structural studies that NDO-O9816-4 binds 3-nitrotoluene in a different orientation from that for nitrotoluene 2,3-dioxygenase. This difference in orientation supports the hypothesis that substrate orientation through active-site interactions controls product regioselectivity.
MATERIALS AND METHODS
Protein expression and purification.
The expression plasmid for the Phe-352-Val NDO-O9816-4 protein was generated by swapping a portion of the Phe-352-Val NDO-O9816-4 gene from pDTG141F352V (32) into pDTG121 (37), using the KpnI and Bsu36I restriction sites. The DNA fragments were ligated using Quick T4 DNA ligase (New England Biolabs, Ipswich, MA). Clones were screened using Escherichia coli DH5α cells, and plasmid DNAs were purified from colonies by use of a plasmid mini-prep kit (QIAGEN, Valencia, CA). E. coli BL21(DE3)Star protein expression cells (Invitrogen, Carlsbad, CA) were transformed with the Phe-352-Val pDTG121 expression vector. Protein expression was carried out in a manner similar to the method described by Lee et al. (23). Transformed cells were grown in mineral salts medium (36) with 150 μg/ml ampicillin, 10 mM glucose, and 0.6 mM thiamine in 1.5-liter batches at 37°C until an optical density at 600 nm of 0.5 to 0.7 was reached. The cells were then cooled to 15°C, and protein expression was induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Upon induction, cells were supplemented with 15 ml of 1 M glucose and 0.25 mM ferrous ammonium sulfate. The cells were grown for 16 h with shaking at 150 rpm at 15°C. After 16 h, cells were harvested by centrifugation. Cell pellets were resuspended in BTDG (50 mM bis-Tris [pH 6.8], 1 mM sodium dithiothreitol, 5% glycerol) column buffer at a ratio of 1 ml/gram and then frozen at −80°C.
Protein purification was performed as previously described (23). Cells were lysed using a French press and then centrifuged at ∼180,000 × g for 30 min to remove cellular debris. The clarified lysate was loaded onto a 600-ml Q-Sepharose FF (GE Amersham, Piscataway, NJ) column, and a 0 to 2 M potassium chloride gradient was used for elution. Fractions containing the reddish-brown protein were pooled and concentrated using an Amicon N2 concentrator with a YM100 membrane. Ammonium sulfate (4 M) was added to the concentrated protein to a final concentration of 1 M ammonium sulfate. The protein was loaded onto a 150-ml octyl Sepharose FF (GE Amersham, Piscataway, NJ) column and eluted with a 1.0 to 0.0 M ammonium sulfate gradient. The reddish-brown fractions were again pooled and concentrated. The concentrated protein was buffer exchanged into 1 mM potassium phosphate, pH 6.8. The protein was loaded onto a 40-μm, type 1 hydroxyapatite ion-exchange column (Bio-Rad, Hercules, CA) and eluted using a 0.001 to 1.0 M potassium phosphate gradient. The fractions containing NDO-O9816-4 were concentrated and then buffer exchanged with a 50 mM morpholineethanesulfonic acid, pH 6.8, solution. The protein was concentrated to 60 mg/ml using Amicon Ultra centrifugal filter units with a 100-kDa cutoff. Protein droplets were flash-frozen in liquid nitrogen and stored at −80°C until used for crystallization.
Protein crystallization and complex formation.
Crystallization of the purified protein was performed using the hanging-drop method. Two microliters of protein solution was added to 2 μl of crystallization solution on a siliconized glass coverslip and placed above a well of 0.5 ml of crystallization solution. Crystals formed in drops containing 1.9 to 2.2 M ammonium sulfate, 4 to 6% dioxane, and 100 mM morpholineethanesulfonic acid, pH 5.0 to 5.8. Drops were incubated at 6°C, and crystals formed after 48 to 72 h.
Substrates were soaked into crystals in a manner similar to that reported previously for the protein-naphthalene complex (19). One microliter of ethanol saturated with substrate was added to 19 μl of crystallization solution, and crystals were transferred to the soak solution for 2 to 4 weeks to allow the substrate to bind.
Data collection, processing, structure solution, and refinement.
X-ray diffraction data for the wild-type NDO-O9816-4 phenanthrene complex were collected on an IMCA-CAT beamline 17-ID station at Advanced Photon Source in Argonne National Laboratories, using an ADSC Quantum 210 charge-coupled device detector. X-ray diffraction data for all other structures were collected on an MBC beamline 4.2.2 station at Advanced Light Source in Lawrence Berkeley National Laboratories, using a Noir-1 charge-coupled device detector. Data collection and processing statistics are reported in Table 1. Data were processed using d*trek (33). A previous structure of wild-type NDO-O9816-4 (Protein Data Bank no. 1O7H) was used directly as a starting model for refinement. Refinement was performed using Refmac5 (27) in the CCP4 6.0.0 (2) software collection. The molecular visualization programs O (17) and Coot (8) were used for model building and visualization. The ligands were modeled into the electron density, and then the position was refined along with the protein in Refmac5. Solvent molecules were found using Arp/Warp (21), verified by a composite omit map using Brown and Ramaswamy's solvent omit software (4), and modeled where electron density was present. Images were rendered and root mean square deviation (RMSD) calculations were performed using PyMOL (6). Heats of formation were calculated using the MOPAC module in Chem3D Ultra (CambridgeSoft, Cambridge, MA).
TABLE 1.
Summary of crystallographic data collection and refinement statistics
| Parameter | Value or characteristic for enzyme-substrate pair (Protein Data Bank no.)
|
|||||
|---|---|---|---|---|---|---|
| Phe-352-Val mutant (2HMJ) | Wild type- phenanthrene (2HMK) | Phe-352-Val mutant- phenanthrene (2HML) | Wild type- anthracene (2HMM) | Phe-352-Val mutant- anthracene (2HMN) | Wild type-3- nitrotoluene (2HMO) | |
| Data collection statistics | ||||||
| Beamline | MBC 4.2.2 | IMCA 17-ID | MBC 4.2.2 | MBC 4.2.2 | MBC 4.2.2 | MBC 4.2.2 |
| Space group | R32 (155) | R32 (155) | R32 (155) | R32 (155) | R32 (155) | R32 (155) |
| a (Å) | 139.9 | 140.0 | 139.9 | 139.7 | 139.5 | 140.2 |
| b (Å) | 139.9 | 140.0 | 139.9 | 139.7 | 139.5 | 140.2 |
| c (Å) | 208.6 | 208.5 | 208.4 | 208.1 | 208.2 | 208.3 |
| α (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 90 | 90 | 90 | 90 |
| γ (°) | 120 | 120 | 120 | 120 | 120 | 120 |
| Wavelength (Å) | 1.04021 | 1.0000 | 1.04021 | 1.04021 | 1.04021 | 1.04021 |
| Resolutiona (Å) | 15.99-1.50 (1.55-1.50) | 39.51-1.65 (1.71-1.65) | 17.97-1.80 (1.86-1.80) | 15.97-1.60 (1.66-1.60) | 16.91-1.70 (1.76-1.70) | 15.95-1.60 (1.66-1.60) |
| No. of observed reflectionsa | 541,766 (33,775) | 666,754 (62,473) | 337,248 (33,610) | 318,594 (29,139) | 472,166 (46,415) | 723,659 (25,950) |
| No. of unique reflectionsa | 123,779 (11,687) | 93,678 (9,354) | 72,381 (7,201) | 100,884 (9,731) | 85,038 (8,437) | 102,411 (9,895) |
| Completenessa (%) | 99.2 (94.4) | 99.6 (100.0) | 99.9 (100.0) | 98.5 (95.5) | 99.7 (100.0) | 99.2 (96.6) |
| Rsyma,b (%) | 0.078 (0.518) | 0.065 (0.306) | 0.108 (0.464) | 0.069 (0.449) | 0.072 (0.481) | 0.100 (0.430) |
| Avg <I>/<σ(I)> | 9.6 (2.0) | 13.3 (4.5) | 9.4 (3.1) | 10.0 (2.3) | 11.1 (2.9) | 11.8 (2.1) |
| Data refinement statistics | ||||||
| Rfactorc (%) | 15.5 | 15.8 | 16.4 | 16.4 | 15.9 | 16.1 |
| Rfreec (%) | 18.6 | 18.3 | 19.0 | 19.8 | 19.4 | 19.7 |
| RMSD (bond lengths) (Å) | 0.012 | 0.010 | 0.010 | 0.012 | 0.012 | 0.011 |
| RMSD (bond angles) (°) | 1.418 | 1.342 | 1.322 | 1.422 | 1.381 | 1.377 |
| No. of protein atomsd | 5,095 | 5,112 | 5,084 | 5,093 | 5,084 | 5,094 |
| No. of nonprotein atomsd | 84 | 119 | 107 | 126 | 111 | 108 |
| No. of solvent moleculesd | 754 | 651 | 550 | 731 | 581 | 716 |
| Avg temp factorse | ||||||
| α subunit | 18.56 (5.84) | 20.23 (5.63) | 18.43 (5.33) | 19.66 (5.81) | 23.97 (7.16) | 20.79 (5.16) |
| β subunit | 20.95 (7.67) | 22.19 (7.96) | 20.59 (8.06) | 21.45 (8.33) | 26.30 (9.30) | 22.72 (7.26) |
| Solvent | 35.32 (12.03) | 11.05 (3.42) | 30.00 (11.02) | 34.52 (11.92) | 10.85 (3.08) | 31.64 (2.76) |
| Heteroatoms | 40.31 (17.32) | 12.04 (2.97) | 35.49 (13.37) | 36.99 (11.19) | 13.01 (4.27) | 38.17 (10.54) |
| Substrate | Not applicable | 33.83 (3.17) | 16.79 (0.28) | 53.78 (3.38) | 26.29 (0.45) | 31.64 (2.76) |
Values in parentheses are for the highest-resolution shell.
 where Ii(hkl) is the ith measurement of reflection hkl and I(hkl) is the average for that reflection.
where Ii(hkl) is the ith measurement of reflection hkl and I(hkl) is the average for that reflection.
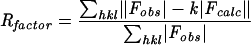 where Fobs and Fcalc are the observed and calculated structure factors, respectively. Rfree is the same, but for a test set of reflections not used in refinement.
where Fobs and Fcalc are the observed and calculated structure factors, respectively. Rfree is the same, but for a test set of reflections not used in refinement.
Nonhydrogen atoms.
Numbers in parentheses are standard deviations.
RESULTS AND DISCUSSION
Structures of NDO-O9816-4 Phe-352-Val mutant.
Previous studies of NDO-O9814-6 have shown that mutations in the active site can change the regio- and stereoselectivity of product formation (29, 31, 32, 41). At that time, it was unclear how these mutations altered the enzyme to change product formation. Phe-352 was chosen as the residue to investigate because it was shown to have a relatively large impact on both the regio- and stereoselectivity of product formation in mutation studies. The structure of the Phe-352-Val mutant was determined by single-crystal X-ray diffraction studies. Crystallographic statistics are reported in Table 1. Overall, no large variations in the secondary and tertiary structures of the α and β subunits were observed compared to wild-type NDO-O9816-4. Mononuclear iron ligand distances are reported in Table 2. The RMSD for the α subunits of the Phe-352-Val mutant compared to the wild type is 0.087 Å (for 416 C-α atoms). Regions in both the wild type and the Phe-352-Val mutant that exhibit higher RMSDs than average are the loop region covering the entrance to the active site, residues 205 to 235 in the alpha subunit, and residues 90 to 99 in the loop region of the Rieske domain. These regions also exhibit higher B factors and lower electron densities. Analogous regions are disordered in other RO structures. Rieske cluster-coordinating residues did not have significant differences in position from those in wild-type NDO-O9816-4.
TABLE 2.
Mononuclear iron distances
| Residue or group | Distance (Å) to mononuclear iron in enzyme-substrate structure (Protein Data Bank no.)
|
|||||
|---|---|---|---|---|---|---|
| Phe-352-Val mutant (2HMJ) | Wild type- phenanthrene (2HMK) | Phe-352-Val mutant- phenanthrene (2HML) | Wild type- anthracene (2HMM) | Phe-352-Val mutant- anthracene (2HMN) | Wild type-3- nitrotoluene (2HMO) | |
| Asp-205 (OD1) | 2.33 | 2.58 | 2.12 | 2.22 | 2.22 | 2.26 |
| Asp-205 (OD2) | 2.52 | 2.15 | 2.40 | 2.44 | 2.35 | 2.55 |
| His-208 (NE2) | 2.11 | 2.13 | 2.26 | 2.14 | 2.17 | 2.16 |
| His-213 (NE2) | 2.04 | 2.04 | 2.08 | 2.04 | 2.06 | 2.05 |
| OHa | 1.96 | 2.04 | 1.66 | 1.94 | 1.98 | 1.79, 2.05 |
| Substrateb | NAd | 4.17, 4.72 | 3.36, 3.67 | 4.14, 4.26 | 4.47, 4.54 | 4.46 (5.81)c |
Hydroxide ion(s) ligated to iron.
Distance to the hydroxylated substrate carbon(s).
Distance to the methyl carbon in the nonproductive conformation.
NA, not applicable.
In the Phe-352-Val mutant, the side chain beta carbon and one gamma carbon are in similar positions to those of the beta and gamma carbons of Phe-352 in wild-type NDO-O9816-4 (Fig. 3A). The other gamma carbon of Val-352 is positioned toward the hydrophobic side chains of Val-312 and Val-348. The side chains of Val-348 and Val-312 do not significantly deviate from their positions in wild-type NDO-O9816-4.The active-site cavity is significantly larger than that of NDO-O9816-4 due to the decreased bulk of the position 352 side chain. This change creates a larger area near the mononuclear iron. The Phe-352 side chain in NDO-O9816-4 also shields the active site from more hydrophilic areas behind the phenyl ring. The mutation to valine removes this shielding, exposing the active site to the backbone nitrogen and side chain carbons of Glu-359. Two additional water molecules are seen in the active site, occupying a portion of the cavity created by the mutation.
FIG. 3.

(A) Cartoon of Phe-352-Val NDO-O9816-4. The figure shows the valine substitution (yellow balls and sticks) and the two water molecules (small red spheres). Electron density (1.0 σ 2Fo-Fc) is shown as a blue wire mesh. The position of phenylalanine in wild-type NDO-O9816-4 is shown as a gray ball-and-stick model. The mononuclear iron (large brown sphere), iron-coordinating residues (yellow balls and sticks), and water (small red sphere) are also shown. (B) Wild-type NDO-O9816-4 in complex with phenanthrene (orange balls and sticks). Electron density (1.0 σ 2Fo-Fc) is shown as a blue wire mesh. The mononuclear iron (large brown sphere), iron-coordinating residues (yellow balls and sticks), and water (small red sphere) are also shown. (C) Phe-352-Val NDO-O9816-4 in complex with phenanthrene (orange balls and sticks). Electron density (1.0 σ 2Fo-Fc) is shown as a blue wire mesh. The mononuclear iron (large brown sphere), iron-coordinating residues (yellow balls and sticks), and water (small red sphere) are also shown. (D) Overlay of wild-type and Phe-352-Val NDO-O9816-4 in complex with phenanthrene. Phenanthrene is shown in the orientations observed in the wild type (gray balls and sticks) and Phe-352-Val (orange balls and sticks). Residue 352, phenanthrene (gray balls and sticks), and valine (yellow balls and sticks) are also shown in the cartoon model. The mononuclear iron (large brown sphere), iron-coordinating residues (yellow balls and sticks), and water (small red sphere) are also shown.
Structures of Phe-352-Val NDO-O9816-4 bound to phenanthrene and anthracene.
In the structure of wild-type NDO-O9816-4, the naphthalene ligand is oriented in the active site with carbons 1 and 2 of the first ring closest to the mononuclear iron (19). This orientation explains the extreme regio- and stereoselectivity of the cis-dihydrodiol produced by the enzyme (14, 15). Similarly, the structure of NDO-O9816-4 with phenanthrene shows that the carbons closest to the mononuclear iron are hydroxylated by the enzyme to form the major product observed in biotransformation assays, (3S,4R)-3,4-dihydrophenanthrene-3,4-diol (31, 32) (Fig. 3B). As in other structures of NDO-O9816-4 bound to substrates, there is no significant side chain rearrangement observed in the active site due to substrate binding.
Unlike wild-type NDO-O9816-4, the Phe-352-Val mutant catalyzes the formation of (−)-cis-1,2-dihydroxy-1,2-dihydrophenanthrene (Fig. 2) as the major product (31, 41). The crystal structure of the Phe-352-Val mutant echoes this change, showing that phenanthrene is oriented differently in the active site compared to the case in the wild type. The increased volume due to the mutation of Phe-352 allows phenanthrene to bind in a new orientation, thus allowing a different set of carbon atoms to be positioned near the mononuclear iron for hydroxylation (Fig. 3C). The new orientation of the ligand in the active site allows a different pair of symmetry-related atoms access to the mononuclear iron (Fig. 3D). This other pair of symmetry-related atoms also forms a 1,2-diol, but with the opposite stereochemistry. Because the mutation at Phe-352 does not create any steric restrictions with respect to the orientation observed in the wild type, it is possible that a small percentage of molecules will bind in orientations different from that observed in the mutant complex. The possibility for other, less populated orientations explains why a mixture of regio- and stereoisomers is observed in biotransformations, rather than an enantiomerically pure product (32).
In contrast, the position of anthracene did not differ significantly between the Phe-352-Val mutant and wild-type complex structures (Fig. 4). In both structures, anthracene bound in a position similar to that for naphthalene. This is not surprising, since the chemical structures of anthracene and naphthalene are very similar. Residues Val-209 and Leu-307 position the first and second rings of the substrate. This is also observed in the NDO-O9816-4 naphthalene complex structure. However, in both of the anthracene complex structures, the third ring (farthest from the iron) has poor electron density and higher temperature factors. This is most likely due to the ability of the third ring to move more than the other two rings. Because residues Val-209 and Leu-307 “anchor” the first and second rings, anthracene can act as a lever, with small movements near the iron being exaggerated in the third (distal) ring. Side chain density for the enzyme is also poor in this region, most likely due to the flexible nature of the loop that covers the active-site entrance. The ligand was modeled as partially occupied based on temperature factors of surrounding residues. The partial occupancy of the ligand is most likely due to the low solubility of anthracene under the high-salt conditions of the crystallization mother liquor.
FIG. 4.

Overlay of wild-type (yellow cartoon) and Phe-352-Val NDO-O9816-4 (blue cartoon) in complex with anthracene. Val-209 and Leu-307 are seen “anchoring” the ligand in the active site. Naphthalene bound to wild-type NDO-O9816-4 (Protein Data Bank no. 1O7G) is shown with red balls and sticks. The mononuclear iron (large brown sphere), iron-coordinating residues (balls and sticks), and waters (small red spheres) are also shown.
Biotransformation assays have shown previously that wild-type NDO-O9816-4 converts anthracene to enantiomerically pure (+)-(1R,2S)-cis-dihydroxy-1,2-dihyroanthracene (1, 16). In previous studies, the Phe-352-Val mutant was tested for comparison, and over 96% of the product formed was also (+)-(1R,2S)-cis-dihydroxy-1,2-dihyroanthracene. However, unlike the case for the wild-type enzyme, a trace amount of the (1S,2R) stereoisomer was also detected (31). These results can be rationalized through comparison of the structures. While similar positions were observed for anthracene in both wild-type and mutant NDO-O9816-4, the increased volume of the active site created near the mononuclear iron allows anthracene an alternate orientation, placing the first ring in a position similar to that of the hydroxylated phenanthrene ring observed in the Phe-352-Val mutant. This would allow the symmetry-related pair of carbon atoms in anthracene to be attacked, forming the same regioisomer (1, 2), but with opposite stereochemistry (1S,2R).
The same type of mechanism can be proposed for creation of the (1S,2R) enantiomer of naphthalene observed in Phe-352-Val NDO-O9816-4 biotransformations but not produced by the wild type (14, 15, 31, 32). Attempts to determine the structure of the Phe-352-Val mutant bound to naphthalene resulted in electron density maps suggesting that naphthalene is disordered in the active site. This also suggests that the mutant allows naphthalene to adopt alternative binding conformations.
NDO-O9816-4 and NBDO-O have differences in 3-nitrotoluene binding.
NDO-O9816-4 and nitrobenzene dioxygenase from Comamonas sp. strain JS765 (NBDO-OJS765) have been shown to form different products from nitroarene compounds, such as nitrobenzene and 3-nitrotoluene. In the case of NDO-O9816-4, nitrobenzene has been shown not to be a substrate and 3-nitrotoluene is converted to 3-nitrobenzyl alcohol (38); however, NBDO-OJS765 converts nitrobenzene to catechol and nitrite (28) and converts 3-nitrotoluene to 4-methylcatechol (25). While these differences in substrate selectivity and product regioselectivity exist, NBDO-OJS765 shares high sequence similarity with NDO-O9816-4, with ∼82% similarity for the alpha subunits, and differs by only 6 of the 22 residues that create the topology of the active site (10). The structures of NBDO-OJS765 demonstrate that the substrates bind in the active site with the hydroxylated carbons closest to the mononuclear iron, similar to the case for other ROs (10). The structures also suggest that the orientation of substrates is mediated by a hydrogen bond between the side chain of Asn-258 and the nitro group on the substrate. Interestingly, Asn-258 is one of the active-site residues that is not conserved between NBDO-OJS765 and NDO-O9816-4; however, it is conserved between NBDO-OJS765 and other dinitrobenzene dioxygenases (18).
In order to compare the differences in handling of nitroarene substrates between NDO-O9816-4 and NBDO-OJS765, we determined the structure of NDO-O9816-4 bound to 3-nitrotoluene. The electron density in the active site showed that 3-nitrotoluene binds in the active site at two positions (Fig. 5). In the first position, the nitro group of the substrate sits closest to the mononuclear iron. One nitro group oxygen hydrogen bonds to the waters/hydroxides coordinating the iron, 2.5 Å from the closest water/hydroxide, and to the side chain of Asn-201, 3.1 Å from the side chain nitrogen atom. The other oxygen hydrogen bonds to the main chain oxygen of Asn-201, 3.1 Å apart, and the side chain nitrogen of Asn-297, 3.4 Å apart. This appears to be a nonproductive orientation, with the closest carbon to the mononuclear iron being 6.2 Å away. This distance suggests that carbon atoms are too far away from the site of catalysis to be oxidized. However, the second binding position does position the methyl group of 3-nitrotoluene 4.5 Å from the mononuclear iron, which is close enough for catalysis. The position is controlled by a hydrogen bond between oxygen atoms of the substrate and the side chain of His-295. The substrate, 3-nitrotoluene, is oriented in the NDO-O9816-4 active site with the benzyllic carbon near the mononuclear iron, which is monohydroxylated to form the alcohol. This is different from the NBDO-OJS765 structure, which shows the aromatic ring positioned near the mononuclear iron (Fig. 6). The structure of NDO-O9816-4 bound to 3-nitrotoluene agrees with the hypothesis that product selectivity is a result of substrate orientation and that substrate orientation is controlled by interactions with active-site residuzes.
FIG. 5.

NDO-O9816-4 bound to 3-nitrotoluene. Wild-type NDO-O9816-4 (yellow balls and sticks) is shown in complex with 3-nitrotoluene, which is modeled in two positions. Position 1, shown with green balls and sticks, is the nonproductive conformation. Position 2, shown with orange balls and sticks, is the productive conformation.
FIG. 6.

NDO-O9816-4 bound to 3-nitrotoluene. Wild-type NDO-O9816-4 (yellow balls and sticks) is shown in complex with 3-nitrotoluene (productive conformation). Overlaid is 3-nitrotoluene from NBDO-OJS765 (Protein Data Bank no. 2BMR), shown with pink balls and sticks. This is a different orientation than that observed for NDO-O9816-4. Three residues from NBDO-OJS765, Asn-258, Phe-293, and Ile-350 (pink balls and sticks), which are believed to help control nitroarene orientation in NBDO-OJS765, are overlaid.
Orientation hypothesis and structural implications.
In this study, we tested the hypothesis that active-site residues not involved in coordinating the mononuclear iron are responsible for substrate selection and orientation. This, in turn, controls product regio- and stereoselectivity. While this is a simple premise on a general level, an understanding of the structural impact of alterations in the active site is required for more complex rational engineering of the enzyme. A major assumption in our hypothesis is that hydroxylated positions are equivalent with respect to reactivity. If multiple orientations of substrate are allowed in the active site with equal favorability, but one orientation places the more reactive atom(s) closer to the mononuclear iron, there may be only one product observed. This can be demonstrated in the structure of wild-type NDO-O9816-4 bound to phenanthrene. In this case, there are two positions that are approximately equidistant from the mononuclear iron, i.e., the 1,2 and 2,3 positions; however, only the 1,2-diol is observed in biotransformation assays (31, 32). In this situation, the chemistry of the reaction dictates which product will be formed. The calculated heat of formation for the 1,2-diol is −30.1 kcal/mol, and that for the 2,3-diol is −13.8 kcal/mol. While the enzyme orientation does not favor the formation of the 1,2-diol versus the 2,3-diol, the formation of the 1,2-diol is much more favorable chemically.
Structural studies of NDO-O9816-4 with compounds that contain noncarbon atoms are currently being performed to investigate how the enzyme handles substrates with heteroatoms. Other mutations in the NDO-O9816-4 active site have also been shown to alter stereo- and regioselectivity (41). These changes most likely change the shape and/or flexibility of the active site, allowing an alternative orientation of substrate in the active site. Structural investigations of other mutations are also currently under way.
Previous reports demonstrated that NDO-O9816-4 Phe-352 mutants have decreased activity in vivo (31, 32, 41). Wild-type NDO has been shown to uncouple oxygen consumption from product formation, slowly forming hydrogen peroxide from oxygen (22). Hydrogen peroxide-induced damage in the active site was believed to inactivate the enzyme. Because the 2-His-1-carboxy motif at the mononuclear iron was unchanged in the mutants, it is likely that the mutation did not alter the mechanism of oxygen activation. Instead, the decreased activity in the mutants could be due to increased oxygen uncoupling and subsequent inactivation of the enzyme. Alternatively, the decreased activity may be a result of a loss of affinity for iron at the mononuclear iron site.
While there has been success in altering enzyme product regio- and stereoselectivity through directed evolution (reviewed in references 11 and 39), this method does not offer details on a molecular level about the enzyme mechanism. Because of this, there may be certain limitations to enzyme substrate acceptance or product selectivity that are never observed but readily discernible through structure-based methods. The results presented here demonstrate that RO stereo- and regioselectivity is controlled, at least in part, by substrate orientation in the active site. This information will enable further rational engineering of the enzyme to change the topology of the active site, directly or indirectly, and adjust product regio- and stereoselectivity.
Acknowledgments
We thank David Gibson and Hans Eklund for their great support in this work. Rebecca Parales has been a wonderful collaborator throughout the work carried out in S.R.'s laboratory. We thank Chi-Li Yu, Heather Hanson, Wensheng Liu, Nathan Coussens, Eric Brown, and Emmanuel Quiazon for assistance with molecular biology and protein preparation. We thank Juan Parales for assistance with protein crystallization. We thank Jay Nix, Ed Westbrook, and Darren Sherrell for their help with data collection at beamline 4.2.2. We thank Lisa Keefe, Irina Koshelev, and Kevin Battaile for help with data collection at the IMCA-CAT beamline.
Use of the Molecular Biology Consortium beamline 4.2.2 at the Advanced Light Source at Lawrence Berkeley National Laboratories was made possible through the University of Iowa institutional membership in the MBC. The ALS is supported by U.S. DOE under contract no. DE-AC03-76SF00098. Use of the IMCA-CAT beamline 17-ID (or 17-BM) at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with the Center for Advanced Radiation Sources at the University of Chicago. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31-109-Eng-38. A.L.O. and D.J.F. are supported by Center for Environmentally Beneficial Catalysis and Center for Biocatalysis and Bioprocessing fellowships, respectively. S.R. acknowledges financial support from USPHS grant GM62904.
REFERENCES
- 1.Akhtar, M. N., D. R. Boyd, N. J. Thompson, M. Koreeda, D. T. Gibson, V. Mahadevan, and D. M. Jerina. 1975. Absolute stereochemistry of the dihydroanthracene-cis- and -trans-1,2-diols produced from anthracene by mammals and bacteria. J. Chem. Soc. (Perkin 1) 1975:2506-2511. [PubMed] [Google Scholar]
- 2.Anonymous. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 3.Boyd, D. R., and T. D. Bugg. 2006. Arene cis-dihydrodiol formation: from biology to application. Org. Biomol. Chem. 4:181-192. [DOI] [PubMed] [Google Scholar]
- 4.Brown, E. N., and S. Ramaswamy. 2006. Verification of solvent molecules through composite-omit electron density maps. University of Iowa, Iowa City.
- 5.Carredano, E., A. Karlsson, B. Kauppi, D. Choudhury, R. E. Parales, J. V. Parales, K. Lee, D. T. Gibson, H. Eklund, and S. Ramaswamy. 2000. Substrate binding site of naphthalene 1,2-dioxygenase: functional implications of indole binding. J. Mol. Biol. 296:701-712. [DOI] [PubMed] [Google Scholar]
- 6.DeLano, W. L. 2000. The PyMOL molecular graphics system, 0.98 ed. DeLano Scientific, San Carlos, Calif.
- 7.Eaton, R. W., and P. J. Chapman. 1992. Bacterial metabolism of naphthalene: construction and use of recombinant bacteria to study ring cleavage of 1,2-dihydroxynaphthalene and subsequent reactions. J. Bacteriol. 174:7542-7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emsley, P., and K. Cowtan. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60:2126-2132. [DOI] [PubMed] [Google Scholar]
- 9.Ferraro, D. J., L. Gakhar, and S. Ramaswamy. 2005. Rieske business: structure-function of Rieske non-heme oxygenases. Biochem. Biophys. Res. Commun. 338:175-190. [DOI] [PubMed] [Google Scholar]
- 10.Friemann, R., M. M. Ivkovic-Jensen, D. J. Lessner, C. L. Yu, D. T. Gibson, R. E. Parales, H. Eklund, and S. Ramaswamy. 2005. Structural insight into the dioxygenation of nitroarene compounds: the crystal structure of nitrobenzene dioxygenase. J. Mol. Biol. 348:1139-1151. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa, K., H. Suenaga, and M. Goto. 2004. Biphenyl dioxygenases: functional versatilities and directed evolution. J. Bacteriol. 186:5189-5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furusawa, Y., V. Nagarajan, M. Tanokura, E. Masai, M. Fukuda, and T. Senda. 2004. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. J. Mol. Biol. 342:1041-1052. [DOI] [PubMed] [Google Scholar]
- 13.Gakhar, L., Z. A. Malik, C. C. Allen, D. A. Lipscomb, M. J. Larkin, and S. Ramaswamy. 2005. Structure and increased thermostability of Rhodococcus sp. naphthalene 1,2-dioxygenase. J. Bacteriol. 187:7222-7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeffrey, A. M., H. J. Yeh, D. M. Jerina, T. R. Patel, J. F. Davey, and D. T. Gibson. 1975. Initial reactions in the oxidation of naphthalene by Pseudomonas putida. Biochemistry 14:575-584. [DOI] [PubMed] [Google Scholar]
- 15.Jerina, D. M., J. W. Daly, A. M. Jeffrey, and D. T. Gibson. 1971. cis-1,2-Dihydroxy-1,2-dihydronaphthalene: a bacterial metabolite from naphthalene. Arch. Biochem. Biophys. 142:394-396. [DOI] [PubMed] [Google Scholar]
- 16.Jerina, D. M., H. Selander, H. Yagi, M. C. Wells, J. F. Davey, V. Mahadevan, and D. T. Gibson. 1976. Dihydrodiols from anthracene and phenanthrene. J. Am. Chem. Soc. 98:5988-5996. [DOI] [PubMed] [Google Scholar]
- 17.Jones, T. A., J. Y. Zou, S. W. Cowan, and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 18.Ju, K. S., and R. E. Parales. 2006. Control of substrate specificity by active-site residues in nitrobenzene dioxygenase. Appl. Environ. Microbiol. 72:1817-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlsson, A., J. V. Parales, R. E. Parales, D. T. Gibson, H. Eklund, and S. Ramaswamy. 2003. Crystal structure of naphthalene dioxygenase: side-on binding of dioxygen to iron. Science 299:1039-1042. [DOI] [PubMed] [Google Scholar]
- 20.Kauppi, B., K. Lee, E. Carredano, R. E. Parales, D. T. Gibson, H. Eklund, and S. Ramaswamy. 1998. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6:571-586. [DOI] [PubMed] [Google Scholar]
- 21.Lamzin, V. S., and K. S. Wilson. 1993. Automated refinement of protein models. Acta Crystallogr. D 49:129-147. [DOI] [PubMed] [Google Scholar]
- 22.Lee, K. 1999. Benzene-induced uncoupling of naphthalene dioxygenase activity and enzyme inactivation by production of hydrogen peroxide. J. Bacteriol. 181:2719-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee, K., B. Kauppi, R. E. Parales, D. T. Gibson, and S. Ramaswamy. 1997. Purification and crystallization of the oxygenase component of naphthalene dioxygenase in native and selenomethionine-derivatized forms. Biochem. Biophys. Res. Commun. 241:553-557. [DOI] [PubMed] [Google Scholar]
- 24.Lee, K. S., J. V. Parales, R. Friemann, and R. E. Parales. 2005. Active site residues controlling substrate specificity in 2-nitrotoluene dioxygenase from Acidovorax sp. strain JS42. J. Ind. Microbiol. Biotechnol. 32:465-473. [DOI] [PubMed] [Google Scholar]
- 25.Lessner, D. J., G. R. Johnson, R. E. Parales, J. C. Spain, and D. T. Gibson. 2002. Molecular characterization and substrate specificity of nitrobenzene dioxygenase from Comamonas sp. strain JS765. Appl. Environ. Microbiol. 68:634-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martins, B. M., T. Svetlitchnaia, and H. Dobbek. 2005. 2-Oxoquinoline 8-monooxygenase oxygenase component: active site modulation by Rieske-[2Fe-2S] center oxidation/reduction. Structure 13:817-824. [DOI] [PubMed] [Google Scholar]
- 27.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53:240-255. [DOI] [PubMed] [Google Scholar]
- 28.Nishino, S. F., and J. C. Spain. 1995. Oxidative pathway for the biodegradation of nitrobenzene by Comamonas sp. strain JS765. Appl. Environ. Microbiol. 61:2308-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parales, R. E. 2003. The role of active-site residues in naphthalene dioxygenase. J. Ind. Microbiol. Biotechnol. 30:271-278. [DOI] [PubMed] [Google Scholar]
- 30.Parales, R. E., N. C. Bruce, A. Schmid, and L. P. Wackett. 2002. Biodegradation, biotransformation, and biocatalysis (b3). Appl. Environ. Microbiol. 68:4699-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parales, R. E., K. Lee, S. M. Resnick, H. Jiang, D. J. Lessner, and D. T. Gibson. 2000. Substrate specificity of naphthalene dioxygenase: effect of specific amino acids at the active site of the enzyme. J. Bacteriol. 182:1641-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parales, R. E., S. M. Resnick, C. L. Yu, D. R. Boyd, N. D. Sharma, and D. T. Gibson. 2000. Regioselectivity and enantioselectivity of naphthalene dioxygenase during arene cis-dihydroxylation: control by phenylalanine 352 in the alpha subunit. J. Bacteriol. 182:5495-5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pflugrath, J. W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D 55:1718-1725. [DOI] [PubMed] [Google Scholar]
- 34.Que, L., Jr. 2000. One motif—many different reactions. Nat. Struct. Biol. 7:182-184. [DOI] [PubMed] [Google Scholar]
- 35.Resnick, S. M., K. Lee, and D. T. Gibson. 1996. Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp. strain NCBI 9816. J. Ind. Microbiol. Biotechnol. 17:438-457. [Google Scholar]
- 36.Stanier, R. Y., N. J. Palleroni, and M. Doudoroff. 1966. The aerobic pseudomonads: a taxonomic study. J. Gen. Microbiol. 43:159-271. [DOI] [PubMed] [Google Scholar]
- 37.Suen, W. C., and D. T. Gibson. 1994. Recombinant Escherichia coli strains synthesize active forms of naphthalene dioxygenase and its individual alpha and beta subunits. Gene 143:67-71. [DOI] [PubMed] [Google Scholar]
- 38.Suen, W. C., B. E. Haigler, and J. C. Spain. 1996. 2,4-Dinitrotoluene dioxygenase from Burkholderia sp. strain DNT: similarity to naphthalene dioxygenase. J. Bacteriol. 178:4926-4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wackett, L. P. 1998. Directed evolution of new enzymes and pathways for environmental biocatalysis. Ann. N. Y. Acad. Sci. 864:142-152. [DOI] [PubMed] [Google Scholar]
- 40.Wackett, L. P. 2002. Mechanism and applications of Rieske non-heme iron dioxygenases. Enzyme Microb. Technol. 31:577-587. [Google Scholar]
- 41.Yu, C. L., R. E. Parales, and D. T. Gibson. 2001. Multiple mutations at the active site of naphthalene dioxygenase affect regioselectivity and enantioselectivity. J. Ind. Microbiol. Biotechnol. 27:94-103. [DOI] [PubMed] [Google Scholar]