

Abstract
Alzheimer’s Disease (AD) is characterized by the accumulation of aggregated amyloid β-peptide (Aβ) in the brain. The physiological mechanisms and factors that predispose to Aβ aggregation and deposition are not well understood. In this report, we show that calcium can predispose to Aβ aggregation and fibril formation. Calcium increased the aggregation of early forming protofibrillar structures and markedly increased conversion of protofibrils to mature amyloid fibrils. This occurred at levels 20-fold below the calcium concentration in the extracellular space of the brain, the site at which amyloid plaque deposition occurs. In the absence of calcium, protofibrils can remain stable in vitro for several days. Using this approach, we directly compared the neurotoxicity of protofibrils and mature amyloid fibrils, and demonstrate that both species are inherently toxic to neurons in culture. Thus, calcium may be an important predisposing factor for Aβ aggregation and toxicity. The high extracellular concentration of calcium in the brain, together with impaired intraneuronal calcium regulation in the aging brain and AD, may play an important role in the onset of amyloid-related pathology.
Amyloid β-peptide (Aβ) is the primary constituent of amyloid plaques in Alzheimer’s disease (AD). Cleavage of the amyloid β precursor protein (APP) leads to the production of Aβ peptides of varying lengths, of which the 40 amino acid peptide is the major species (Aβ1–40) (1). Mutations that cause familial Alzheimer’s disease generally lead to an increase in the level of the more fibrillogenic 42 amino acid peptide Aβ1–42 (1,2). The aggregation of Aβ into mature amyloid fibrils occurs through a number of intermediate structural forms, variously refered to as oligomers or protofibrils. Soluble oligomeric species and protofibrillar structures may affect neuronal function and viability (3–5), and mature amyloid fibrils are toxic to neurons in vitro and in vivo (6,7). Identifying factors that promote Aβ aggregation may thus be important for understanding the pathophysiology of AD and developing therapeutic strategies.
The mechanism by which Aβ aggregates in the brain is not fully understood, although there is increasing evidence that metal ions may play a role. Physiological levels of copper and zinc have been shown to accelerate Aβ aggregation (8–10), and trace levels of copper and zinc may initiate seeding and oligomerization of Aβ (11). It has also been suggested that the binding of Aβ to the membrane sialoglycolipid GM-1 ganglioside can lead to a conformational change in Aβ and seeding of aggregation (12). Recent evidence suggests that Aβ1–42 is the Aβ species required for the seeding of amyloid fibril formation and plaque deposition in vivo, as mice over-expressing only Aβ1–42 developed amyloid plaques, whereas mice over-expressing only Aβ1–40 did not (13). In this report we show that calcium can accelerate aggregation of Aβ1–42, with marked effects on the formation of early protofibrillar structures, as well as the protofibril to fibril conversion event. Both protofibrillar and fibrillar forms of Aβ are neurotoxic. These effects of calcium on Aβ aggregation occurred at concentrations 20-fold lower than the 2 mM calcium concentration present in the extracellular space of the brain, implicating a potential role for calcium in Aβ aggregation and toxicity in the AD brain.
EXPERIMENTAL PROCEDUIRES
Chemicals and reagents
All cell culture reagents were purchased from Invitrogen, chemicals were from Sigma-Aldrich, unless stated otherwise.
Preparation of Aβ
Aβ1–42 was from either rPeptide, Biosource, or Bachem and all gave similar results (Aβ1–42 was used for all the experiments described and will be referred to as Aβ from here on). Aβ was solubilized in 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP) at a concentration of 1 mM. HFIP was allowed to evaporate off in a fume hood, followed by drying under vacuum in a Savant SpeedVac. The Aβ HFIP film was stored at −20 °C until use. For experiments in which Aβ was aggregated in the presence of 150 mM NaCl or in Dulbecco’s Modified Eagle Medium (DMEM), Aβwas resuspended at 200 μM in reverse-osmosis purified (RO) water (MP Biomedicals), and taken to pH 11.5 with 1N NaOH to solubilize the peptide. Aβ was then filtered through a 30 kD Ultrafree-MC with Ultracel-PL membrane centrifugal filter (Millipore). The concentration of Aβ post filtration was determined using the Bio-Rad Dc Protein assay kit, with unfiltered Aβ as a standard.
Aβ aggregation
Aβ solubilized in HFIP was resuspended at 2 mM in dimethyl sulfoxide (DMSO). 2 mM Aβ in DMSO was diluted to 100 μM in 75 mM MOPS pH 7.4 buffer, and metal ions added as described in the text. pH was confirmed to be 7.4 by using an Orion Micro pH electrode (Thermo Electron Corporation) or by the addition of phenol red. Aggregation was at 37 °C without stirring. For investigation of Aβ aggregation at early time-points, Aβ was brought to 100 μM in MOPS buffer as described above, and immediately centrifuged at 320,000 × g for 1.5 hours in an Optima TLX desktop ultracentrifuge (Beckman Coulter) to remove pre-existing aggregates. Aβ concentration was determined using the Bio-Rad Dc Protein assay kit, with non-centrifuged Aβ as a standard, and the Aβ aged at 50 μM at 37 °C without stirring. For aging in the presence of 150 mM NaCl or in DMEM, 30 kD filtered Aβ was brought to pH 7.4 with 75 mM MOPS buffer, and aged at 50 μM at room temperature, or 10 μM at 37 °C, without stirring. All aggregation experiments were performed at least 3 times per condition.
Congo red binding assay
The Congo red assay was performed according to Klunk et al (14). Aβ and Congo red (dye content 97%) were diluted to 8–10 μM and 25 μM respectively in phosphate buffered saline (PBS) to a final volume of 150 μL. Aβ aged at 10 μM was used without dilution. Absorbance was read at 405 nm and 540 nm in a Titertek Multiskan Plus 96 well plate spectrophotometer. μM Congo red binding by Aβ fibrils was calculated using equation 13 of Klunk et al (14).
Thioflavin T binding assay
The thioflavin T binding assay was performed according to Naiki and Nakakuki (15). 0.5 μM Aβ and 5 μM thioflavin T diluted in 50 mM glycine buffer pH 8.5 in a total volume of 2 mL were analyzed in a F-4500 fluorescence spectrophotometer (Hitachi). Excitation was set at 446 nm, and emission data collected at 485 nm.
Electron microscopy
For protofibril and fibril visualization, Aβ was centrifuged at 14000 rpm in a desktop centrifuge (Eppendorf) at 4 °C for 30 minutes. The supernatant was removed and the pellet resuspended at 10 μM in RO water. For preformed fibrils and protofibrils that had been added to culture medium for 24 hours at 20 μM, the medium was centrifuged as above and the pellet resuspended at 100 μM for use in the Congo red assay, and 10 μM for electron microscopy. Aβ aged at 10 μM was diluted 1:1 with RO water. For electron microscopy of early forming Aβ species no spin was performed and samples were diluted 1:1 with RO water. All samples were then prepared for negative staining as follows: 2–5 μL of the sample was adsorbed to a carbon coated grid that was glow discharged in an Edwards Auto 306 vacuum evaporator for 30 seconds. Excess liquid was removed with a filter paper (Whatman #1) and the samples were stained with 1% uranyl acetate or 1% uranyl formate for one minute. The grids were examined using a JEOL 1200EX transmission electron microscope.
Inductively coupled plasma mass spectrometry
Samples were acidified to 5% HNO3 using trace-metal grade concentrated acid (JT Baker). The amount of metal in each sample was quantified by inductively coupled plasma mass spectrometry (ICP-MS; Perkin-Elmer Elan 6100). Standards were prepared from a concentrated multi-element standard (SPEX CertiPrep Inc, Metuchen, NJ) that was serially diluted in 5% HNO3 to obtain working standards in the range of 0.5 μg/L to 25 μg/L. Indium (2 μg/L) was used as an internal standard.
SDS PAGE and Western blotting
0.5 μL of a 100 μM Aβ sample was diluted with RO water and 2X sample buffer to a final volume of 20 μl and 5 μL run on a 10–20% tricine gel (Invitrogen), transferred to PVDF (Whatman) and probed with monoclonal antibody 6E10 (Signet) for the detection of Aβ.
Neuronal cultures
E18 rat hippocampal neurons were plated at 1.25 × 105 per well on coverslips coated with lysine and laminin in 24 well plates, or at 2 × 104 in 96 well plates coated with lysine. Neurons were plated in DMEM supplemented with 10% bovine calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate and penicillin-streptomycin (100 units/mL and 100 μg/mL respectively). After 2 days in culture the medium was replaced with Neurobasal supplemented with B27-AO, and a half media change performed every 3–4 days. Neurons were treated after 6–7 days in vitro (DIV).
Detection of cell death
Apoptosis was detected by the characteristic shrunken and blebbed morphology of neuronal nuclei observed by Hoechst staining. Cell death due to loss of membrane integrity was measured by the release of lactate dehydrogenase (LDH) into the cell culture medium. Aβ was aged at 100 μM in the presence or absence of 1 or 2 mM calcium for 24–48 hours at 37°C and fibril and protofibril content measured by Congo red and thioflavin T as described. Aβ was then added to 6–7 DIV neurons on coverslips in 24-well plates for Hoechst staining, or to 96-well plates for LDH measurement for 24 hours. For Hoechst staining, the culture medium was removed and the neurons fixed in 4% paraformaldehyde for 15 minutes at room temperature, washed in PBS for 5 minutes, stained with Hoechst dye for 5 minutes in PBS, washed in PBS and then water and the coverslips mounted onto glass slides. At least 500 cells were counted per well by an investigator blind to the treatment, and triplicate wells used per condition. The results shown are the average of at least 3 independent experiments. For LDH measurement the CytoTox Non-Radioactive Cyotoxicity Assay kit (Promega) was used according to the manufacturers instructions. Three independent experiments were performed using 6 wells per condition.
RESULTS
Calcium accelerates Aβ protofibril to fibril conversion
We observed that Aβ aggregates more rapidly in tap water than reverse osmosis purified water, and that calcium was the main agent in tap water that correlated with this effect (Supplementary Fig. 1). We therefore decided to investigate the effects of calcium on Aβ aggregation. Aβ was incubated at 100 μM for up to 120 hours at 37 °C in 75 mM MOPS buffer in the presence or absence of 2 mM calcium. The formation of fibrils, as determined by Congo red binding, was significantly accelerated in the presence of calcium. Fibrils start to form within 6 hours in the presence of calcium, but require 48 hours for a similar amount of fibril formation in the absence of calcium (Fig 1A). In contrast, thioflavin T binding did not show a difference in Aβ aggregation kinetics in the presence or absence of calcium (Fig 1B). This suggested that calcium was accelerating the formation of an Aβ species that was recognized by Congo red, but not thioflavin T. To further investigate this difference in Aβ structure, Aβ was Western blotted after incubation for 24 hours in either the presence or absence of 2 mM calcium. A species of approximately 80–120 kD was identified only in the absence of calcium, and Aβ species of 12 kD and 16 kD were also increased in the absence of calcium (Fig 2A). The sample aged in the presence of calcium had little Aβ signal due to the formation of high molecular weight aggregated protein that was unable to enter the gel. We performed electron microscopy on the Aβ species produced after 48 hours in the presence or absence of calcium. When Aβ was incubated with calcium, mature amyloid fibrils formed (Fig 2B). When Aβ was incubated without calcium for 48 hours, small curvilinear structures formed that resembled previously described protofibrils (16,17), as well as a few longer fibrils (Fig 2B). The protofibrillar structures had lengths less than 200 nm, also consistent with the dimensions of previously described protofibrils (16,17). Therefore, it appeared that protofibrils were forming in either the presence or absence of calcium, giving rise to a positive thioflavin T signal and the presence of the 80–120 kD Aβ species identified by immunoblotting. However, in the presence of calcium, protofibrils were rapidly converted to mature amyloid fibrils. To confirm this sequence of events, an electron microscopy time course was performed. After 6 hours of incubation in the presence of calcium, both protofibrils and fibrils were present, but by 24 hours only mature amyloid fibrils were detected (Fig 3). In contrast, Aβ incubated in the absence of calcium became protofibrillar by 6 hours and remained protofibrillar for up to 72 hours of incubation before converting to fibrils (Fig 3). These results suggest that calcium promotes the conversion of protofibrils to mature amyloid fibrils.
Fig. 1. Calcium accelerates the formation of Congo red but not thioflavin T positive Aβ fibrils.
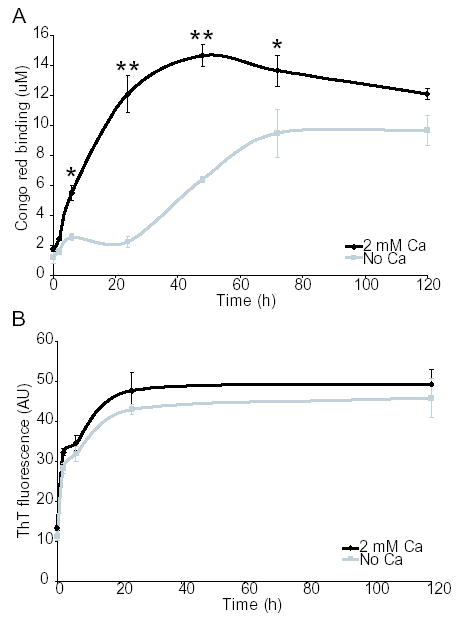
Aβ was incubated at 100 μM in 75 mM MOPS at 37 °C in the presence or absence of 2 mM calcium. A, Congo red binding. B, thioflavin T binding. *, P<0.01, **, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. Each data point comprises 3–5 independent experiments, error bars are SEM.
Fig. 2. Calcium accelerates protofibril to fibril conversion.
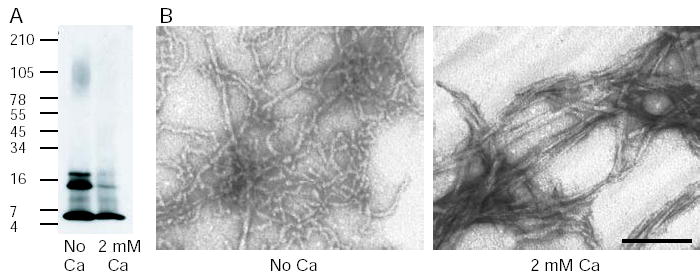
Aβ was incubated at 100 μM in 75 mM MOPS at 37 °C in the presence or absence of 2 mM calcium for 24 – 48 hours. A, immunoblotting with an anti-Aβ antibody (6E10) reveals a species of approximately 80–120 kD when Aβ is incubated in the absence of calcium, presumably protofibrils that bind thioflavin T but not Congo red. This species is not present when Aβ is incubated in the presence of calcium, due to the formation of mature fibrils that do not enter the gel. B, electron microscopy shows small curvilinear protofibrils form when Aβ is incubated in the absence of calcium and mature amyloid fibrils form when calcium is present. The scale bar is 100 nm.
Fig. 3. Time course of fibril formation in the presence and absence of calcium.
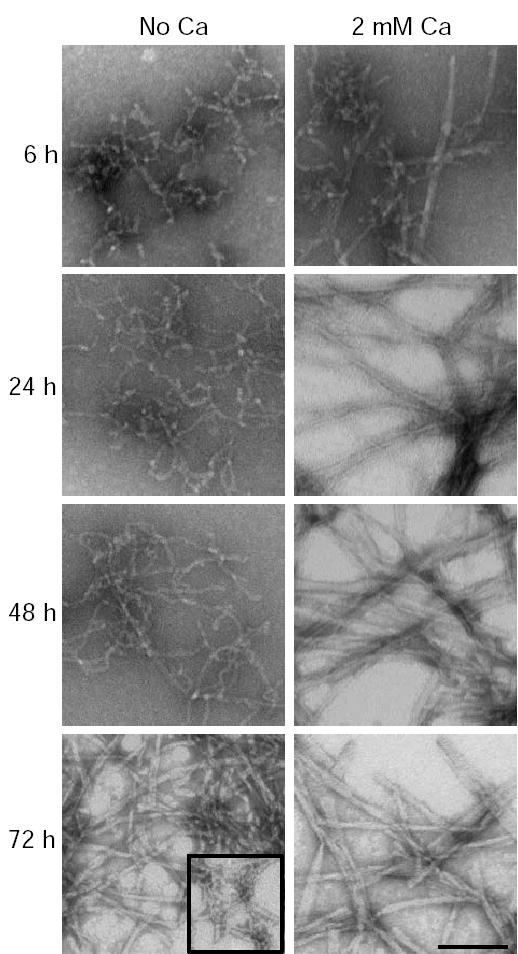
Electron microscopy was performed on Aβ incubated at 37 °C in 75 mM MOPS in the presence or absence of 2 mM calcium for 2, 6, 24, 48 and 72 hours. At 6 hours with calcium the Aβ is a mixture of protofibrils and fibrils. By 24 hours only mature fibrils are present. In the absence of calcium, protofibrils are present at 6 and 24 hours. At 48 hours the majority of the Aβ is protofibrillar with some longer fibril like structures. At 72 hours most Aβ is fibrillar, but some protofibrils remain, shown in the boxed inset. The scale bar is 100 nm.
Zinc and copper do not accelerate protofibril to fibril conversion
It has previously been shown that physiological levels of copper and zinc can accelerate Aβ aggregation (8,9). We therefore tested whether these concentrations of copper and zinc had an effect on protofibril to fibril conversion. Aβ was aged for 24 hours in the absence of metal ions, or in the presence of either 2 mM calcium, 20 μM copper or 20 μM zinc. Congo red and thioflavin T binding showed that only calcium was able to accelerate protofibril to fibril conversion (Fig 4). We also measured the levels of copper and zinc in our 2 mM calcium solution by inductively coupled plasma mass spectrometry to ensure that contaminating trace copper and zinc were not having an effect. Copper was below our detection limits of 0.5 μg/L, and zinc was at the level of our detection limits, 0.5 μg/L, ± std dev 0.039. This is equivalent to an upper limit of 140 nM zinc. These results suggest that copper and zinc do not have an effect on protofibril to fibril conversion in this Aβ aging paradigm.
Fig. 4. Zinc and copper do not accelerate protofibril to fibril conversion.
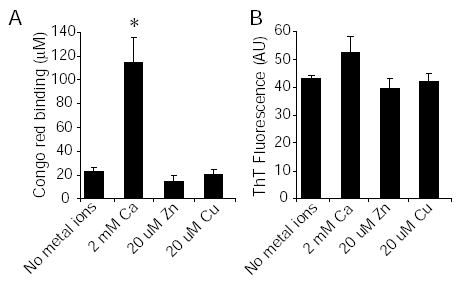
Aβ was incubated at 100 μ M in 75 mM MOPS at 37 °C with either no metal ions, 2 mM calcium, 20 μM zinc or 20 μM copper for 24 hours. A, Congo red binding. B, thioflavin T binding. *, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. Each data point comprises 3 independent experiments, error bars are SEM.
Calcium accelerates Aβ aggregation at a range of calcium and Aβ concentrations
We then determined the concentration range for calcium-induced acceleration of protofibril to fibril conversion. A standard curve shows that the EC50 for calcium-mediated fibril formation was approximately 4 mg/l (100 μM) (Fig 4). This is 20-fold lower than the 2 mM calcium concentration in the extracellular space of the brain, the site at which amyloid plaque deposition occurs. Thus, calcium in the extracellular space of the brain may provide a favorable milieu for amyloid fibril formation and plaque deposition.
The effects of calcium were then assessed in the presence of a physiological concentration of sodium chloride at lower Aβ concentrations. Aβ was first passed through a 30 kD filter to remove preformed larger aggregates. Aβ was aged at 50 μM at room temperature, or at 10 μM at 37 °C. Congo red binding, thioflavin T binding and electron microscopy showed that under these conditions calcium accelerated protofibril to fibril conversion (Fig 6A–D). These experiments show that calcium accelerates Aβ aggregation in the presence of physiological concentrations of salt.
Fig. 6. Calcium accelerates protofibril to fibril conversion in the presence of a physiological concentration of sodium chloride.

Aβ was incubated in 75 mM MOPS with 150 mM NaCl at 50 μM at room temperature (A–C) or 10 μM at 37 °C (D–E). A, Congo red binding. B, thioflavin T binding. C, electron microscopy at 38 hours. D, Congo red binding. E, thioflavin T binding. F, electron microscopy at 12 hours. The scale bar is 100 nm. *, P<0.05, **, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. Each data point comprises 3–4 independent experiments, error bars are SEM.
Calcium accelerates the formation of early Aβ aggregates
To determine if calcium can also accelerate the formation of early Aβ aggregates, we analyzed the aggregation state of Aβ in the presence or absence of calcium during the first 2 hours of incubation. Freshly solubilized Aβ was centrifuged for 1.5 hours at 320,000 × g to remove preformed aggregates. Electron microscopy performed following centrifugation showed that just a few, small spherical Aβ aggregates remained (Fig 7B), which did not lead to a thioflavin T positive signal (Fig 7A). After 30 minutes of incubation, there was a small increase in thioflavin T-positive species in the presence of calcium (Fig 7A). By electron microscopy, the effect of calcium was more striking. Short individual protofibrils were observed as well as aggregates of protofibrils (Fig 7B). After one hour of incubation in the presence of calcium, thioflavin T binding was significantly greater than that observed in the absence of calcium (Fig 7A), and protofibrillar and fibrillar structures were apparent by electron microscopy (Fig 7B). These results suggest that calcium accelerates the formation of protofibrillar as well as fibrillar Aβ aggregates.
Fig. 7. Calcium accelerates the formation of early Aβ aggregates.
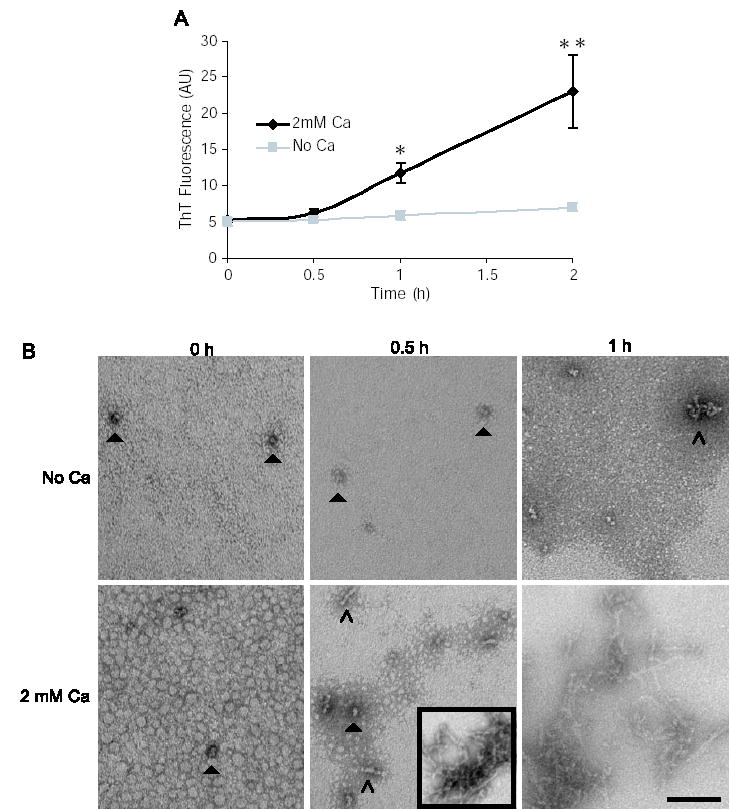
Aβ was incubated at 50 μM in 75 mM MOPS at 37 °C with or without 2 mM calcium. A, thioflavin T binding was measured at 0, 0.5, 1 and 2 hours. B, electron microscopy was performed at 0, 0.5, and 1 hour. Small spherical structures (closed arrows) are observed at 0 and 0.5 hours with and without calcium. Short protofibrils (open arrows) and larger protofibrillar aggregates (boxed inset), are observed at 0.5 hours only in the presence of calcium. The scale bar is 100 nm. *, P<0.05, **, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. Each data point comprises 4–6 independent experiments, error bars are SEM.
Protofibrils and fibrils are both neurotoxic
To investigate the neurotoxicity of fibrils and protofibrils, these two forms were selectively generated by incubating Aβ in the presence or absence of calcium, respectively. Preformed fibrils and protofibrils were then added to primary rat hippocampal neuronal cultures. The extent of protofibril to fibril conversion in the culture medium of neurons after a 24 hour incubation was determined by Congo red binding and electron microscopy (Fig 8A, B). There was no significant conversion to fibrils during this time period suggesting that protofibrils are stable in the cell culture medium. Thus, Neurobasal/B27-AO medium, which contains 1.8 mM calcium (18), did not promote the conversion of protofibrils to fibrils within 24 hours. In contrast, Dulbecco’s Modified Eagle Medium (DMEM) containing calcium significantly accelerated Aβ aggregation and fibril formation, as determined by Congo red binding, thioflavin T binding and electron microscopy (Fig 8C–E). It is unclear why our preformed protofibrils are stable in Neurobasal/B27-AO. However, this enabled us to examine the neurotoxicity of stable Aβ protofibrils.
Fig. 8. Aβ protofibrils and fibrils are both toxic to neurons in culture.
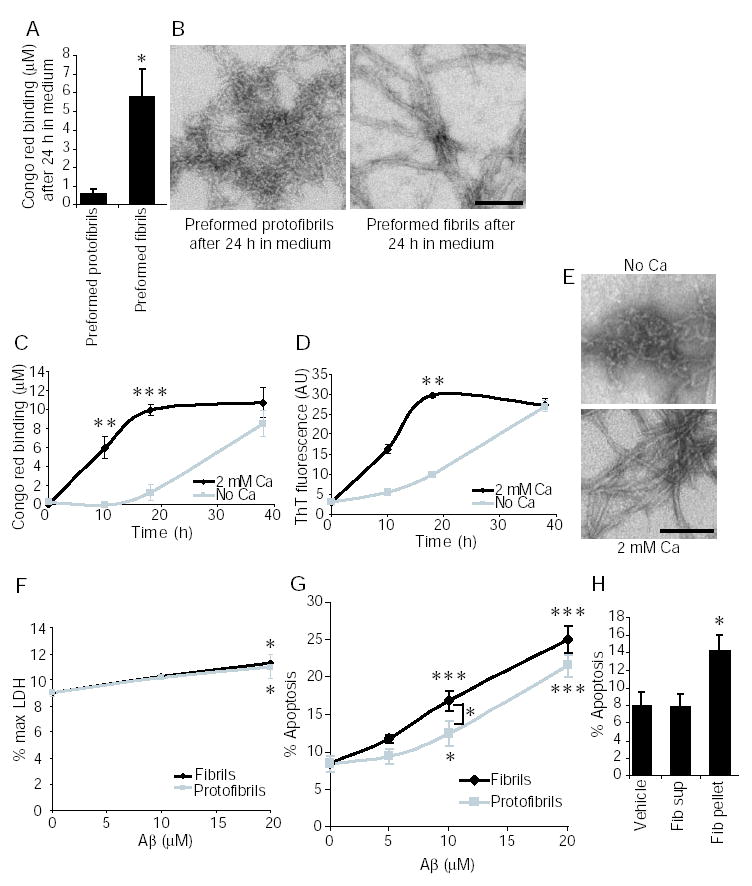
Aβ was incubated at 100 μM in the presence of 1 or 2 mM calcium to generate preparations of fibrils or protofibrils which were then added to neuronal cell cultures (A,B,F–G). Aβ was also aggregated at 50 μM at room temperature in DMEM in the presence and absence of calcium, to examine the effect of calcium on Aβ aggregation in cell culture medium (C and D). A, Congo red binding of Aβ in the conditioned medium was measured after a 24 hour incubation with neurons. *, P<0.05, t test. B, conditioned medium from protofibril and fibril treated cultures was removed at 24 hours and the Aβ species present in the medium analyzed by negative staining electron microscopy. C, Congo red binding of Aβ aggregated in DMEM. D, thioflavin T binding of Aβ aggregated in DMEM. E, electron microscopy of Aβ aged in DMEM for 18 hours. F, LDH assay after a 24 hour treatment with protofibrils and fibrils. G, neuronal cell death after a 24 hour treatment with protofibrils or fibrils was determined by counting apoptotic nuclei as determined by morphology using Hoechst staining. At 10 μM fibrils are significantly more toxic than protofibrils. H, Fibrillar Aβ was prepared by aging in the presence of 1 mM calcium. The volume of Aβ required for a 10 μM treatment was centrifuged at 18,000 × g for 30 minutes. The pellet (pel) and supernatant (sup) were then tested for neurotoxicity after 24 hours as described above. The scale bars in panels B and E are both 100 nm. For panels C–H, *, P<0.05; **, P<0.01, ***, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. The number of independent experiments that comprise each data point for each panel are: A, 5. C and D, 4. F, 3, G, 5–7. H, 3. Error bars are SEM in all panels.
Cell death was measured after 24 hours to ensure that the protofibrils had not converted to fibrils. Hoechst dye was used to identify the characteristic nuclear morphology of apoptotic cells. The LDH assay was used to measure cell death via loss of membrane integrity through release of lactate dehydrogenase (LDH) into the cell culture medium. The LDH assay gave a very small but statistically significant increase in cell death with both protofibrils and fibrils at 20 μM (Fig 8F). The insensitivity of the LDH assay for measuring protofibril and fibril toxicity over the relatively short time period of 24 hours has been previously described, with 2–3 days required to see significant cell death (4). We therefore focused on the more sensitive method of scoring apoptotic cells using Hoechst dye. Both protofibrils and fibrils induced neuronal apoptosis, with a trend towards increased toxicity of fibrils versus protofibrils that was statistically significant at a concentration of 10 μM (Fig 8G). To ensure that small amounts of soluble Aβ species that might remain in our fibrillar preparations after in vitro aggregation were not responsible for the observed toxicity, we centrifuged the fibrillar Aβ preparation and compared the toxicity of the pellet and supernatant. Only the pelletable insoluble Aβ was neurotoxic while the supernatant did not elicit any neurotoxicity, indicating that small amounts of contaminating soluble Aβ are not responsible for the observed toxicity (Fig 8H). These results suggest that both protofibrils and fibrils are inherently neurotoxic.
DISCUSSION
The aggregation of Aβ is a characteristic pathological feature of AD that may contribute to neurodegeneration. These experiments indicate that physiological concentrations of calcium markedly accelerate Aβ aggregation. Calcium had significant effects on the generation of both early protofibrillar forms of Aβ, as well as the conversion of protofibrils to mature amyloid fibrils. The concentration of calcium in the extracellular space of the brain is approximately 2 mM, which is 20-fold higher than the calcium concentration required to promote fibril formation in vitro. Thus, calcium may play an important role in Aβ aggregation and amyloid deposition in AD. Moreover, the increased formation of early protofibrillar aggregates suggests that calcium may play a role in the initiation of Aβ aggregation.
A role for metal ions in accelerating Aβ aggregation has previously been reported for zinc and copper (8,9). We did not observe an effect of zinc and copper on protofibril to fibril conversion under the conditions used. The effects of copper and zinc may occur at earlier stages of Aβ aggregation. This would be consistent with evidence that trace levels of copper and zinc can initiate Aβ aggregation and increase oligomer formation (11). The studies on zinc and copper were performed with Aβ40 (8,9), while this study used Aβ42, which could also explain the differences observed. Further, the copper effect required a slightly acidic pH, which was not required for the effects of calcium, which occurs at physiological pH (8). In addition, the effect of zinc on Aβ aggregation is salt dependant and reversible by zinc chelation (10). In contrast, the effects of calcium are independent of salt concentration and were not reversed by calcium chelation (data not shown). This difference in the stability of zinc- and calcium-induced aggregates suggests that they may have different conformations. The conformation of different Aβ species clearly has an effect on their ability to bind the dyes Congo red and thioflavin T. The inability of protofibrils to bind Congo red that we observed is consistent with another report (4). However, a different study reported variable binding of Congo red to protofibrils (19).
The stability of protofibrils in the absence of calcium enabled an assessment of their effects on cultured neurons, providing evidence that both protofibrils and fibrils are neurotoxic, consistent with previous reports (4,6,7). The toxicity of protofibrils appears to be inherent to the protofibrillar structure, as we did not detect significant conversion to fibrils in the conditioned medium of cells treated with protofibrils. This lack of conversion in tissue culture medium appears to be a property of Neurobasal/B27-AO medium, as protofibril conversion did occur in DMEM. The reason for this is presently unclear, but may relate to the presence of albumin in B27 medium (18), which has been shown to bind Aβ(33). Fibrils also appear to be inherently neurotoxic, as we did not detect protofibrillar contamination in fibrillar preparations generated in the presence of calcium. As a further control, we centrifuged fibrillar Aβ and compared the toxicity of the peptide in the pellet and supernatant. Only the insoluble pelleted fraction that contained the fibrils was toxic, indicating that fibrils alone can mediate toxicity. The supernatant is likely to contain very small amounts of soluble Aβ, as most of the Aβ converts to fibrils during aging in the presence of calcium. Therefore, the lack of neurotoxicity of the supernatant does not suggest that soluble Aβ per se is not toxic, but that it cannot explain the toxicity of the fibrils in this case. Previous reports suggest that soluble oligomeric Aβ can be neurotoxic (3,4). Another study, however, reported that neither fibrils nor soluble Aβ were toxic unless they were combined, suggesting that ongoing polymerization of Aβ is required for Aβ toxicity (34). While this is likely to be an important mechanism, our results suggest it is not required, as Aβ fibrils alone are neurotoxic.
Several lines of evidence suggest that calcium homeostasis may become altered in the aging brain. Using microarray analysis, we have shown that mRNA levels of the calcium binding proteins calbindin and calretinin decrease significantly in the aging human brain, as does PMCA2, a plasma membrane calcium ATPase responsible for pumping calcium out of the cell (21). Decreases in calbindin and calretinin have also been detected by immunohistochemistry in the brains of aged humans, monkeys and rodents (22–25). These changes would be expected to lead to an increase in intracellular free calcium, which could potentially initiate Aβ aggregation. Calcium homeostasis may also be altered by AD-causing mutations in presenilin 1 (PS1) (26,27), and several lines of evidence suggest that Aβ itself can increase intracellular calcium levels (28–31). Moreover, memantine, an NMDA receptor antagonist that reduces calcium influx, can improve cognition in moderate to severe AD patients (32). Thus, calcium dysregulation may contribute to the pathogenesis of AD by increasing the formation of neurotoxic aggregated forms of Aβ.
Fig. 5. Standard curve for calcium induced protofibril to fibril conversion.

A, Aβ was incubated at 100 μM in 75 mM MOPS at 37 °C for 48 hours in the presence of varying amounts of calcium. Fibril formation was determined by Congo red binding. *, P<0.01, **, P<0.001 using ANOVA and Student-Neuman-Keuls post hoc test. Each data point comprises 3 independent experiments, error bars are SEM.
Acknowledgments
We thank Maria Ericsson for assistance with electron microscopy, Vernon Ingram and Atta Ahmad for advice on Aβ filtration, and Raj Gopalakrishnan, Matthew Hass, Patrick Loerch and Tao Lu for helpful discussion. This work was supported by National Institutes of Health Grants AG17573 and NS030352 (to B.A.Y), and fellowships from the Harvard Center for Neurodegeneration and Repair and the Lefler Foundation (to A.M.I.).
References
- 1.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 2.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 3.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 6.Lorenzo A, Yankner BA. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 8.Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI. J Biol Chem. 1998;273:12817–12826. doi: 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- 9.Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 10.Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel JP, Tanzi RE, Bush AI. J Biol Chem. 1997;272:26464–26470. doi: 10.1074/jbc.272.42.26464. [DOI] [PubMed] [Google Scholar]
- 11.Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. J Biol Inorg Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, Yamamoto N, Michikawa M, Yoshikawa Y, Terao K, Matsuzaki K, Lemere CA, Selkoe DJ, Naiki H, Yanagisawa K. J Neurosci. 2004;24:4894–4902. doi: 10.1523/JNEUROSCI.0861-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klunk WE, Jacob RF, Mason RP. Anal Biochem. 1999;266:66–76. doi: 10.1006/abio.1998.2933. [DOI] [PubMed] [Google Scholar]
- 15.Naiki H, Nakakuki K. Lab Invest. 1996;74:374–383. [PubMed] [Google Scholar]
- 16.Harper JD, Wong SS, Lieber CM, Lansbury PT. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 17.Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- 18.Brewer GJ, Torricelli JR, Evege EK, Price PJ. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 19.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen MS, Vorum H, Lindersson E, Jensen PH. J Biol Chem. 2001;276:22680–22684. doi: 10.1074/jbc.M101181200. [DOI] [PubMed] [Google Scholar]
- 21.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 22.Fahandejsaadi A, Leung E, Rahaii R, Bu J, Geula C. Neuroreport. 2004;15:443–448. doi: 10.1097/00001756-200403010-00012. [DOI] [PubMed] [Google Scholar]
- 23.Wu CK, Nagykery N, Hersh LB, Scinto LF, Geula C. Neuroscience. 2003;120:249–259. doi: 10.1016/s0306-4522(03)00248-3. [DOI] [PubMed] [Google Scholar]
- 24.Bu J, Sathyendra V, Nagykery N, Geula C. Exp Neurol. 2003;182:220–231. doi: 10.1016/s0014-4886(03)00094-3. [DOI] [PubMed] [Google Scholar]
- 25.Villa A, Podini P, Panzeri MC, Racchetti G, Meldolesi J. Eur J Neurosci. 1994;6:1491–1499. doi: 10.1111/j.1460-9568.1994.tb01010.x. [DOI] [PubMed] [Google Scholar]
- 26.Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM. J Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack–Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 28.Arispe N, Rojas E, Pollard HB. Proc Natl Acad Sci U S A. 1993;90:567–571. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 30.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mark RJ, Hensley K, Butterfield DA, Mattson MP. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Jama. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- 33.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. J Biol Chem. 1996;271:32916–32922. doi: 10.1074/jbc.271.51.32916. [DOI] [PubMed] [Google Scholar]
- 34.Wogulis M, Wright S, Cunningham D, Chilcote T, Powell K, Rydel RE. J Neurosci. 2005;25:1071–1080. doi: 10.1523/JNEUROSCI.2381-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]