

Abstract
Nitric oxide (NO) was found to inhibit the copper-dependent induction of the yeast CUP1 gene. This effect is attributable to an inhibition of the copper-responsive CUP1 transcriptional activator Ace1. A mechanism is proposed whereby the metal binding thiols of Ace1 are chemically modified via NO- and O2-dependent chemistry, thereby diminishing the ability of Ace1 to bind and respond to copper. Moreover, it is proposed that demetallated Ace1 is proteolytically degraded in the cell, resulting in a prolonged inhibition of copper-dependent CUP1 induction. These findings indicate that NO may serve as a disrupter of yeast copper metabolism. More importantly, considering the similarity of Ace1 to other mammalian metal-binding proteins, this work lends support to the hypothesis that NO may regulate/disrupt metal homeostasis under both normal physiological and pathophysiological circumstances.
Nitric oxide (NO) is a molecule of extreme interest because of its numerous physiological functions (for a review, see ref. 1). For example, NO generated by vascular endothelial cells is a messenger molecule involved in the maintenance of vascular tone via interaction with the heme enzyme guanylate cyclase (2). Nitric oxide is also involved in central nervous system function, where it serves as an activator of guanylate cyclase (3). Thus, in its role as a mediator of vascular tone and as a signal transduction agent in the central nervous system, NO appears to act primarily by activating guanylate cyclase, leading to increased levels of the cellular messenger cGMP. However, NO generated from activated macrophages during an immune response is thought to have cytotoxic and/or cytostatic properties against target organisms in a largely cGMP-independent fashion (4). One important cGMP-independent function of NO is as a regulator of cellular iron metabolism (5), as NO has been found to alter the activity of iron regulatory protein, an important protein involved in the maintenance in iron homeostasis (6). It is likely that alterations in metal metabolism elicited by NO may represent an important aspect of its pathophysiology.
It appears that one important aspect of NO physiology and pathophysiology involves its interactions with protein thiols. That is, the activation and inactivation of thiol-containing proteins by NO are thought to be responsible for some of the biological actions of NO (for reviews, see refs. 7–9). Recent and profound discoveries in the yeast Saccharomyces cerevisiae indicate that the intracellular transport, sequestration, and uptake of metals occurs through the actions of many thiol-containing proteins. For example, the yeast copper chaperones Lys7, Atx1, Cox17, and Ccc2 all appear to bind copper via thiol ligation (ref. 10 and references therein). Also, the yeast metallothioneins sequester “free” copper by using thiol-rich metal binding domains (11). Finally, the yeast metal-responsive transcription factors Ace1 and Mac1 are responsible for the expression of a variety of metal regulatory proteins and appear to respond to copper by binding it in an analogous fashion to metallothionein (12–16). Considering the known chemical and biochemical interactions of NO and NO-derived species (collectively referred to as NOx and described chemically later) with thiols, it is not unreasonable to suspect that NO may be capable of regulating/disrupting yeast metal metabolism by reacting with the metal-binding thiol ligands in proteins. Furthermore, because of the increasing evidence that a high degree of homology exists between the metal metabolizing systems in yeast and mammalian cells, it is likely that effects observed in yeast will serve as a model for similar effects in mammalian cells. Therefore, we have utilized S. cerevisiae to examine specifically the interaction of NO with the thiol-containing and metal-responsive yeast transcription factor Ace1. The interactions between Ace1 and NO are representative of how NO may effect metal-binding thiol proteins and how this interaction may alter cellular metal metabolism.
Experimental Procedures
Chemicals and Solutions.
Diethylammonium-1-(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA/NO) was synthesized by the method of Drago and Paulik (17). Stock solutions of DEA/NO were prepared in double distilled water (DDW) immediately before use. Diphenylthiocarbazone and o-nitrophenyl-β-d-galactopyranoside were purchased from Sigma, and bathocuproine disulfonic acid was purchased from Aldrich. The Cu(I)-acetonitrile complex (CuI(CH3CN)2ClO4) was synthesized according to the method of Hemmerich and Sigwart (18). All other chemicals and solutions were purchased from commercial suppliers and were of the highest purity available.
Yeast Strain, Plasmid, and Transformation.
The parental strain in this study is the wild-type strain EG217 (MATa leu2-3, 112 trp1-1 his3-11, 15 ura3-1 ade2-1). A yeast high copy number reporter plasmid harboring CUP1-β-galactosidase (lacZ) fusion gene YEpCUP-HSE-M-lacZ was constructed as described (19). An Ace1 over-expression plasmid (YEpACE1) and an ace1Δ mutant (KKY7, W303 1a containing the ace1Δ 255 complete deletion of the ACE1 ORF) were constructed as described (ref. 20; D. Koch and D.J.T., unpublished work).
Transformations of yeast were performed according to the method of Elble (21) with modification (22). Transformants of EG217 with YEpCUP-HSE-M-lacZ and YEpACE1 are referred to as 217CUP and 217ACE, respectively.
Overnight cultures of each strain was inoculated in yeast extract/peptone/dextrose medium or synthetic dextrose medium (SD) supplemented with the appropriate drop-out (OD600 = 0.1) and were grown at 30°C with shaking (275 rpm) (23). Late log phase cultures (10 ml, OD600 = 3–4) were utilized for all experiments unless otherwise noted.
β-Galactosidase Activity.
Decomposed DEA/NO, made by incubation of DEA/NO in DDW at room temperature overnight, were used as control solutions. Copper in the SD −ura medium was removed by the method of Holmquist (24) by using diphenylthiocarbazone to give SD −ura −Cu. To prevent the contamination by copper of the 217CUP preculture, bathocuproine disulfonic acid (BCS) was used to chelate Cu(I) in the medium. Thus, preculture of 217CUP was performed in 10 ml of SD −ura −Cu containing 100 μM BCS. Treatment regimens with DEA/NO and/or 2-mercaptoethanol (2-ME) are indicated in the figure legends. To induce the CUP1 promoter-linked β-galactosidase activity, the cells were washed with DDW twice and were resuspended with SD −ura containing 100 μM CuSO4. After incubation at 30°C for 0–8 hours with shaking (275 rpm), cells were washed with DDW, were frozen with liquid nitrogen, and were stored at −80°C until use. The cells were defrosted on ice, and 2 volumes of acid-treated glass beads (0.5 mm diameter) and 2–4 volumes of Z buffer (100 mM NaPi, pH 7/10 mM KCl/1 mM MgSO4/50 mM 2-ME) were added. The mixture was vigorously mixed by vortexing six times for 30 seconds at 1-min intervals. The supernatant was clarified by centrifugation at 15,000 × g for 5 min at 4°C. Enzyme activity was assayed by the general method of Thorvaldsen et al. (25) with slight modifications. In brief, the supernatant of the cell homogenate (5–10 μg of protein) was incubated in 1 ml of Z buffer containing 0.63 mg/ml o-nitrophenyl-β-d-galactopyranoside at 30°C for 5 min. The reactions were stopped by addition of 0.3 ml of 1 M Na2CO3, and the absorbance at 420 nm was measured. Protein concentrations were determined by the method of Bradford by using BSA as a standard (26). Units of β-galactosidase are defined as absorbance at 420 nm × 103⋅min−1⋅mg protein−1. 5′-TCGAGAGCGATGCGTCTTTTCCGCTGAACCGTTCCAGCAAAAAAGACTAC-3′ 3′- CTCGCTACGCAGAAAAGGCGACTTGGCAAGGTCGTTTTTTCTGATG-5′
Anaerobic Experiments.
Late log phase cultures of 217CUP (10 ml) were transferred to 25-ml Erlenmeyer flasks, some of which were equipped with gas tight rubber septa. For the flasks to be made anaerobic, nitrogen gas was swept through the flask headspace, using syringe needles placed through the septa, at 30°C with shaking for at least 30 min. The concentration of oxygen in a representative solution was monitored by using a Yellow Springs Instruments oxygen monitor. When the O2 in the medium decreased to <1–2% of the air saturation value, an anaerobic stock solution of DEA/NO (0 to 1 mM, final concentration) was injected into the flask using a gas tight syringe. The mixture was then incubated at 30°C for 60 min with shaking. Comparative experiments run under aerobic conditions were carried out identically except that the flasks were not swept with N2 gas and were left open to air. To assure that no NO remained in solution before workup of the anaerobic experiments, solution levels of NO in the flasks were monitored by using a previously described procedure (27). Thus, anaerobic experiments were worked up only after NO was no longer detected in the reaction solutions. After exposure to DEA/NO under either anaerobic or aerobic condition, cells were washed with DDW twice and were incubated in SD −ura plus 0.1 mM CuSO4 for 1 hour at 30°C with shaking. Then, β-galactosidase activity in the cells was measured as described above.
Electrophoretic Mobility-Shift Assay (EMSA).
Yeast cell extraction and EMSA were performed according to the methods of Fürst et al. (13). KKY7 and 217ACE were grown in 800 ml of synthetic dextrose complete medium (SDC) and SD −leu, respectively. Cells were collected, were pelleted on a clinical centrifuge, and were washed twice with homogenization buffer (0.2 M Tris⋅HCl, pH 8.0/0.4 M ammonium sulfate/10% glycerol/5 mM EDTA/7 mM 2-ME/1 mM PMSF). The final pellet was then resuspended with 0.5 packed cell volume of homogenization buffer. The suspensions were vortexed with acid-treated glass beads (0.5-mm diameter) for 5 min at 4°C. After centrifugation at 4°C, an equal amount of saturated ammonium sulfate was added to the supernatant and was incubated on ice for 30 min. The precipitate was collected by centrifugation at 20,000 × g for 30 min at 4°C, and the pellet was resuspended in 20 ml of Na-Hepes (pH 8.0) containing 10% glycerol. The aliquots were stored at −80°C until use.
The copper-dependent upstream activated sequence (UASc) to which Ace1 specifically binds was amplified by PCR using the CLONTECH Advantage -HF PCR kit and YEpCUP-HSER-M-lacZ as a template. The 62-bp PCR product was designed to have specific restriction sites, XhoI and SnaB I at each end. The PCR fragment was subcloned into pCR2.1 (Invitrogen), and the sequence was confirmed by using a T7 sequencing primer (data not shown). Large scale preparation of the plasmid was performed by standard methods (28). After cleaving the plasmid with XhoI and SnaB I, the 50-bp fragment of UASc DNA was separated by using 1.5% agarose gel electrophoresis. Centrifuge tubes (0.6 ml, plastic) were then prepared by perforating a small hole into their bottom with a needle. The bands containing the fragment were then cut out and transferred to the modified 0.6-ml centrifuge tubes. These tubes were then placed into 1.5-ml centrifuge tubes and were centrifuged at 12,000 × g for 10–30 min at 4°C. After centrifugation, to the gel (now in the outer tube) was added an equal volume of neutralized phenol. The mixture was vortexed vigorously to form an emulsion. The emulsion was frozen at −80°C for 30 min. After defrosting at room temperature, the mixture was then centrifuged at 15,000 × g for 5 min. The aqueous layer was then separated, and the DNA was precipitated by using ethanol. The fragment was confirmed by 12% polyacrylamide gel electrophoresis. The UASc DNA obtained was used for EMSA as a probe. The oligonucleotide has the following sequence:
In the EMSA experiments, CuI(CH3CN)2ClO4 was used to activate Ace1. Stock solutions (1 mM) of CuI(CH3CN)4ClO4 were made just before use by dissolution into 1 M acetonitrile. The Ace1-DNA binding reaction (10 μl) contained 0.01 pmol (60,000 cpm) of oligonucleotide labeled with a klenow fragment (Promega) and [α-32P]TTP, 0.3 μg/10 μl of poly (dI-dC), 10 mM Hepes buffer (pH 8), and 5% glycerol. The treatment regimen for CuI(CH3CN)4ClO4, thiol compounds, potassium cyanide (KCN), BCS, and DEA/NO are indicated in the figure legends. After reaction at room temperature, the mixtures were applied onto 6% native polyacrylamide gel and were run at 20 mA for 45–50 min in 25 mM Tris⋅borate (pH 8.3) and 2.5 mM EDTA. The gels were dried on a filter paper and were exposed to x-ray film (Fuji) for 12–24 hours.
Results
To determine the effects of NO on the copper-dependent induction of CUP1, a CUP1 promoter and β-galactosidase gene fusion (CUP1-lacZ) reporter was utilized. Addition of CuSO4 to late log phase cultures of 217CUP resulted in an increase in time- and concentration-dependent β-galactosidase activity (data not shown). These results indicate that the reporter gene system for monitoring Ace1 activation and CUP1 induction was operational.
Examination of the effects of NO on CUP1 induction was carried out by using diethylammonium-1-(N,N-diethylamino) diazen-1-ium-1,2-diolate (DEA/NO, t1/2 = 4–8 min under the conditions of our experiments) as an NO donor. Thus, cells were exposed to 1 mM DEA/NO for 0, 5, 10, 30, and 60 min. After DEA/NO exposure, cells were washed with DDW and were exposed to CuSO4 for 1 hour. β-galactosidase activity was determined as a measure of CUP1 induction. As shown in Fig. 1A, as little as a 5 min preincubation with 1 mM DEA/NO resulted in a near complete inhibition of copper-mediated CUP1 induction.
Figure 1.

Inhibition of copper-dependent CUP1 induction by DEA/NO. Late log-phase cultures of 217CUP in SD −ura −Cu plus 0.1 mM BCS were used. (A) Time course of inhibition of CUP1 induction by DEA/NO. The cells were incubated with (closed circle) or without (open circle) 1 mM DEA/NO for indicated time at 30°C with shaking (275 rpm). The cells were then homogenized and assayed for β-galactosidase activity. (B) Dose-dependent inhibition of CUP1 induction by DEA/NO. The cells were exposed to 0–1 mM of DEA/NO (closed square) or decomposed DEA/NO (open square) for 10 min at 30°C with shaking (275 rpm). After washing with DDW twice, the cells were resuspended in SD −ura plus 0.1 mM CuSO4 to start induction of CUP1-lacZ and were incubated at 30°C for 60 min. Representative experiments are shown.
Inhibition of CUP1 induction by NO was also observed with simultaneous exposure of yeast to DEA/NO and copper. For example, simultaneous exposure of yeast to 1 mM DEA/NO and 0.1 mM CuSO4 resulted in near complete inhibition of CUP1 induction even after 2 hours of incubation (data not shown).
Dose-dependent inhibition of copper-mediated CUP1 induction by DEA/NO was also observed. Yeast were exposed to 0–1 mM DEA/NO for 10 min followed by a wash with DDW and exposure to 0.1 mM CuSO4. Increasing concentrations of DEA/NO gave increasing levels of inhibition of CUP1 induction (Fig. 1B). Near complete inhibition of CuSO4-mediated CUP1 induction was observed at 0.4 mM DEA/NO.
The results shown in Fig. 1A indicated that the NO-mediated inhibition of copper-dependent CUP1 induction persisted for up to 1 hour after 1 mM DEA/NO administration. Because of the short half-life of DEA/NO and the instability of NO under the experimental incubation conditions, virtually no NO is present in the incubation solutions after 30 min of exposure to an initial 1 mM DEA/NO exposure (data not shown). Clearly, NO-mediated inhibition of CUP1 induction persists even in the near absence of NO. To test whether NO inhibition of the copper-dependent induction of CUP1 was reversible at longer times, a long term time course experiment was carried out. Thus, 217CUP cells were exposed to 1 mM DEA/NO for 10 min, were washed, and were incubated with CuSO4 for up to 8 hours. β-galactosidase activity was determined at 2-hour intervals. One hour after exposure to DEA/NO, the activity started to increase and recovered to 61% of control after 8 hours (Fig. 2). This result indicates that NO inhibition of copper-dependent CUP1 induction is slowly reversible.
Figure 2.

Time-dependent recovery of CUP1 induction after inhibition by DEA/NO. Late log phase 217CUP cells were incubated with (closed circle) or without (open circle) DEA/NO (1 mM) at 30°C for 10 min with shaking (275 rpm). After washing, the cells were resuspended in SD −ura plus 0.1 mM CuSO4 to induced CUP1-lacZ for 0–8 hours. β-galactosidase activity was then determined. Representative experiments are shown.
Copper-dependent induction of CUP1 occurs via the activation of the copper-responsive transcription factor Ace1. Thus, Ace1 is a likely target for the actions of NO in inhibiting copper-dependent CUP1 induction. To evaluate whether interaction of NO with Ace1 is responsible for the inhibition of copper-mediated CUP1 induction by NO, the effect of DEA/NO on the binding activity of Ace1 to the CUP1 upstream activating sequence, UASc DNA, was examined by using an electrophoretic mobility-shift assay (EMSA). As shown in Fig. 3A, lane 5, an Ace1 adduct with UASc DNA forms in the presence of 0.1 mM Cu(I), and, as expected, copper-dependent Ace1-UASc DNA adduct formation is inhibited by the copper binding agents KCN and BCS (lanes 3 and 4) and is absent in the ace1Δ mutant (lane 2). The multiple bands observed in the EMSA study have been seen in other studies as well (13) and are, as yet, unidentified. Identification of the Ace1-UASc DNA adduct was based on the following observations; (i) only one band is both copper- and Ace1-dependent, (ii) the Ace1-UASc DNA-assigned band was the only one to exhibit a Cu(I) concentration-dependent binding to UASc that reached a binding plateau at 50 μM Cu(I) (data not shown), (iii) the presence of this band was completely inhibited in the presence of either 0.5 mM KCN or 0.5 mM BCS, (iv) incubation of UASc with the cell extract from ace1Δ complete delete mutant in the presence of Cu(I) showed no band at the assigned position.
Figure 3.

Inhibition of Ace1 binding to CUP1 promoter DNA by DEA/NO. Electrophoretic mobility-shift assays were performed using 6% polyacrylamide gel. (A) Cell extracts (3 μg of protein/lane) from KKY7 (lane 2) or 217ACE (lanes 3, 4, and 5) were incubated with 0.5 mM KCN (lane 3) or 0.5 mM BCS (lane 4) at room temperature for 10 min, followed by incubation with 32P-labeled CUP1 UASc DNA (60,000 dpm) and 0.1 mM CuI(CH3CN)2ClO4 (lanes 1, 2, and 5) or 0.1 M acetonitrile (lane 3 and 4) for 10 min at room temperature. Lane 1 contains no cell extract. (B) Cell extracts (217ACE) were incubated with the indicated concentration of DEA/NO (lanes 1–5) or 4 mM decomposed DEA/NO (lane 6) for 10 min at room temperature. To the mixtures were then added 32P-labeled CUP1 UASc DNA (60,000 dpm) and 0.1 mM CuI(CH3CN)2ClO4, and they incubated for 10 min at room temperature. Electrophoresis and exposure to x-ray film are described in Experimental Procedures. Representative experiments are shown.
After validation of the EMSA assay, the effect of NO on the copper-dependent binding of Ace1 to UASc DNA was examined. As shown in Fig. 3B, NO was found to inhibit the copper-dependent binding of Ace1 to the UASc DNA. Under the conditions of this experiment, inhibition of the binding of Ace1 by DEA/NO was observed at DEA/NO concentrations between 1 and 2 mM (Fig. 3B, lanes 2 and 3). At a concentration of 4 mM DEA/NO and above, near complete inhibition of Ace1 binding was observed (Fig. 3B, lanes 4 and 5). The inhibition of Ace1 binding by DEA/NO was attributable to NO because decomposed DEA/NO (4 mM) exhibited no effect (Fig. 3B, lane 6).
Copper binding to Ace1 occurs through thiol ligation. The N-terminal region of Ace1 has been shown to bind four Cu(I) ions through eight cysteine thiols. Thus, Ace1 thiol modification by NO, and NO-derived species, may be responsible for the observed inhibition of activity. To test this idea, the inhibition of copper-dependent Ace1 activity by NO was examined by using exogenously added 2-mercaptoethanol (2-ME). 2-ME was chosen because other thiols, such as N-acetyl cysteine or DTT, altered the copper-dependent activity of Ace1 when added to cells. Exposure of 217CUP cells to DEA/NO in the presence of 10 mM 2 ME showed no inhibition of copper-dependent CUP1 induction. Thus, 2-ME is capable of blocking NO-mediated inhibition of copper -dependent Ace1 activation. However, treatment of cells with 10 mM 2-ME after exposure to DEA/NO did not reverse the inhibition of CUP1 induction (Fig. 4A). These results indicate that 2-ME can protect against NO-mediated inhibition of Ace1, if present at the time of NO exposure. However, 2-ME added after exposure to NO was incapable of reversing NO-mediated inhibition of Ace1.
Figure 4.

Reversibility of Ace1 inhibition by DEA/NO in vivo and in vitro. (A) 217CUP late log phase cells were incubated with (open bars) or without (closed bars) DEA/NO (1 mM) for 10 min at 30°C with shaking (275 rpm) in the absence (left pair of columns) or presence (middle pair of columns) of 10 mM 2-ME. The pair of columns on the right represents experiments in which exposure to 10 mM 2-ME (10 min) occurred after DEA/NO exposure. The cells were induced (CUP1-lacZ) for 60 min and were assayed for β-galactosidase activity as described in Experimental Procedures. (B) Cell extracts of 217ACE were incubated with (lanes 2, 4, and 6) or without (lanes 1, 3, and 5) DEA/NO (4 mM). To the mixtures were added 10 mM 2-ME either during (lanes 3 and 4) or after (lanes 5 and 6) exposure to DEA/NO, and they incubated for 10 min. The EMSA was performed as described in Experimental Procedures. Representative experiments are shown.
The effect of thiols on NO-mediated inhibition of Ace1 activity was further examined by using the EMSA system described previously. As observed earlier (Fig. 3B), 4 mM DEA/NO completely blocked the binding of Ace1 to UASc DNA (Fig. 4B, lane 1 vs. lane 2). Interestingly, 2-ME (10 mM) alone slightly enhanced the binding of Ace1 to UASc DNA (Fig. 4B, lanes 3 and 5 versus lane 1). This may be attributable to either an increase in reduced copper or 2-ME-mediated reduction of metal-binding oxidized Ace1 thiols. Under the conditions of this experiment, 10 mM 2-ME also prevented inhibition of Ace1 binding activity by DEA/NO when the cell preparations were exposed to DEA/NO simultaneously with the thiol compound (Fig. 4B, lane 4). However, treatment with 10 mM 2-ME after exposure to DEA/NO partially reversed the effect of NO (Fig. 4B, lane 6). Thus, inhibition of Ace1 binding to CUP1 by NO using this in vitro assay could be partially reversed by the addition of 2-ME (as measured by EMSA).
It is essential to note that there are important differences between the EMSA and reporter gene systems used to elucidate the effects of exogenous thiols on NO-mediated inhibition of Ace1 activity. The experiments involving the use of a reporter gene to monitor CUP1 induction (Fig. 4A), required an extensive (1 hour) incubation of whole cells after the exposure to DEA/NO for 10 min. This is required to allow time for β-galactosidase expression. However, the EMSA experiments (Fig. 4B) required only a 10-min incubation of cell homogenates after a 10-min exposure to DEA/NO. Furthermore, the EMSA experiment was carried out in the presence of a protease inhibitor. Thus, Ace1 degradation as a result of NO exposure is possible in the reporter gene assay and less likely in the EMSA study (discussed in more detail later).
The above results implicate an interaction between NO and thiols in the NO-mediated inhibition of Ace1 activity. Much of the potential chemistry of thiol modification by NO is known to depend on molecular oxygen (O2) (discussed later). To test the idea that NO-O2 chemistry leads to the modification of Ace1 thiols, leading to subsequent loss of copper-dependent activation, the O2-dependency of the inhibitory effect of DEA/NO on CUP1 induction was examined. Under aerobic conditions, dose-dependent inhibition of CUP1 induction by DEA/NO was observed, and 0.4 mM of DEA/NO completely inhibited the induction (Fig. 5). Much less inhibition by DEA/NO was observed under anaerobic conditions in which 0.4 mM DEA/NO showed no significant inhibition and even 1 mM of DEA/NO inhibited significantly less compared with aerobic conditions (Fig. 5). Thus, the majority of inhibition of CUP1 induction by NO clearly occurs in an oxygen-dependent manner.
Figure 5.

Oxygen-dependency of the inhibition of CUP1 induction by DEA/NO. Late log phase cultures of 217CUP were exposed to DEA/NO (0–1 mM) under aerobic (closed circle) or anaerobic (open circle) conditions for 60 min. The cells were induced (CUP1-lacZ) for 60 min and were assayed for β-galactosidase activity as described in Experimental Procedures. Representative experiments are shown.
Discussion
One of the many important aspects of nitrogen oxide physiology is their potential interaction with thiols and thiol-containing proteins. It is likely that modification of the actions of thiol functional groups in proteins by NOx species represents both normal physiological and potential pathophysiological occurrences. The importance of thiol proteins to metal metabolism (i.e., the transport, sequestration, uptake, and utilization) is well established, especially in the yeast S. cerevisiae. Thus, it would not be unexpected to find that NO could alter or disrupt metal metabolism in yeast by interacting with these thiol-containing proteins. One of the many metal regulatory proteins in yeast that utilize thiols to ligate metals is the copper-responsive transcriptional activator Ace1. In S. cerevisiae, Ace1 acts as a sensor for intracellular copper. Under conditions of high concentrations of copper, Ace1 up-regulates gene expression for CUP1 (12, 13), CRS5 (29), and SOD1 (30). Cup1, a yeast metallothionein, plays an important role in copper detoxification, presumably because of its ability to sequester copper. Sod1 may also be involved in copper sequestration because the mammalian homologue of Cu, Zn-superoxide dismutase has been reported to have a role in copper buffering. However, Cup1 is thought to be the dominant protein for protection against copper toxicity because CUP1 delete mutants are extremely sensitive to copper toxicity (31). Furthermore, an ACE1 gene delete mutant, ace1Δ, has been shown to be extremely sensitive to copper, indicating that the CUP1 and Ace1 pathway is essential for copper resistance (32).
Herein, we observed that exposure of the yeast S. cerevisiae to the NO-donor DEA/NO resulted in a dramatic and rapid inhibition of the activity of the metal-responsive transcription factor Ace1 (Fig. 1). DEA/NO is representative of a class of NO-donor referred to as the NONOates and are the most kinetically and chemically defined NO-donors available (33). Evidence implicating Ace1 as the target for the actions of NO in inhibiting CUP1 induction was obtained by using EMSA to monitor the effect of NO on Ace1 binding to UASc DNA (Fig. 3).
Ace1 binds copper through eight cysteinyl thiols to form a tetracopper cluster that is similar to the copper-thiol cluster in metallothionein (34, 35). Significantly, metal release from metallothionein by NO has been reported and is thought to be attributable to S-nitrosation of the cysteine thiols (36, 37). Thus, considering the reported similarity between Ace1 and metallothionein and the previous observation that NO was capable of releasing metals from metallothionein, it was not surprising that NO was able to inhibit the activity of Ace1. Analogous to metallothionein, the mechanism of inhibition of Ace1 by NO was likely to be the result of modification of the copper-binding thiol residues on the protein. Further indication that the thiols of Ace1 are the targets of NO action, 2-ME was able to protect Ace1 from NO-mediated inactivation (Fig. 4).
Thiol modification by NO can occur via an oxygen-dependent generation of reactive nitrogen oxides such as dinitrogen trioxide (N2O3) (reactions 1–3) (38–41). Nitrogen dioxide (NO2) has also been proposed to be capable of oxidizing thiolate species as well (ref. 42 and references therein). Thus, N2O3 and NO2 constitute possible NOx species referred to throughout this paper.
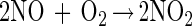 |
1 |
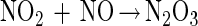 |
2 |
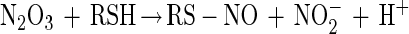 |
3 |
It has also been proposed that thiols can directly react with NO to generate an adduct that subsequently reacts with O2 to give O2− and the corresponding S-nitrosothiol (43) (reactions 4 and 5).
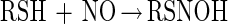 |
4 |
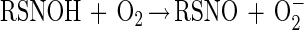 |
5 |
Regardless of the exact mechanism, S-nitrosothiol formation from exposure to NO is O2-dependent. However, thiol modification by NO can also occur anaerobically, although this is a relatively slow reaction (of questionable physiological relevance) and will not generate an S-nitrosothiol product. For example, reaction of thiols with NO under anaerobic conditions can lead to the generation of disulfide products and reduced nitrogen oxides (44, 45).
From our results, it appears that the mechanism of inhibition of Ace1 activity by NO primarily involves an O2-mediated process (Fig. 5). This finding implicates S-nitrosothiol formation in the mechanism of NO inhibition of Ace1 activity. Previous studies on S-nitrosothiol stability indicate that they can be converted to disulfides when exposed to other thiols (reaction 6) (47).
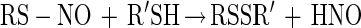 |
6 |
Thus, inhibition of thiol protein activity by NO can result from thiol conversion to either an S-nitrosothiol or a disulfide.
Regardless of whether the inhibition of Ace1 activity by NO was caused by conversion of the metal-binding thiols to S-nitrosothiols or disulfides, addition of an exogenous reducing thiol (such as 2-ME) after NO exposure should reverse the inhibition by NO by converting the oxidized protein thiols back to the reduced forms. Interestingly, this was found not to be the case in in vivo (whole cell preparations) when CUP1 induction was measured by using a reporter gene (Fig. 4). However, in in vitro (cell homogenates) EMSA studies that measured the actual binding of Ace1 to the CUP1 promoter, 2-ME was capable of partially reversing the NO-mediated inhibition (Fig. 4). A reasonable explanation for this apparent discrepancy is that in vivo modification of Ace1 thiols leads to complete demetallation that, in turn, results in a proteolytic instability of the apo-Ace1 protein. This is not unreasonable because it has been shown that apo-Ace1 is more sensitive to proteolysis than the Cu-Ace1 and Cu-Ace1-DNA complexes (13, 46, 47). Metallothionein is also known to exhibit similar proteolytic instability when demetallated (for example, see refs. 48 and 49). If, indeed, NO-mediated demetallation leads to proteolytic degradation and complete loss of constitutive Ace1 protein, it may be expected that Ace1 activity could return via de novo synthesis once NO dissipates. This appears to be the case as Ace1 activity began to slowly return after 1 hour (Fig. 2). Significantly, it has been suggested that Ace1 has zinc (II) in the copper binding domain under conditions of low copper (49), which may protect Ace1 from proteolytic degradation at low copper concentrations. Thus, it was likely that demetallation of Ace1 by NO involved the loss of both copper and zinc from the metal binding domains before its proteolytic degradation. In the in vitro EMSA studies, however, there was less likelihood of apo-Ace proteolytic degradation because of the shorter timescale of the experiment, the addition of a protease inhibitor, and the semipurified nature of the Ace1 preparation. Thus, the inhibition of Ace1 DNA-binding activity by NO was partially reversed by the addition of the exogenous thiol, 2-ME.
Because of the fact that no Ace1 homologue has yet to be discovered in mammalian systems, the relevance of this work to mammalian physiology may not be immediately obvious. However, Ace1 is representative of a series of metal binding thiol proteins, in both yeast and human, that are involved in metal metabolism (for review, see ref. 50). For example, the yeast copper chaperones Cox17 (51), Lys7 (52), and Atx1 (53) and the intracellular copper transporters Ccc2 (54) and Sco1 (55) have all been shown to have copper-thiol clusters and may also be susceptible to NO-mediated disruption. Many of these yeast proteins have mammalian homologues. The yeast Ccc2, Atx1, Lys7, and Cox17 all have homologous human proteins (Wilson disease protein, HAH1, CCS, and human COX17, respectively) (for reviews, see refs. 50, 56, and 57). Thus, it would not be surprising to find that NO is capable of disrupting metal metabolism in human cells by altering the metal-thiol interactions of these proteins in an analogous fashion to that found here for Ace1. Moreover, the ability of exogenous thiols to protect this type of disruption implicates physiologically relevant thiols, such as glutathione, as being important for the maintenance of the actions of these proteins in the presence of NO.
The effect of NO on metal metabolism and metal-responsive systems has also been examined previously by others. For example, Hibbs and coworkers found that NO can dramatically alter the iron status of cells by disrupting Fe-S cluster proteins (for example, see ref. 58). Similarly, it has been reported that NOx species are capable of disrupting the Fe-S-containing iron response protein, altering its activity (59). Interestingly, the interaction of NO with iron-response proteins resembles that elicited by iron deficiency (60, 61). As mentioned previously, metallothionein can be demetallated by NO, which could result in a disruption of heavy metal homeostasis and toxicity (36, 37). Finally, gene expression can be altered by NO-mediated destruction of zinc finger proteins (37, 62). In all of these cases, NO is disrupting the specific interactions of thiol (or sulfur) ligands with a metal. It remains to be seen whether metal binding thiols represent a class of biological thiols that are particularly susceptible to alteration via NO-mediated chemistry. Regardless, it is clear that a potential role for NO in both normal physiology and pathophysiology may be as a regulator/disrupter of metal metabolism via its interactions with metal bonding proteins.
As is evidenced by this work, the yeast S. cerevisiae represents a valuable tool for identifying and characterizing possible effects of NO on metal metabolism and metal-dependent transcription. It should be emphasized that it remains to be established whether the effects of NO in the yeast model are relevant to mammalian systems. Homologous pathways in mammalian cells and yeast, however, would predict that yeast will serve to establish possible targets for NO activity. Regardless, the observed effects of NO on metal-thiol proteins in yeast point to a physiological target in mammalian cells worthy of further investigation.
Abbreviations
- NO
nitric oxide
- DEA/NO
diethylammonium-1-(N,N-diethylamino) diazen-1-ium-1,2-diolate
- 2-ME
2-mercaptoethanol
- EMSA
electrophoretic mobility-shift assay
- DDW
double distilled water
- SD
synthetic dextrose medium
- BCS
bathocuproine disulfonic acid
- UASc
upstream activated sequence
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050586597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050586597
References
- 1.Moncada S, Palmer R M J, Higgs E A. Pharmacol Rev. 1991;43:109–141. [PubMed] [Google Scholar]
- 2.Ignarro L J. Semin Hematol. 1989;26:63–76. [PubMed] [Google Scholar]
- 3.Garthwaite J. Trends Neurosci. 1991;14:60–67. doi: 10.1016/0166-2236(91)90022-m. [DOI] [PubMed] [Google Scholar]
- 4.MacMicking J, Xie Q, Nathan C. Annu Rev Immunol. 1997;14:323–350. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 5.Domachowske J B. Biochem Mol Med. 1997;60:1–7. doi: 10.1006/bmme.1996.2557. [DOI] [PubMed] [Google Scholar]
- 6.Drapier J-C. Methods Companion Methods Enzymol. 1997;11:319–329. [Google Scholar]
- 7.Stamler J S. Curr Top Microbiol Immunol. 1995;196:19–36. doi: 10.1007/978-3-642-79130-7_4. [DOI] [PubMed] [Google Scholar]
- 8.Butler A R, Rhodes P. Anal Biochem. 1997;249:1–9. doi: 10.1006/abio.1997.2129. [DOI] [PubMed] [Google Scholar]
- 9.Williams D L H. Acc Chem Res. 1999;32:869–876. [Google Scholar]
- 10.Eide D J. Annu Rev Nutr. 1998;18:441–469. doi: 10.1146/annurev.nutr.18.1.441. [DOI] [PubMed] [Google Scholar]
- 11.Winge D R, Nielson K B, Gray W R, Hamer D H. J Biol Chem. 1985;260:14464–14470. [PubMed] [Google Scholar]
- 12.Thiele D J. Mol Cell Biol. 1988;8:2745–2752. doi: 10.1128/mcb.8.7.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fürst P, Hu S, Hackett R, Hamer D. Cell. 1988;55:705–717. doi: 10.1016/0092-8674(88)90229-2. , and erratum (1989) 56, 321. [DOI] [PubMed] [Google Scholar]
- 14.Dameron C T, Winge D R, George G N, Sansone M, Hu S, Hamer D. Proc Natl Acad Sci USA. 1991;88:6127–6131. doi: 10.1073/pnas.88.14.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jungmann J, Reins H-A, Lee J, Romeo A, Hassett R, Kosman D, Jentsch S. EMBO J. 1993;12(13):5051–5056. doi: 10.1002/j.1460-2075.1993.tb06198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamaguchi-Iwai Y, Serpe M, Haile D, Yang W, Kosman D J, Klausner R D, Dancis A. J Biol Chem. 1997;272:17711–17718. doi: 10.1074/jbc.272.28.17711. [DOI] [PubMed] [Google Scholar]
- 17.Drago R S, Paulik F E. J Am Chem Soc. 1960;82:96–98. [Google Scholar]
- 18.Hemmerich P, Sigwart C. Experientia. 1963;19:488–489. [Google Scholar]
- 19.Santoro N, Johansson N, Thiele D J. Mol Cell Biol. 1998;18:6340–6352. doi: 10.1128/mcb.18.11.6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huibregtse J M, Engelke D R, Thiele D J. Proc Natl Acad Sci USA. 1989;86:65–69. doi: 10.1073/pnas.86.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elble R. Biotechniques. 1992;13:18–20. [PubMed] [Google Scholar]
- 22.Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics. Plainview, New York: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 23.Longo V D, Gralla E B, Valentine J S. J Biol Chem. 1996;271:12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- 24.Holmquist B. Methods Enzymol. 1988;158:6–12. doi: 10.1016/0076-6879(88)58042-4. [DOI] [PubMed] [Google Scholar]
- 25.Thorvaldsen J L, Sewell A K, McCowen C L, Winge D R. J Biol Chem. 1993;268:12512–12518. [PubMed] [Google Scholar]
- 26.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Fukuto J M, Hobbs A J, Iganrro L J. Biochem Biophys Res Commun. 1993;196:707–713. doi: 10.1006/bbrc.1993.2307. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. In: Molecular Cloning. Ford N, Nolan C, Ferguson M, Ockler M, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 29.Culotta V C, Howard W R, Liu X F. J Biol Chem. 1994;269:25295–25302. [PubMed] [Google Scholar]
- 30.Gralla E B, Thiele D J, Silar P, Valentine J S. Proc Natl Acad Sci USA. 1991;88:8558–8562. doi: 10.1073/pnas.88.19.8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen L T, Howard W R, Strain J J, Winge D R, Culotta V C. J Biol Chem. 1996;271:18514–18519. doi: 10.1074/jbc.271.31.18514. [DOI] [PubMed] [Google Scholar]
- 32.Tamai K T, Gralla E B, Ellerby L M, Valentine J S, Thiele D J. Proc Natl Acad Sci USA. 1993;90:8013–8017. doi: 10.1073/pnas.90.17.8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keefer L K, Nims R W, Davies K M, Wink D A. Methods Enzymol. 1996;268:281–293. doi: 10.1016/s0076-6879(96)68030-6. [DOI] [PubMed] [Google Scholar]
- 34.Narula S S, Mehra R K, Winge D R, Armitage I M. J Am Chem Soc. 1991;113:9354–9358. [Google Scholar]
- 35.Peterson C W, Narula S S, Armitage I M. FEBS Lett. 1996;379:85–93. doi: 10.1016/0014-5793(95)01492-6. [DOI] [PubMed] [Google Scholar]
- 36.Misra R R, Hochadel J F, Smith G T, Cook J C, Waalkes M P, Wink D A. Chem Res Toxicol. 1996;9:326–332. doi: 10.1021/tx950109y. [DOI] [PubMed] [Google Scholar]
- 37.Kroncke K D, Fehsel K, Schmidt T, Zenke F T, Dasting I, Wesener J R, Bettermann H, Breunig K D, Kolb-Bachofen V. Biochem Biophys Res Commun. 1994;200:1105–1110. doi: 10.1006/bbrc.1994.1564. [DOI] [PubMed] [Google Scholar]
- 38.Goldstein S, Czapski G. J Am Chem Soc. 1996;118:3419–3425. [Google Scholar]
- 39.Wink D A, Nims R W, Darbyshire J F, Christodoulou D, Hanbauer I, Cox G W, Laval F, Laval J, Cook J A, Krishna M C, et al. Chem Res Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 40.Kharitonov V G, Sundquist A R, Sharma V S. J Biol Chem. 1995;270:28158–28164. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- 41.Keshive M, Singh S, Wishnok J S, Tannenbaum S R, Deen W M. Chem Res Toxicol. 1996;9:988–993. doi: 10.1021/tx960036y. [DOI] [PubMed] [Google Scholar]
- 42.Huie R E. Toxicology. 1994;89:193–216. doi: 10.1016/0300-483x(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 43.Gow A J, Buerk D G, Ischiropoulos H. J Biol Chem. 1997;272:2841–2845. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- 44.Pryor W A, Church D F, Govindan C K, Crank G. J Org Chem. 1982;47:159–161. [Google Scholar]
- 45.Wong P S-Y, Hyun J, Fukuto J M, Shirota F N, DeMaster E G, Shoeman D W, Nagasawa H T. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 46.Fürst P, Hamer D. Proc Natl Acad Sci USA. 1989;86:5267–5271. doi: 10.1073/pnas.86.14.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu S, Fürst P, Hamer D. New Biol. 1990;2:544–555. [PubMed] [Google Scholar]
- 48.Winge D R, Miklossy K-A. J Biol Chem. 1982;257:3471–3476. [PubMed] [Google Scholar]
- 49.Winge D R. Prog Nucleic Acid Res Mol Biol. 1998;58:165–195. doi: 10.1016/s0079-6603(08)60036-7. [DOI] [PubMed] [Google Scholar]
- 50.Vulpe C D, Packman S. Annu Rev Nutri. 1995;15:293–322. doi: 10.1146/annurev.nu.15.070195.001453. [DOI] [PubMed] [Google Scholar]
- 51.Glerum D M, Shtanko A, Tzagoloff A. J Biol Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 52.Culotta V C, Klomp L W, Strain J, Casareno R L, Krems B, Gitlin J D. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- 53.Lin S J, Pufahl R A, Dancis A, O'Halloran T V, Culotta V C. J Biol Chem. 1997;272:9215–9220. [PubMed] [Google Scholar]
- 54.Yuan D S, Stearman R, Dancis A, Dunn T, Beeler T, Klausner R D. Proc Natl Acad Sci USA. 1995;92:2632–2636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glerum D M, Shtanko A, Tzagoloff A. J Biol Chem. 1996;271:20531–20535. doi: 10.1074/jbc.271.34.20531. [DOI] [PubMed] [Google Scholar]
- 56.Valentine J S, Gralla E B. Science. 1997;278:817–818. doi: 10.1126/science.278.5339.817. [DOI] [PubMed] [Google Scholar]
- 57.Askwith C, Kaplan J. Trends Biochem Sci. 1998;23:135–138. doi: 10.1016/s0968-0004(98)01192-x. [DOI] [PubMed] [Google Scholar]
- 58.Hibbs J B, Jr, Taintor R R, Vavrin Z, Rachlin E M. Biochem Biophys Res Commun. 1988;157:87–94. doi: 10.1016/s0006-291x(88)80015-9. [DOI] [PubMed] [Google Scholar]
- 59.Lipinski P, Drapier J-C. J Biol Inorg Chem. 1997;2:559–566. [Google Scholar]
- 60.Pantopoulos K, Weiss G, Hentze M W. Mol Cell Biol. 1996;16:3781–3788. doi: 10.1128/mcb.16.7.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hentze M W, Kuhn L C. Proc Natl Acad Sci USA. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berendji D, Kolb-Bachofen V, Meyer K L, Grapenthin O, Weber H, Wahn V, Kroncke K-D. FEBS Lett. 1997;405:37–41. doi: 10.1016/s0014-5793(97)00150-6. [DOI] [PubMed] [Google Scholar]