

Abstract
We present the first steps in applying transverse relaxation-optimized spectroscopy (TROSY) techniques to the study of allosterism. Each β-chain of the hemoglobin (Hb) tetramer has 17 valine residues. We have 15N-labeled the β-chain Val residues and detected 16 of the 17 1H-15N correlation peaks for β-chain Val of the R state CO-Hb structure by using the TROSY technique. Sequence-specific assignments are suggested, based mainly on analysis of the 1H pseudocontact-shift increments produced by oxidizing the diamagnetic R state HbCO to the paramagnetic R state fluoromet form. When possible, we support these assignments with sequential nuclear Overhauser effect (NOE) information obtained from a two-dimensional [1H,1H]-NOESY-TROSY experiment (NOESY, NOE spectroscopy). We have induced further the R-T conformational change by adding the allosteric effector, inositol hexaphosphate, to the fluoromet-Hb sample. This change induces substantial increments in the 1H and 15N chemical shifts, and we discuss the implication of these findings in the context of the tentative sequence assignments. These preliminary results suggest that amide nitrogen and amide proton chemical shifts in a selectively labeled sample are site-specific probes for monitoring the allosteric response of the ensemble-averaged solution structure of Hb. More important, the chemical-shift dispersion obtained is adequate to permit a complete assignment of the backbone 15N/13C resonances upon nonselective labeling.
Protein–protein interactions are the basis for allosteric regulation, a phenomenon that is of fundamental importance to the basic metabolic processes in all living organisms. Not only the paradigmatic allosteric protein, hemoglobin (Hb), but all other allosteric proteins are oligomeric assemblies with multiple substrate (ligand)-binding sites that achieve positive cooperativity by linking small, substrate-induced changes in local tertiary structure to large, global changes in quaternary structure. The textbook view on how allosteric proteins work derives in most part from the postulate of a concerted transition between two forms of an allosteric protein that differ significantly in ligand/substrate affinity/reactivity as a result of their differences in tertiary and quaternary structure. However, structural studies on such diverse systems as hemoglobin (Hb) (1–3), glycogen phosphorylase (4), fructose-1,6-bisphosphatase (5), and a cyclic nucleotide-gated ion channel (6) show that each exhibits several stable and distinct structures, which suggests the possible contribution of an ensemble of structures to the allosteric-reaction pathway. Such studies set the task of determining the actual contributions of the ensemble members and of how these shift in response to the introduction of ligand/substrates and effectors, such as protons and organic phosphates in the case of Hb.
High-resolution NMR should play a central role in such studies, and in the case of Hb (molecular mass, 65,000 Da), the observation of a small number of spins with outstanding spectral properties has yielded a continuing flow of biologically relevant information since as early as 1969 (7). However, the applicability of NMR has been restricted by the high molecular masses of the oligomeric allosteric proteins.
Recent developments in NMR spectroscopy give promise of monitoring spins from all residues of high-molecular-mass proteins. Specifically, triple resonance experiments with 2H decoupling can yield backbone assignments for uniformly 13C,15N-labeled and highly deuterated proteins up to 60 kDa (8). Recent implementation of transverse relaxation-optimized spectroscopy (TROSY) in triple resonance experiments has improved sensitivity severalfold for 2H/13C/15N-labeled proteins (9–11), and complete backbone assignments of proteins up to 110 kDa has been shown to be feasible (9, 10).
In this report, we present the first step in applying these techniques to the study of the allosteric transition within the hemoglobin tetramer. We have begun by using the expression system of Hernan et al. (12) [which yields functionally and structurally faithful β(V1M)-chains] to 15N-label the backbone amide nitrogens of the β-chain Val residues (3, 13). Valine is an ideal residue for such a site-specific labeling experiment because it is one of the most abundant amino acid residues in both the α- and β-chains of Hb (Fig. 1), which permits incorporation of a large number of probes for monitoring structural changes dispersed throughout the entire molecule.
Figure 1.

Cartoon of the T structure of deoxy-Hb (βV1M) (1dxu.pdb), with the 17 valine residues in the β-chain indicated and classified according to the calculated theoretical pseudocontact shift for R state fluoromet-Hb, as described in the text: blue, helices B, D, and E; yellow, helices A, C, and F; green, helices G and H.
Each of the β(V1M)-chains of the Hb tetramer has 17 valine residues, and we have detected 16 of the 17 1H-15N correlation peaks for the valines of the R state HbCO structure by using the TROSY technique. Sequence-specific assignments are suggested, based mainly on analysis of the 1H pseudocontact-shift increments produced by oxidizing the diamagnetic R state HbCO to the paramagnetic R state fluoromet form. When possible, we support these assignments with sequential nuclear Overhauser effect (NOE) information obtained from a two-dimensional (2D) [1H,1H]-NOESY-TROSY experiment (NOESY, NOE spectroscopy) (14). We then induced the R-T conformational change by adding the allosteric effector, inositol hexaphosphate (IHP), to the fluoromet-Hb sample. This change produces substantial increments in the 1H and 15N chemical shifts, and we discuss the implication of these findings in the context of the tentative sequence assignments.
These preliminary results suggest that amide nitrogen and amide proton chemical shifts in a selectively labeled sample are site-specific probes for monitoring the allosteric response of the ensemble-averaged solution structure of Hb. More important, the chemical-shift dispersion obtained is adequate to permit a complete assignment of the backbone 15N/13C resonances upon nonselective double labeling. Thus, the ability to monitor the response of the entire Hb structure to ligands and allosteric effectors is within our grasp.
Materials and Methods
Preparation of Hb Containing 15N-Val-Labeled β-Subunits.
The β-globin gene was overexpressed in Escherichia coli by using the expression system of Hernan et al. (12). The resulting β-globin is insoluble, does not incorporate heme, and has a methionine in place of the normal NH2-terminal valine. However, when combined with native α-subunits and hemin, the purified β-globin chains are incorporated into tetrameric Hb that is functionally and structurally very similar to native HbA (12, 13, 15). Specific isotopic enrichment of valine backbone nitrogen atoms was achieved by supplementing the LB with 60 mg/liter 15N-labeled valine (Cambridge Isotope Laboratories, Cambridge, MA). Approximately 90 mg of purified Hb tetramer was obtained from 6 liters of bacterial culture. As part of our efforts to investigate incorporation efficiencies, we used only singly labeled valine, but the yields indicate that this procedure can be extended to incorporate the more costly, 13C/15N-labeled valine.
Analysis of a sample of 15N-Val HbCO by electrospray MS (quattro ii; Micromass, Manchester, U.K.) gave two peaks with molecular masses corresponding to those of the α-chain (15,124 Da compared with the expected mass of 15,126 Da) and the unenriched β-chain (15,898 Da compared with an expected mass of 15,899 Da). Because the growth medium contains unlabeled valine, the growth protocol is expected to give, at best, only 50% enrichment per residue; examination of the breadth of the signal for the β-chain suggests that the enrichment per residue is actually no more than ≈10%.
NMR Spectroscopy.
Oxyhemoglobin (oxy-Hb) was dissolved in 90% H2O/10% D2O buffered with 100 mM potassium phosphate that was adjusted to pH 6.0. The pH was not corrected to compensate for the deuterium isotope effect. Carboxyhemoglobin was prepared from oxy-Hb by flushing with CO for ≈30 min. The TROSY spectrum of HbCO in 50 mM Tris⋅Cl buffer (pH 8) is similar to the spectrum of HbCO in 100 mM phosphate buffer (pH 6). R state fluoromet-Hb was prepared from HbO2 by the addition of a slight excess of potassium ferricyanide, followed by the addition of 0.2 M KF. The fluoromet sample then was converted to the T state by the addition of a 2-fold excess of IHP (Sigma). Sample concentrations were ≈4 mM in heme, as determined spectrophotometrically (8451A diode array spectrophotometer; Hewlett–Packard): ɛ(HbCO) = 208 mM−1 cm−1 at 418 nm (16); ɛ(Hb+F−) = 144 mM−1 cm−1 at 403 nm (17).
NMR measurements were performed at 37°C on 15N-labeled protein on a Varian INOVA-600 spectrometer. [1H,15N]-TROSY spectra (18–20) were recorded as a matrix of 128 × 512 complex points in each of t1(15N) and t2(1H) with spectral widths of 1,200 Hz and 9,000 Hz in F1 and F2, respectively. 2D [1H,1H]-NOESY-TROSY spectra (14) were recorded as a matrix of 64 × 256 complex points in each of t1(1H) and t2(1H) with spectral widths of 8,000 Hz and 7,000 Hz in F1 and F2, respectively. An NOE mixing time of 200 ms was used. All spectra were processed with the program nmrpipe (21) by using a sinebell window function shifted by 90°. Data analysis was performed by using pipp (22).
Pseudocontact-Shift Calculations.
The total observed chemical shift can be factored into contributions from diamagnetic and paramagnetic terms (23), the latter of which has origins in pseudocontact and Fermi contact contributions. For the high-spin heme of fluoromet-Hb, the pseudocontact term dominates. Because the g tensor of the heme of fluoromet-Hb is axial (g∥ = 2, along the normal to the heme plane; g⊥ = 6), the pseudocontact-shift contribution (δPT) for the amide 15N and 1H of the 17 Val residues in the β-chain of Hb were calculated by using Eq. 1,
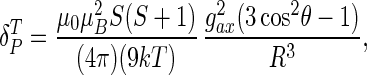 |
1 |
where μ0 is the magnetic field constant, μB is the Bohr magneton, k is the Boltzmann constant, S is the spin number, T is the absolute temperature, gax2 = (g∥2 − g⊥2) <0 for fluoromet-Hb, R is the distance from the amide 15N or 1H to Fe, and θ is the angle between the (15N or 1H)-Fe vector and the heme normal. For the HbO2 R structure, the heme normal is taken as the Fe-O vector, and for the deoxy-Hb T structure, the heme normal is taken as the Fe-Nɛ vector. These were obtained from the x-ray coordinates of the HbO2 (1hhb.pdb) and deoxy-Hb (1dxu.pdb) crystal structures. Protons were added to both structures by using the program reduce (24).
Results and Discussion
Comparison of Heteronuclear Single Quantum Coherence and TROSY Experiments.
TROSY utilizes interference between dipole–dipole coupling and chemical-shift anisotropy to suppress transverse relaxation effects in multidimensional NMR (25). This technique, designed to extend NMR structure determination to larger molecular sizes, correlates the 15N and 1H signals of the backbone amide N—H groups. Fig. 2 shows the [1H,15N]-TROSY spectra of HbCO, fluoromet-Hb, and fluoromet-Hb in the presence of a 2-fold excess of IHP. In all three cases, 16 signals were clearly resolved, accounting for all but 1 of the 17 Val residues in the β-subunit of Hb. Given the low level of enrichment and the observation of only 16 signals, it is not plausible that the 15N-isotope was scrambled into other types of amino acid residues by in vivo biodegradation.
Figure 2.

TROSY NMR spectra of 15N-β-Val-labeled samples of HbCO (green), HbF (red), and HbF with a 2-fold excess of IHP (blue). Conditions: 37°C, 100 mM potassium phosphate buffer at pH 6.
For all three Hb states, the chemical shifts are well dispersed in both the 1H and the 15N dimensions; e.g., for HbCO, the 1H chemical shifts vary between 7.34 ppm and 9.05 ppm, whereas the 15N chemical shifts vary from 115.9 ppm and 124.9 ppm. For comparison, the dispersion for the 1H chemical shifts of the seven Val residues of MbCO (sperm whale) is comparable to that of HbCO, but the 15N chemical shifts are much less disperse, spanning only 2 ppm (26).
As Wuthrich and colleagues reported for uniformly 15N-labeled ftz homeodomain complexed with a 14-bp DNA duplex (25), the TROSY spectra of all three derivatives show reduced line widths in comparison with the conventional heteronuclear single quantum coherence experiment. Because the TROSY technique had not been applied previously to a paramagnetic system, we made a detailed comparison for the sample of fluoromet-Hb having IHP. The decreased TROSY line widths give rise to a signal enhancement (Table 1) for most of the 16 signals, with an average enhancement of 12%, but a largest enhancement of 48% (peak L); only two peaks (B and C) are less intense in the TROSY spectrum. Although the observed TROSY enhancement for the 15N-Val Hb samples seems to be modest, a much more significant enhancement will be attainable when we apply the same technique to perdeuterated samples.
Table 1.
Intensity enhancement factors and chemical-shift increments for the amide correlation peaks obtained from TROSY NMR spectra of 15N-β-Val-labeled Hb derivatives
| Peak | ITROSY/ IHSQC |
1H (ppm)
|
15N (ppm)
|
||
|---|---|---|---|---|---|
| δP | δA | δP | δA | ||
| A | 1.03 | −0.35 | 0.04 | 1.5 | 0.1 |
| B | 0.86 | −0.16 | 0.02 | −0.4 | −0.2 |
| C | 0.80 | −0.13 | 0.09 | 0.0 | 0.1 |
| D | 1.24 | −0.11 | −0.01 | −0.1 | 0.0 |
| E | 1.18 | −0.07 | 0.05 | −0.2 | 0.1 |
| F | 1.06 | −0.04 | 0.00 | 0.1 | −0.1 |
| G | 0.99 | −0.03 | 0.04 | −0.9 | −0.2 |
| H | 1.25 | −0.02 | −0.13 | 0.1 | −0.1 |
| I | 1.21 | 0.03 | 0.07 | −0.0 | −0.3 |
| J | 0.94 | 0.03 | 0.07 | 0.1 | −0.1 |
| K | 1.40 | 0.03 | 0.04 | 0.1 | 0.0 |
| L | 1.48 | 0.13 | 0.20 | 1.4 | 0.0 |
| M | 1.08 | 0.16 | −0.03 | 0.1 | 0.3 |
| N | 1.22 | 0.20 | 0.17 | 0.2 | −0.2 |
| O | 1.18 | 0.22 | 0.04 | 1.4 | 0.1 |
| P | 0.98 | 0.26 | 0.10 | 0.6 | 0.1 |
Intensity enhancement factors were measured for HbF in the presence of a 2-fold excess of IHP. Spectral conditions: 37°C, 100 mM potassium phosphate buffer at pH 6. δp, Chemical shift increment between HbCO and fluoromet-Hb; δΔ, increment upon adding IHP to fluoromet-Hb.
Pseudocontact Shifts.
The 1H chemical-shift increments induced by converting HbCO to fluoromet-Hb (δP in Table 1) were calculated by subtraction. These range from −0.35 ppm to 0.26 ppm; five peaks shift upfield and five shift downfield; the chemical-shift differences of the remaining six peaks are too small to be considered significant (|δP| ≥ 0.05 ppm). For the purpose of this discussion, the peaks in Fig. 2 are labeled A–P, according to the magnitude and direction of the 1H chemical-shift difference between the reduced HbCO state and the oxidized fluoromet-Hb state, so that peak A shows the strongest negative (upfield) shift in the 1H dimension. We analyze these increments below in terms of the paramagnetism of the fluoromet-Hb heme.
Although such 1H chemical-shift increments are well known upon oxidation/reduction between diamagnetic and paramagnetic states of metalloproteins (23, 27, 28), relatively few examples of such effects have been reported for 15N (27, 29–31). For 10 of the backbone Val signals, the chemical shift in the 15N dimension moves significantly (|δP| ≥ 0.1 ppm) in response to the oxidation state of the heme (Table 1); the signals for peaks B, E, and G move upfield, whereas the signals for peaks A and K–P move downfield. Contrary to the prediction of Eq. 1, the chemical-shift increments in the 15N dimension span a larger range than for the 1H dimension, varying from a maximum upfield shift of −0.93 ppm to a maximum downfield shift of 1.54 ppm. All five of the peaks that showed strong downfield shifts in the 1H dimension also show significant downfield shifts in the 15N direction. However, only two (peaks B and E) of the five peaks that showed significant upfield shifts in the 1H spectrum shift upfield in the 15N dimension, and one peak (peak A) actually shows a very strong shift downfield. In addition, peak G, which does not show a significant shift in the 1H dimension, shows a strong upfield shift in the 15N dimension. Clearly, the 15N chemical shifts, which are more sensitive to the local environment than those of 1H, are responding not only to paramagnetic effects, but in some cases these are superseded by changes in the local environment, even though both HbCO and fluoromet-Hb nominally have the R structure. This observation alone is suggestive that the two Hb forms represent slightly different members of the R state ensemble of structures.
Peak Assignments.
We have made tentative assignments of the Val 1H15N peaks by adopting the zeroth order approximation that the R structures of all liganded forms of Hb are equivalent and that the 1H chemical-shift increments induced by converting HbCO to HbF can be assigned to a pseudocontact contribution that is present in the latter, but absent in the diamagnetic HbCO state. The relative pseudocontact shifts (δPT), calculated according to Eq. 1, for each of the 17 Val residues in the β-chain by using the x-ray coordinates of the HbO2 crystal structure are given in Fig. 3. Because the x-ray coordinates of fluoromet-Hb are not available, we used the HbO2 structure to represent that of R structure fluoromet-Hb.
Figure 3.

Correlation between experimental and calculated pseudocontact shifts for the amide groups of the 17 valine residues in the β-chain of the R state structure of Hb. Assignments are based primarily on the alignment of the calculated and experimental chemical shift increments but incorporate results of a [1H,1H]-NOESY-TROSY experiment (Fig. 4) as described in the text. Because only Val-98 is close enough to the α-subunit for its heme to influence δPT, the calculations ignore contributions to the paramagnetism from hemes other than the β-heme. The peaks are classified according to the structural clusters discussed in the text and identified in Fig. 1. The solid line is the best-fit line through the data for the 10 peaks that show a significant experimental shift (slope = 0.92; R2 = 0.97). Cluster II (peaks F–K) falls between the vertical lines that indicate the ±0.05 ppm threshold, below which a paramagnetic-shift increment is not significant, and was excluded from the fit.
Initial examination shows that the pseudocontact calculations correctly describe the global behavior of the oxidation-induced 1H chemical-shift increments. The 17 Val residues distribute themselves into three clusters based on their predicted 1H pseudocontact shift: 7 Val residues are predicted to show a significant negative shift (cluster I), 5 are predicted to be unshifted (cluster II), and 5 are predicted to show a positive shift (cluster III). The direction of the chemical-shift increment is determined by the sign of the angle-dependent term of Eq. 1, and, in fact, the three classes of residues map directly onto the structure of Hb (Fig. 1). In particular, the seven Val residues (positions 23, 33, 34, 54, 60, 67, and 98) that show a significant negative pseudocontact shift are all located in either the B, D, or E helices of the β-chain, with a positive angular factor (θ < 54° or θ > 125°), whereas the five Val residues in cluster III (positions 109, 111, 133, 134, and 137) are all located in helices G or H and have negative angular factors (54° < θ < 125°). The five remaining Val residues comprising cluster II (positions 11, 18, 20, 113, and 126) are so far from the heme-iron that they are not expected to show a significant pseudocontact shift. Repeating these calculations for several other R state structures, including HbCO and aquomet-Hb, showed the same three classes of residues. Thus, the direction of the pseudocontact shift allows us to immediately assign each peak to a particular structural cluster.
To assign the peaks within a given cluster, we ranked the Val 1H signals according to their theoretical chemical-shift increment and also incorporated results from 2D [1H,1H]-NOESY-TROSY experiments discussed below. The result was that we were able to deduce provisional assignments for most of the 16 signals. In making these assignments, we assigned Val-67 as the one whose resonance is not observed. Val-60 and Val-67 are predicted to show the largest upfield shifts, but because Val-67 is closest to the heme, we took its signal to have been broadened beyond detection and thus assigned the peak with the largest observed upfield shift (peak A) to Val-60. The rest of the Val residues in cluster I are assigned to the peaks with substantial negative increments (> −0.05 ppm) and those of cluster III are assigned to the peaks with substantial positive increments (<0.05 ppm). Peaks G–K, whose chemical shifts are not appreciably affected by oxidation, represent the cluster II Val residues that are far from the heme iron. We were able to verify and/or extend some of these assignments with a 2D [1H,1H]-NOESY-TROSY experiment, discussed immediately below. The resulting correlation between theoretical and experimental paramagnetic shifts is shown in Fig. 3.
A 2D [1H,1H]-NOESY-TROSY spectrum (Fig. 4) exhibited correlated signals for 11 of the 16 valine residues detected in the TROSY experiment. Among these, the nine most intense valine signals (peaks A, D, E, H–K, M, and N) show intraresidue, methyl cross-peaks, whereas peaks B and F show cross-peaks only for their neighboring side chains.§ In addition, we detected a highly unusual methyl signal at −1.17 ppm that is not correlated with 1 of the 16 valine peaks observed in the TROSY experiment. This signal likely arises from the methyl group of the 17th valine, which was not seen by TROSY and which we have assigned to Val-67. This assignment is supported by the fact that a peak with a similar chemical shift was observed in the one-dimensional spectrum of HbCO, and, by using natural mutants, it has been assigned to Val-67 (32). It is not surprising that peaks C, G, L, O, and P do not show any cross-peaks in the [1H,1H]-NOESY-TROSY experiment, because they all give broad signals in the TROSY spectrum.
Figure 4.

[1H, 1H]-NOESY-TROSY spectrum of fluoromet-Hb with a 2-fold excess of IHP. Conditions: 37°C, 100 mM potassium phosphate buffer at pH 6.
To test the assignments obtained from the pseudocontact calculations, we examined the cross-peaks to neighboring side chains. Cross-peaks from neighboring side chains were observed in the [1H,1H]-NOESY-TROSY spectrum for six valines (B/K, D, F, H, I, and N). These observed cross-peaks are consistent with the majority of the assignments based on the pseudocontact calculations. For example, peaks I and N (assigned to Val-18 and Val-133, respectively) are adjacent to Lys residues and show cross-peaks (≈2.68 ppm and 2.98 ppm, respectively) that can be attributed to Hɛ of these neighboring Lys residues. Similarly, peak D (assigned to Val-54) has a cross-peak at 1.91 ppm, consistent with the chemical shift of Hβ from Ala-53. Peak F, which did not show any cross-peaks assignable to valine, does show a peak with chemical shift of 1.93 ppm that can be assigned to the Hγ of E22, in agreement with the assignment of peak F to Val-23.
For one pair of valines, H and J, the 2D measurements led us to reverse the paramagnetic-shift assignment. Peak H is assigned to Val-126, not Val-20, because the cross-peak at 1.94 ppm can be associated with Pro-125 and cannot be attributed to Asn-19, whereas peak J is assigned to Val-20 because it does not show a cross-peak, as expected with neighboring Asn-19. Although this puts the predicted pseudocontact shifts in the “wrong order,” the differences in both the predicted and the experimental pseudocontact shifts are small.¶
Fig. 3 compares the normalized experimental and theoretical pseudocontact-shift increments. Excellent correlation (slope = 0.92 with R2 = 0.97) between the experimental and calculated pseudocontact increments is observed. The overall agreement between the experimental and calculated shifts, including the deviation of the slope from unity, is typical for 1H chemical shifts up to a distance of ≈18 A. When the resonances had been assigned independently, such correlations have been used to test and refine the structural models, as for rubredoxin (27). In contrast, we have used the x-ray structures of Hb to obtain preliminary assignments.
Effects of the R-T Quaternary Transition.
In the crystalline state as well as in solution, addition of the allosteric effector, IHP, to fluoromet-Hb is known to produce structural changes that mimic the R-T quaternary change that occurs when O2 is released from oxy-Hb (33, 34). This structural change is manifested in changes in the 1H NMR chemical shifts of the resonances assigned to the hydrogen-bonding interactions in the dimer–dimer interface (34) and in UV resonance Raman spectra that monitor the marker bands of the α42Tyr and β37Trp residues in the dimer–dimer interface (35). Unlike the UV resonance Raman and one-dimensional NMR experiments, which monitor only a small (albeit very specific) segment of the protein structure, the 1H chemical shifts of the backbone amide signals for the Val residues provide probes for structural changes that might occur throughout the entire β-chain, and we find that they are sensitive markers of the quaternary changes that accompany IHP binding.
Fig. 2 compares the TROSY NMR spectra of R-HbF with that of T-HbF prepared by adding IHP; Table 1 summarizes the chemical-shift increments (δA) caused by the R-T transition. Appreciable chemical-shift increments (|δA| ≥ 0.1 ppm), with magnitudes of up to 0.2 ppm, are observed in the 1H dimension for five peaks, one from cluster I (peak C), one from cluster II (peak H), and three from cluster III (peaks L, N, and P). The signals in clusters I and III (peaks C, L, N, and P) move downfield, whereas the peak from cluster II (peak H) shifts upfield. These increments do not arise solely from a change in the paramagnetic contribution. For example, peak H of cluster II has a negligible paramagnetic shift increment upon oxidation, but exhibits the second biggest quaternary shift, −0.13 ppm. In fact, changes in the paramagnetic contribution appear to make a minimal contribution to the quaternary shifts. This was substantiated by a calculation of the expected quaternary-induced paramagnetic increments, as defined as the difference between the paramagnetic shifts calculated for an R structure Hb and for a T structure Hb.
It is not surprising that several of the peaks belonging to cluster III show significant chemical shift changes upon addition of IHP, because most of the Val residues comprising this cluster are located close to the β1β2 interface, where IHP binding is known to occur. Consistent with calculations of the expected quaternary-induced paramagnetic increments, none of the observed peaks assigned to Val residues close to the heme show significant quaternary shifts upon addition of IHP.
The 15N chemical shift is slightly more sensitive to allosteric perturbations than the 1H chemical shift: the R-T transition causes 10 peaks to show significant quaternary increments of their 15N chemical shift |δA| ≥ 0.1 ppm). Of these, four are in cluster I (peaks A, B, E, and F), three are in cluster II (peaks G, H, and I), and three are in cluster III (peaks M, N, and P). Peak I of cluster II shows the largest negative quaternary chemical-shift increment (−0.32 ppm), and peak M of cluster III shows the largest positive quaternary shift (0.30 ppm). Peak L, which shows the largest quaternary increment in the 1H direction, does not show a significant change in its 15N position, whereas seven peaks (peaks A, B, E, F, G, I, and M) show changes in their 15N chemical shift but do not show increments in their 1H chemical shift. In fact, only three peaks show significant shifts in both the 1H and the 15N dimensions. Clearly, the 1H and 15N probes are sensitive to different structural features, and the most complete picture of the structural changes accompanying the quaternary-state transition will emerge only after analyzing both the 1H and the 15N quaternary-shift increments.
Summary.
We have found that advances in multidimensional NMR make it possible to obtain well resolved NMR spectra from a specifically labeled Hb and have used paramagnetic shift measurements, supplemented by NOE techniques, to achieve tentative assignments of the 1H15N peaks of 16 of the 17 β-chain valine residues. We have demonstrated further that the backbone amide groups can be used to monitor structural changes accompanying the allosteric transition in Hb. These results suggest that well resolved spectra now might be attainable with globally labeled Hb, as well, and they have encouraged us to initiate such studies with the aim of achieving complete assignments.
Acknowledgments
We thank Dr. Jeffrey Kavanaugh and Ben Davis (University of Iowa) and Professor Robert Noble and Hilda Hui (State University of New York, Buffalo), Program Project Group collaborators, for preparation of the labeled Hb. This work has been supported by the National Institutes of Health Grant P01 GM58890 (Professor Arthur Arnone, Principal Investigator) and also by Grant HL63203 (B.M.H.).
Abbreviations
- Hb
hemoglobin
- TROSY
transverse relaxation-optimized spectroscopy
- NOESY
nuclear Overhauser effect spectroscopy
- 2D
two-dimensional
- IHP
inositol hexaphosphate
Footnotes
In addition to the methyl cross-peaks, we have observed α-H cross-peaks for four valines (peaks B/K, D, H, and I) and diagonal N—H peaks for three valines (peaks D, I, and M).
There are two places in the sequence at which there are pairs of adjacent Val residues (positions 33 and 34 and positions 133 and 134) that are expected to show off-diagonal N—H-N—H correlations that could support our assignments. Unfortunately, peak O (assigned to Val-134) is too weak to show any cross-peaks in the 2D NOESY-TROSY experiment, and peak E (assigned to Val-34) shows only methyl cross-peaks.
References
- 1.Dickerson R E, Geis I. Hemoglobin: Structure, Function, Evolution, and Pathology. Menlo Park, CA: Benjamin/Cummings; 1983. [Google Scholar]
- 2.Silva M M, Rogers P H, Arnone A. J Biol Chem. 1992;267:17248–17256. [PubMed] [Google Scholar]
- 3.Kavanaugh J S, Weydert J A, Rogers P H, Arnone A. Biochemistry. 1998;37:4358–4373. doi: 10.1021/bi9708702. [DOI] [PubMed] [Google Scholar]
- 4.Johnson L N, Barford D. J Mol Biol. 1990;265:2409–2412. [PubMed] [Google Scholar]
- 5.Villeret V, Huang S H, Zhang Y P, Xue Y F, Lipscomb W N. Biochemistry. 1995;34:4299–4306. doi: 10.1021/bi00013a019. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz M, Karpen J W. Nature (London) 1997;389:389–392. doi: 10.1038/38744. [DOI] [PubMed] [Google Scholar]
- 7.Ho C. Advances in Protein Chemistry. Vol. 43. New York: Academic; 1992. pp. 153–312. [DOI] [PubMed] [Google Scholar]
- 8.Shan X, Gardner K H, Muhandiram D R, Rao N S, Arrowsmith C H, Kay L E. J Am Chem Soc. 1996;118:6570–6579. [Google Scholar]
- 9.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. Proc Natl Acad Sci USA. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. J Biomol NMR. 1999;14:85–88. doi: 10.1023/a:1008346931993. [DOI] [PubMed] [Google Scholar]
- 11.Yang D W, Kay L E. J Biomol NMR. 1999;13:3–10. doi: 10.1023/A:1008329230975. [DOI] [PubMed] [Google Scholar]
- 12.Hernan R A, Hui H L, Andracki M E, Noble R W, Sligar S G, Walder J A, Walder R Y. Biochemistry. 1992;31:8619–8628. doi: 10.1021/bi00151a032. [DOI] [PubMed] [Google Scholar]
- 13.Doyle M L, Lew G, Deyoung A, Kwiatkowski L, Wierzba A, Noble R W, Ackers G K. Biochemistry. 1992;31:8629–8639. doi: 10.1021/bi00151a033. [DOI] [PubMed] [Google Scholar]
- 14.Zhu G, Kong X M, Sze K H. J Biomol NMR. 1999;13:77–81. doi: 10.1023/A:1008398227519. [DOI] [PubMed] [Google Scholar]
- 15.Kavanaugh J S, Rogers P H, Arnone A. Biochemistry. 1992;31:8640–8647. doi: 10.1021/bi00151a034. [DOI] [PubMed] [Google Scholar]
- 16.Banerjee R, Alpert Y, Leterrier F, Williams R J P. Biochemistry. 1969;8:2862–2867. doi: 10.1021/bi00835a025. [DOI] [PubMed] [Google Scholar]
- 17.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. Amsterdam: North–Holland; 1971. [Google Scholar]
- 18.Weigelt J. J Am Chem Soc. 1998;120:12706. [Google Scholar]
- 19.Weigelt J. J Am Chem Soc. 1998;120:10778–10779. [Google Scholar]
- 20.Pervushin K V, Wider G, Wuthrich K. J Biomol NMR. 1998;12:345–348. doi: 10.1023/A:1008268930690. [DOI] [PubMed] [Google Scholar]
- 21.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 22.Garrett D S, Powers R, Gronenborn A M, Clore G M. J Magn Reson. 1991;95:214–220. doi: 10.1016/j.jmr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Banci L, Bertini I, Luchinat C. Methods Enzymol. 1994;239:485–514. doi: 10.1016/s0076-6879(94)39019-3. [DOI] [PubMed] [Google Scholar]
- 24.Word J M, Lovell S C, LaBean T H, Taylor H C, Zalis M E, Presley B K, Richardson J S, Richardson D C. J Mol Biol. 1999;285:1711–1733. doi: 10.1006/jmbi.1998.2400. [DOI] [PubMed] [Google Scholar]
- 25.Pervushin K, Riek R, Wider G, Wuthrich K. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theriault Y, Pochapsky T C, Dalvit C, Chiu M L, Sligar S G, Wright P G. J Biomol NMR. 1994;4:491–504. doi: 10.1007/BF00156616. [DOI] [PubMed] [Google Scholar]
- 27.Wilkens S J, Xia B, Weinhold F, Markley J L, Westler W M. J Am Chem Soc. 1998;120:4806–4814. [Google Scholar]
- 28.Ubbink M, Lian L Y, Modi S, Evans P A, Bendall D S. Eur J Biochem. 1996;242:132–147. doi: 10.1111/j.1432-1033.1996.0132r.x. [DOI] [PubMed] [Google Scholar]
- 29.Jain N U, Pochapsky T C. J Am Chem Soc. 1998;120:12984–12985. [Google Scholar]
- 30.Lindstrom T R, Noren I B E, Charache S, Lehmann H, Ho C. Biochemistry. 1972;11:1677–1681. doi: 10.1021/bi00759a023. [DOI] [PubMed] [Google Scholar]
- 31.Boyd J, Dobson C M, Morar A S, Williams R J P, Pielak G J. J Am Chem Soc. 1999;121:9247–9248. [Google Scholar]
- 32.Bertini I, Eltis L D, Kastran D H W, Luchinat C, Piecioli M. Chemistry. 1995;1:598–607. [Google Scholar]
- 33.Fermi G, Perutz M F. J Mol Biol. 1977;114:421–431. doi: 10.1016/0022-2836(77)90259-5. [DOI] [PubMed] [Google Scholar]
- 34.Perutz M F, Sanders J K M, Chenery D H, Noble R W, Pennelly R R, Fung L, Ho C, Giannini I, Porschke D, Winkler H. Biochemistry. 1978;17:3640–3652. doi: 10.1021/bi00610a034. [DOI] [PubMed] [Google Scholar]
- 35.Jayaraman V, Rodgers K R, Mukerji I, Spiro T G. Biochemistry. 1993;32:4547–4551. doi: 10.1021/bi00068a009. [DOI] [PubMed] [Google Scholar]