

Abstract
The distal portion of chromosome 1p is one of the most commonly affected regions in human cancer. In this study of hereditary and sporadic colorectal cancer, a region of frequent deletion was identified at 32.2 centimorgans from 1ptel. Deletion breakpoints clustered in the vicinity of or inside the gene RIZ, which encodes a retinoblastoma protein-interacting zinc finger protein. Sequence analysis revealed frequent frameshift mutations of the RIZ gene. The mutations consisted of 1- or 2-bp deletions of a coding (A)8 or (A)9 tract and were confined to microsatellite-unstable colorectal tumors, being present in 9 of 24 (37.5%) primary tumors and in 6 of 11 (54.5%) cell lines; in 2 cell lines the mutation was homozygous/hemizygous. The mutations apparently were selected clonally in tumorigenesis, because similar poly(A) tracts in other genes were not affected. Two alternative products of the gene exist, RIZ1, which contains a PR (PRDI-BF1-RIZ1) domain implicated in tumor suppressor function, and RIZ2, which is lacking this motif. Furthermore, the C-terminal region, which contains the poly(A) tracts, includes a PR-binding motif, possibly mediating interactions with other proteins or with RIZ itself (oligomerization). Four of eleven microsatellite-unstable colorectal cancer cell lines, three of which had frameshifts, showed reduced or absent mRNA expression of RIZ1. In a cell line that is homozygous/hemizygous for the typical frameshift mutation, immunoblotting showed truncated RIZ protein, whereas adenovirus-mediated RIZ1 expression caused G2/M arrest and apoptosis. We propose that RIZ is a target of the observed 1p alterations, with impairment of the PR domain-mediated function through either frameshift mutation or genomic deletion.
Sporadic cancer arises as a result of a series of somatic mutations and epigenetic changes that silence tumor suppressor genes or activate oncogenes (1). As such, the APC-RAS-P53 pathway in colorectal cancer is well established (2), but many of its components are not fully understood (3). Recently, two pathways, CIN, a chromosomal instability pathway, and MIN, a microsatellite instability pathway, have been proposed. Loss of heterozygosity (LOH) is a hallmark of the CIN pathway, whereas microsatellite instability (MSI) characterizes the MIN pathway (4). In the MIN pathway, mutations or epigenetic silencing of one of the DNA mismatch repair genes lead to widespread accumulation of mutations, followed by clonal selection (5). The MSI(+) phenotype is easily distinguishable by allelic changes of repetitive regions throughout the genome (6).
Some genes contain repetitive regions in their coding sequences that are often targets of MSI. Insertion or deletion in these repetitive regions leads to frameshift and protein truncation. In colorectal cancer, the TGFBRII gene (7) and BAX gene (8) are examples of genes mutated in this way and whose loss of function is believed to contribute to tumorigenesis. Other examples include the DNA mismatch repair genes MSH6 and MSH3, and IGFR2 (9). In this paper, we describe how a high-density LOH analysis led to the identification of a frequently deleted region in 1p36 comprising the RIZ gene. In MSI(+) tumors that did not show LOH, RIZ was found to be affected by frequent frameshift mutations of one of two coding poly(A) tracts. RIZ encodes a retinoblastoma-interacting zinc finger protein, and the RIZ1 isoform (but not RIZ2, an alternative product of the same gene) is commonly lost or expressed at reduced levels in cancer cells. Forced expression of RIZ1 causes cell-cycle arrest at G2/M and/or apoptosis and suppresses tumorigenicity in nude mice (10–12). Our observation of a high rate of involvement of RIZ through frameshift mutations and genomic loss suggests a role in colorectal tumorigenesis.
Materials and Methods
Tissue Samples and Cell Lines.
Among a total of 22 MSI(−) tumors studied, 8 were selected because they previously had been found to display a CIN phenotype, including LOH at two closely linked markers, D1S228 [32.4 centimorgans (cM)] and D1S507 (36.2 cM) (13). In 14 tumors, the LOH status at 1p was unknown. In addition, we studied 24 MSI(+) tumors from hereditary nonpolyposis colorectal cancer (HNPCC) patients (14, 15). Moreover, 11 MSI(+) and 3 MSI(−) cell lines were purchased from the American Type Culture Collection. The MSI(+) lines were DLD1, LS411N, SW48, HCT116, LoVo, HCT15, and LS174T (colorectal cancer); MDAH2774 and SK-OV3 (ovarian cancer); AN3CA (endometrial cancer); and DU145 (prostate cancer). The MSI(−) lines were MDAMB231, MDAMB435S, and SKBR3 (breast cancer), which had been previously characterized for RIZ expression (10).
LOH Analysis.
Primary normal/tumor pairs were investigated by using fluorescently labeled microsatellites. Primer sequences were obtained from the Genome Database (http://gdbwww.gdb.org/). Amplifications of each microsatellite were done in 15-μl volumes with 10 ng of each respective genomic DNA, 8 pmol of each primer (5′ primer, fluorescently labeled), 100 μM each dNTP, 0.6 unit of AmpliTaq Gold DNA Polymerase (PE Biosystems, Foster City, CA), 10 mM Tris⋅HCl (pH 8.3), 50 mM KCl, and 2 mM MgCl2. PCR products were loaded onto a 377XL sequencer (PE Biosystems). Allele size and fluorescent intensity were determined by genescan and genotyper software (PE Biosystems). LOH was analyzed by determining the fluorescent intensity of each allele and calculating the ratio (13). A sample was scored as showing LOH if an allelic ratio of <0.67 or >1.5 was obtained.
Because of a high degree of MSI observed in the HNPCC tumor DNAs, single-nucleotide polymorphisms (SNPs) were also used to determine LOH in the subset of 24 HNPCC normal/tumor DNA pairs. Primer sequences were obtained from the Human SNP database (http://www-genome.wi.mit.edu/SNP/human/index.html). SNPs were amplified in 25-μl volumes with 100 nmol of each of the respective PCR primers, 25 ng of genomic DNA, 100 μM each dNTP, 1.0 unit of AmpliTaq Gold DNA Polymerase (Perkin–Elmer), 10 mM Tris⋅HCl (pH 8.3), 50 mM KCl, and 2 mM MgCl2. PCR products were purified by using exonuclease 1 and shrimp alkaline phosphatase (Amersham Life Sciences) and directly sequenced in one direction with one of the amplification primers and the BigDye Terminator chemistry (PE Biosystems). Samples that failed or sequenced poorly were resequenced in the other direction with the other amplification primer. LOH determination was done by a method similar to the microsatellite analysis.
Mutation Analysis.
Candidate genes were screened for mutations by direct sequencing of genomic PCR products. To facilitate direct sequencing of PCR products, all primers were tailed with M13-forward (TGTAAAACGACGGCCAGT) and M13-reverse (CAGGAAACAGCTATGACC) sequences (primer sequence is available from authors on request). PCRs were done in 25-μl volumes with 100 nmol of each of the respective PCR primers, 25 ng of genomic DNA, 100 μM each dNTP, 1.0 unit of Taq Gold DNA polymerase (Perkin–Elmer), 10 mM Tris⋅HCl (pH 8.3), 50 mM KCl, and 2 mM MgCl2. PCR fragments were purified by using the Exonuclease I/Shrimp Alkaline Phosphatase PCR Product Presequencing Kit (United States Biochemical). After purification according to the manufacturer's protocol, 2 μl of the PCR products were sequenced by using the BigDye Terminator AmpliTaq FS Cycle Sequencing Kit (PE Biosystems).
Expression Analysis.
RNA for expression analysis was isolated by using the RNAeasy Mini Kit (Qiagen, Chatsworth, CA). Reverse transcription of isolated RNA was done by using Superscript RT (Life Technologies, Rockville, MD), and cDNA amplification was done by using the GeneAmp Gold RNA PCR kit (PE Biosystems). Primers used for determination of RIZ1- and RIZ2-specific PCRs were as described (10). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific primers were used as a control (16).
Immunoprecipitation and Immunoblot Analysis.
Immunoprecipitation and immunoblot analysis was performed according to previously described procedures (17). Cell extracts were immunoprecipitated with RIZ antiserum 1637 against the N terminus of RIZ2 or preimmune serum. Immunoprecipitated products were resolved on a 5% SDS gel followed by immunoblot analysis using mouse antiserum KGSE against the N terminus of RIZ2.
Analysis of Cells Expressing Ectopically Introduced RIZ1.
This analysis was performed as described previously (10). Briefly, colon cancer cells were seeded at 2 × 105 cells in 6-cm dishes and infected with recombinant adenovirus at a multiplicity of infection of 100. At 48 hr postinfection, cells were processed for DNA histogram analysis.
Results
Deletion Mapping of 1p.
Most of the sporadic colorectal cancer tumors showed deletion of the entire 1ptel region up to ≈40 cM, where there was a return to heterozygosity in several tumors (Fig. 1). The region of return to heterozygosity is telomeric to several candidate tumor suppressor genes and oncogenes, including PAX7 at 46.2 cM, PLA2 at 46.2 cM, E2F2 at 52.4 cM, and MYCL at 71 cM. In comparison with MSI(−) tumors, the area of shared deletion was more restricted in MSI(+) tumors from HNPCC patients (Fig. 1). Although the majority of the SNP markers closest to 1ptel were not informative, a region of common deletion was identified by the marker WIAF-481 at 32.2 cM, which showed LOH in 8 of 15 (53%) informative HNPCC tumors (Fig. 1). The allelic imbalance values for SNPs were highly reproducible (Fig. 2), indicating the validity of these markers in the deletion mapping of MSI(+) tumors in particular. In comparison, a much lower rate of LOH was observed for nearby markers such as D1S450 (1/11) at 22.9 cM and D1S228 (1/8) at 32.4 cM.
Figure 1.

Deletion mapping of chromosome 1p in HNPCC and sporadic colorectal cancers. Markers highlighted in red indicate LOH (allelic ratios of <0.67 or >1.5). Yellow highlighted markers show MSI. Blue highlighted markers are informative and show no LOH, and markers that are not highlighted are not informative. Δ, Frameshift mutation in poly(A) repeat.
Figure 2.
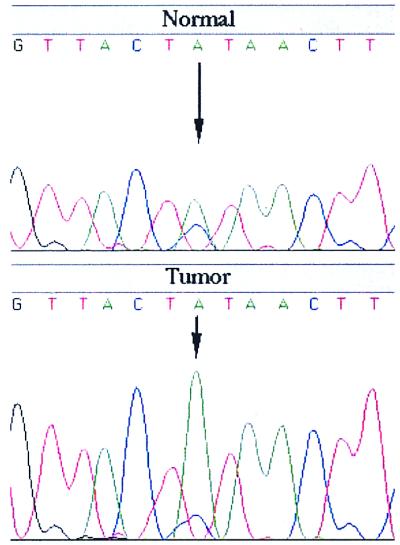
Example of LOH with SNP WIAF-481. A high number (8/15 or 53.3%) of informative HNPCC tumors display LOH with this SNP. This marker maps to 32.2 cM on the Human SNP database (http://www-genome.wi.mit.edu/SNP/human/index.html).
Identification of RIZ as a Candidate Gene for 1p Alterations.
Functional candidate genes in the region of 32.2 cM from 1ptel were examined and screened for mutations. The RIZ gene (retinoblastoma protein-binding zinc finger protein) maps to 32.2 cM on GeneMap 99 (http://www.ncbi.nlm.nih.gov/genemap99/). RIZ is within 370 kb of D1S228, as inferred from yeast artificial chromosome analysis (16). RIZ lies in 3′ to 5′ orientation from the telomere of chromosome 1p. In HNPCC tumors, there was a gradual decline of the LOH rate from WIAF-481 to RIZ 3′ to RIZ 5′. Three intragenic polymorphisms of RIZ were analyzed for LOH, including a codon Pro-704 deletion at exon 8, an intron 4 CA repeat (18), and a SNP flanking exon 4 (GAT to GAC 18 bases 3′ of the coding exon 4 sequence). The LOH rates for these RIZ markers were 21% (3/14), 9% (1/11), and 0% (0/8), respectively. Tumor 5393T showed LOH of exon 8 Pro-704 but retention of the other intragenic markers, consistent with location of a deletion break point inside the RIZ gene. Analysis of MSI(−) tumors that were not preselected for 1p LOH revealed LOH rates of 30% (4/13), 23% (3/13), and 60% (3/5), respectively. Tumor 7T-OSU showed LOH of RIZP704 but retention of heterozygosity of RIZ intron 4 CA, again indicating a deletion breakpoint within RIZ. The location of RIZ in the vicinity of the common LOH region and the observed deletion breakpoints within RIZ suggest that RIZ is a candidate target of 1p36 alterations in both hereditary and sporadic colorectal cancers.
Examination of the coding sequence of RIZ showed potentially hypermutable tracts of (A)8 and (A)9 in exon 8 of the RIZ gene. In 9 of 24 (37.5%) of the MSI(+) HNPCC tumors, frameshift mutations were found in either the (A)8 [one MSI(+) tumor; Fig. 3A] or the (A)9 [eight MSI(+) tumors; Fig. 3B] tract. Furthermore, 6 of 11 MSI(+) cell lines (HCT116, LoVo, LS411N, LS174T, MDAH2774, and AN3CA) showed frameshift mutations in the (A)9 tract. In cell lines HCT116 and AN3CA, the mutation was homozygous/hemizygous, resulting in biallelic involvement (Fig. 3C). None of the 23 tested MSI(−) sporadic colorectal cancers contained mutations in either of the polyadenosine tracts, indicating that these regions were mutational hotspots in MSI(+) tumors only.
Figure 3.

(A) Truncating frameshift mutation in the coding (A)8 repeat of exon 8 of the RIZ gene. MSI(+) tumor sample H2AT (Lower) has a 1-bp deletion in this repeat. (B) Examples of truncating frameshift mutations in the coding (A)9 repeat of exon 8 of the RIZ gene. MSI(+) tumor sample 5381T (Center) has a 1-bp deletion in this repeat. MSI(+) tumor sample 8201T (Lower) has a 2-bp deletion in this repeat. (C) Colon cancer cell line HCT116 is homozygous/hemizygous for a 1-bp deletion in the (A)9 repeat of exon 8 of RIZ, and endometrial cell line AN3CA has both 1- and 2-bp deletions in this repeat. MSI(+) colon cell lines LoVo, LS411N, and LS174T also have 1-bp deletions in this repeat, as does the ovarian cell line MDAH2774.
RIZ Frameshift Mutations Are Clonally Selected in Tumorigenesis.
To investigate whether similar mononucleotide tracts in other genes are involved to a comparable degree, we studied the coding (A)8 tracts of the PMS2 and DNA polymerase α genes. Frameshift mutations in these tracts were completely absent in the MSI(+) tumors (P = 0.0005). MSI(+) tumors were further screened for mutations in the (A)9 tracts of the RECQL, BLM, and KIAA0355 genes. One of 24 MSI(+) tumors had 1-bp deletions in the RECQL and KIAA0355 genes (P = 0.005). MSI(+) tumors were screened further for mutations in the (A)9 tracts of the RECQL, BLM, and KIA0355 genes. Of 24 MSI(+) tumors, 1 had 1-bp deletions in the BLM gene (P = 0.005). Taken together, these results suggested that the RIZ frameshift mutations were specifically selected during the clonal evolution of colorectal tumorigenesis.
Decreased RIZ1 Expression in Cell Lines.
Previous reports have shown that RIZ is expressed in two alternative transcripts, RIZ1 and RIZ2 (10). RIZ1 is commonly lost, whereas RIZ2 is present uniformly in several 1p36-linked cancer types (10–12). To determine whether expression was affected in RIZ-mutated colorectal tumors, we studied expression by the reverse transcription–PCR as described previously (10–12). Because of total overlap between the smaller RIZ2 and the larger RIZ1 transcripts, transcription cannot be measured for RIZ2 alone; hence, one reaction is specific for RIZ1, whereas the other measures RIZ1 + RIZ2. Breast cancer cell line MDAMB435S was used as a control that expresses only the RIZ2 isoform, and MDAMB231 was used as a control cell line that expresses both RIZ1 and RIZ2 isoforms (10). MSI(+) colorectal cancer cell lines (4 of 11) showed reduced or lost mRNA expression of RIZ1 in the presence of abundant RIZ1 + RIZ2 transcript (Fig. 4), supporting previous observations that an imbalance in the amounts of RIZ1 and RIZ2 is associated significantly with malignancy (10–12). In three cell lines with altered RIZ1 expression, frameshift mutations in the polyadenosine tracts were present, including cell line HCT116, which is homozygous or hemizygous for this mutation. Of these cell lines, LS411N was found to express only the RIZ2 isoform, whereas LoVo showed reduced expression of RIZ1. Also, SW48 showed reduced or absent expression of RIZ1, but had no mutations in the adenosine repeats of RIZ. The RIZ sequence is large (nearly 8 kb), and it is possible that there are other areas in the gene or in the promoter region that could be affected, resulting in altered RIZ expression in these cell lines.
Figure 4.

Expression analysis of the RIZ1 and RIZ2 isoforms in breast, colorectal, ovarian, and endometrial cancer cell lines. No RIZ1 transcript at all is detectable in mutation-positive cell line LS411N (colon). Other mutation-positive cell lines show decreased expression of RIZ1, including LoVo (colon) and HCT116 (colon). Breast cancer cell lines MDAMB435S and SKBR3 are used as positive controls, because they have been previously reported to not express RIZ1 or to express it at a reduced level.
RNA isolated from frozen tissue from the mutation-positive HNPCC tumors did not show clear expression changes of RIZ. This finding is likely caused by the contamination of normal tissue in the isolated RNA tumor samples. Titration experiments of cell-line RNA showed that even with a mixture of 90% RIZ2-expressing mRNA to 10% RIZ1- and RIZ2-expressing mRNA, the RIZ1 and RIZ2 isoform is PCR amplified (data not shown).
Frameshift Mutation Leads to Expression of Truncated RIZ Proteins.
The frameshift caused by the deletion of one adenosine at the (A)9 tract (at nucleotide position 4700 of the RIZ1 coding sequence) is expected to cause the fusion of truncated RIZ1 and RIZ2 lacking the C-terminal 219 aa with a novel reading frame of 76 aa. This would lead to the expression of mutant RIZ1 and RIZ2 that are 157 residues shorter than their wild-type counterparts. To confirm that such truncated RIZ1 and RIZ2 proteins indeed were expressed from the mutant allele, immunoprecipitation and immunoblot analysis was performed using the HCT116 cell line that is homozygous or hemizygous for the frameshift mutation. We first confirmed the usual pattern of RIZ protein expression in DLD1 cells that expressed both RIZ1 and RIZ2 mRNAs. In all tumor cell lines we have studied previously, RIZ1 protein was at low levels and difficult to detect, whereas RIZ2 protein was at higher levels and readily detectable (10, 17, 19). Similarly, in DLD1 cells, RIZ2 protein of 250 kDa was detected, and RIZ1 protein was at low or undetectable levels (Fig. 5). In contrast, full-length RIZ2 protein was not detected in the HCT116 cell line, but instead a shorter protein of ≈230 kDa was observed, consistent with truncation of RIZ2 protein by the frameshift mutation (Fig. 5). Although the experiment was not informative for RIZ1 protein, a truncated RIZ1 protein could be inferred from the results on RIZ2.
Figure 5.

Immunoblot analysis of RIZ protein expression. Equal amounts of cell extracts from DLD1 and HCT116 cells were immunoprecipitated by preimmune serum (PI) or anti-RIZ serum (αRIZ) 1637 as indicated at the top of each lane. The immunoprecipitated products were resolved on a 5% SDS gel followed by immunoblot analysis using mouse RIZ antiserum KGSE.
RIZ1 Causes G2/M Arrest, Apoptosis, or Both in Colorectal Cancer Cell Lines.
Adenovirus-mediated RIZ1 expression has been shown to cause G2/M arrest, apoptosis, or both in breast and liver cancer cell lines. We examined the effects of adenovirus-mediated RIZ1 expression on HCT116 and DLD1 colon cancer cell lines. Immunoblot analysis confirmed full-length RIZ1 protein expression in both cell lines upon infection with AdRIZ1 at a multiplicity of infection of 100 (data not shown). The fraction of infected cells in G2/M increased significantly at 48 h after AdRIZ1 infection in HCT116 and DLD1 cells. At 48 and 72 h postinfection with AdRIZ1, HCT116 cells showed sub-G1 DNA content indicating apoptotic cell death (Fig. 6 and Table 1). However, few cells with sub-G1 DNA content were observed in DLD1. The results show that RIZ1 caused G2/M arrest and apoptosis in HCT116 cells, but only G2/M arrest in DLD1 cells.
Figure 6.

Effects of adenovirus (Ad)-mediated RIZ1 expression on colorectal cancer cell lines HCT116 and DLD1. RIZ1 caused G2/M arrest and apoptosis in HCT116 cells, but only G2/M arrest in DLD1 cells, as shown by DNA content analysis.
Table 1.
Effects of RIZ1 expression on colorectal cancer cell lines HCT116 and DLD1
| Cell line | Cells, difference in %
|
|||
|---|---|---|---|---|
| Apoptosis | G1 | S | G2/M | |
| HCT116 | +25 ± 2.0 | −47 ± 1.3 | +6.1 ± 0.7 | +20 ± 0.8 |
| DLD1 | +9.5 ± 1.2 | −28 ± 2.1 | −2.2 ± 0.5 | +26 ± 1.7 |
DNA histogram analysis was performed 72 h after adenovirus (Ad) RIZ1 or AdNull virus infection. The values shown represent the difference in percentage of cell populations between AdRIZ1- and AdNull-infected cells. The increase caused by AdRIZ1 over AdNull is indicated by a plus sign (+), and a decrease is indicated by a minus sign (−). The values represent the mean ± SD of three experiments.
Discussion
RIZ is a protein with two alternative forms of 280 kDa (RIZ1) and 250 kDa (RIZ2). Apart from the N-terminal PR (PRDI-BF1-RIZ1) domain that is present in RIZ1 but not in RIZ2, the gene contains several other motifs of interest. These motifs include an RB-binding motif that is related to that of the adenovirus E1A oncoprotein, eight zinc finger motifs, and a PR-binding motif in the C-terminal region (17, 20, 21). The PR domain is necessary for the negative regulatory function of RIZ (10, 17) and may be involved in chromatin-mediated transcriptional activation or repression (20, 21). The interaction of the PR domain with the C-terminal binding domain might be necessary for homo- or hetero-oligomerization of RIZ, and for interactions with other proteins (21). Database comparison of the presently identified hypermutable regions of the zinc finger region of the RIZ gene shows that this region is highly conserved even among such evolutionarily divergent species as Saccharomyces cerevisiae and Drosophila melanogaster. The (A)8 tract in exon 8 of the RIZ gene is immediately 5′ of the most C-terminal zinc finger domain, and frameshift mutation in the (A)8 tract predicts termination of translation such that this zinc finger would be truncated. The (A)9 tract is 30 nt (10 aa) past this same zinc finger domain. Frameshift mutations in either tract are predicted to lead to the loss of the C-terminal domain of the RIZ protein that is involved in PR binding. Thus, it is likely that these frameshifts would be highly deleterious to the function of the RIZ protein.
Several criteria have been proposed to determine whether an affected gene is a true target of inactivation involved in tumorigenesis (22). These criteria include a high frequency of inactivation, biallelic inactivation, involvement of the candidate MSI target gene in a bona fide growth-suppressor pathway, inactivation of the same growth-suppression pathway in MSI-negative tumors through inactivation of the same gene or genes within the same pathway, and functional suppressor studies of in vitro or in vivo models. RIZ shows a high degree of frameshift mutations that, in some instances, are biallelic and followed by reduced expression. RIZ is potentially involved in the Rb growth-suppressor pathway, and RIZ1 expression causes G2/M cell-cycle arrest and/or apoptosis and suppresses tumorigenicity in nude mice (10–12). Our data show that RIZ1 expression caused G2/M arrest and apoptosis in the HCT116 cell line, which does not have any full-length RIZ, whereas RIZ1 did not cause apoptosis in DLD1 cells, which carry the wild-type RIZ gene. The results suggest that colon cancer cell lines with mutant RIZ may be more susceptible to RIZ1-induced apoptosis.
How does this correlate with the lost or reduced transcription of RIZ1 in most cell lines with a frameshift mutation? We speculate that, in some cases, hemi- or homozygous somatic mutation or loss of RIZ1-specific sequence may occur in the course of clonal evolution. We show that in several tumors with LOH in the region, the return to heterozygosity occurred in the vicinity of or inside of RIZ. Presently available polymorphic markers are not informative enough to provide a full analysis of every tumor. Moreover, LOH is difficult or impossible to assess in many cell lines, because the alleles that were present in the germ line are not known. We entertain the hypothesis that the function of RIZ may be impaired by somatic events in at least two different ways. In MSI(+) tumors (MIN pathway), frameshift mutations in the 3′ end of the gene interfere with the interaction between the C terminus of the protein and its N-terminal PR domain. In MSI(−) tumors (CIN pathway), mutations or deletions of the PR domain of RIZ1 may have similar effects. In this series of tumors, RIZ was affected by either LOH or frameshift mutation, but not both, suggesting that LOH and frameshift mutations had similar, alternative functions in RIZ-associated tumorigenesis.
Previous studies have established that RIZ1 is often underexpressed, whereas RIZ2 is present or overexpressed in cancer cells (10–12), but the exact genetic mechanisms underlying these changes is unknown. Our data suggest more than one mechanism. The fact that RIZ is altered in many cancers, and the fact that it resides in a chromosomal region that is frequently lost in many tumors, calls for closer scrutiny of RIZ in many cancers.
Acknowledgments
We thank Bert Vogelstein for critical reading of the manuscript. We also thank Lauri Aaltonen for help and Julie Meek, Samuel Richards, Marilotta Turunen, and Xiao-Hong Yang for technical assistance. Our work was supported by National Institutes of Health (NIH) Grant P30CA16058 to Ohio State University, Comprehensive Cancer Center; NIH Grant CA67941 and European Commission Grant BMH4CT960772 to A.d.l.C.; NIH Grant CA82282 to P.P.; and NIH Grant R01-CA76146, Tobacco Related Disease Research Program Grant TRDRP-7RT0026, and Cancer Research Program of California Grant CCRP-1II0023 to S.H.
Abbreviations
- MSI
microsatellite-unstable
- PR
PRDI-BF1-RIZ1
- LOH
loss of heterozygosity
- HNPCC
hereditary nonpolyposis colorectal cancer
- SNP
single-nucleotide polymorphism
- cM
centimorgan
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040579497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040579497
References
- 1.Loeb L A, Springgate C F, Battula N. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 2.Fearon E R, Vogelstein B. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Kinzler K W, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 4.Lengauer C, Kinzler K W, Vogelstein B. Nature (London) 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 5.Boland C R. Hosp Pract. 1997;32:87–96. doi: 10.1080/21548331.1997.11443598. [DOI] [PubMed] [Google Scholar]
- 6.Aaltonen L A, Peltomaki P, Leach F S, Sistonen P, Pylkkanen L, Mecklin J P, Jarvinen H, Powell S M, Jen J, Hamilton S R, et al. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 7.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan R S, Zborowska E, Kinzler K W, Vogelstein B, et al. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 8.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed J C, Perucho M. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 9.Souza R F, Appel R, Yin J, Wang S, Smolinski K N, Abraham J M, Zou T-T, Shi Y-Q, Lei J, Cottrell J, et al. Nat Genet. 1996;14:255–257. doi: 10.1038/ng1196-255. [DOI] [PubMed] [Google Scholar]
- 10.He L, Yu J X, Liu L, Buyse I M, Wang M-S, Yang Q-C, Nakagawara A, Brodeur G M, Shi Y E, Huang S. Cancer Res. 1998;58:4238–4244. [PubMed] [Google Scholar]
- 11.Jiang G-L, Liu L, Buyse I M, Simon D, Huang S. Int J Cancer. 1999;83:541–547. doi: 10.1002/(sici)1097-0215(19991112)83:4<541::aid-ijc17>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 12.Jiang G-L, Huang S. Histol Histopathol. 2000;15:109–117. doi: 10.14670/HH-15.109. [DOI] [PubMed] [Google Scholar]
- 13.Canzian F, Salovaara R, Hemminki A, Kristo P, Chadwick R B, Aaltonen L A, de la Chapelle A. Cancer Res. 1996;56:3331–3337. [PubMed] [Google Scholar]
- 14.Aaltonen L A, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomaki P, Chadwick R B, Kaariainen H, Eskelinen M, Jarvinen H, et al. N Engl J Med. 1998;338:1481–1487. doi: 10.1056/NEJM199805213382101. [DOI] [PubMed] [Google Scholar]
- 15.Holmberg M, Kristo P, Chadwick R B, Mecklin J P, Jarvinen H, de la Chapelle A, Nystrom-Lahti M, Peltomaki P. Hum Mutat. 1998;11:482–483. doi: 10.1002/(SICI)1098-1004(1998)11:6<482::AID-HUMU15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 16.Leygue E, Snell L, Dotzlaw H, Hole K, Hiller-Hitchcock T, Roughley P J, Watson P H, Murphy L C. Cancer Res. 1998;58:1348–1352. [PubMed] [Google Scholar]
- 17.Buyse I M, Shao G, Huang S. Proc Natl Acad Sci USA. 1995;92:4467–4471. doi: 10.1073/pnas.92.10.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang, W., Piao, Z., Simon, D., Sheu, J.-C. & Huang, S. (2000) Genes Chromosomes Cancer, in press. [PubMed]
- 19.Liu L, Shao G, Steele-Perkins G, Huang S. J Biol Chem. 1997;272:2984–2991. doi: 10.1074/jbc.272.5.2984. [DOI] [PubMed] [Google Scholar]
- 20.Xie M, Shao G, Buyse I M, Huang S. J Biol Chem. 1997;272:26360–26366. doi: 10.1074/jbc.272.42.26360. [DOI] [PubMed] [Google Scholar]
- 21.Huang S, Shao G, Liu L. J Biol Chem. 1998;273:15933–15939. doi: 10.1074/jbc.273.26.15933. [DOI] [PubMed] [Google Scholar]
- 22.Boland C R, Thibodeau S N, Hamilton S R, Sidransky D, Eshleman J R, Burt R W, Meltzer S J, Rodriguez-Bigas M A, Fodde R, Ranzani G N, Srivastava S. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]