

Abstract
The ability of organisms to evolve resistance threatens the effectiveness of every antibiotic drug. We show that in the nematode Caenorhabditis elegans, simultaneous mutation of three genes, avr-14, avr-15, and glc-1, encoding glutamate-gated chloride channel (GluCl) α-type subunits confers high-level resistance to the antiparasitic drug ivermectin. In contrast, mutating any two channel genes confers modest or no resistance. We propose a model in which ivermectin sensitivity in C. elegans is mediated by genes affecting parallel genetic pathways defined by the family of GluCl genes. The sensitivity of these pathways is further modulated by unc-7, unc-9, and the Dyf (dye filling defective) genes, which alter the structure of the nervous system. Our results suggest that the evolution of drug resistance can be slowed by targeting antibiotic drugs to several members of a multigene family.
Ivermectin is used to treat numerous parasitic infections of humans, pets, and livestock (1). Treatment with ivermectin is the cornerstone of efforts to eradicate river blindness (onchocerciasis). However, reports of resistance to ivermectin in nematodes are increasingly common (2–4). Ivermectin also kills the nematode Caenorhabditis elegans at therapeutic concentrations, making C. elegans a useful model system in which to examine mechanisms of ivermectin toxicity and resistance. Ivermectin activates glutamate-gated chloride channels (GluCls) that contain α-type channel subunits (5–7). In C. elegans, α-type subunits are encoded by a family of genes including: glc-1 (encoding GLC-1/GluClα1), avr-15 (encoding AVR-15/GluClα2), and possibly other uncharacterized genes found in the genome sequence (5–8). Severe loss-of-function mutations in glc-1 or avr-15 do not make worms resistant to ivermectin (6, 7), either because GluCls are not physiologically important targets of ivermectin, or because multiple GluCl genes contribute independently to ivermectin sensitivity. To clarify the role of the GluCls in the nematocidal effects of ivermectin, we had screened for ivermectin-resistant mutants (6). Here we analyze the effects of these and other, previously characterized mutations on ivermectin sensitivity. We show that simultaneous mutation of three genes encoding GluCl α-type subunits confers high-level resistance to ivermectin. Our results suggest that the ability of ivermectin to target several members of a multigene family may decrease the rate at which resistance evolves.
Methods
Genetics.
Unless otherwise indicated the mutant alleles used were: avr-14(ad1302), avr-15(ad1051), gcl-1(pk54∷Tc1), unc-7(e5), unc-9(e101), osm-1(ad1307), osm-5(ad1308), dyf-11(ad1303), and che-3(ad1306). avr-15(ad1051), glc-1(pk54∷Tc1), unc-7(e5), and unc-9(e101) appear to be molecular nulls (refs. 6, 7, and 18; T. Starich, personal communication). Ivermectin-resistant mutants were isolated in a screen for ivermectin resistance in an avr-15(ad1051) background by using the mutagen ethyl methanesulfonate as described (6). All strains were outcrossed twice with N2 and reisolated by selecting for ivermectin-resistant progeny in the F2 generation.
avr-14 was mapped to chromosome I by crossing DA1302 avr-14(ad1302); avr-15(ad1051) males into DA438 bli-4(e937); rol-6(e187); daf-2(e1368) vab-7(e1562); unc-31(e928); dpy-11(e224); lon-2(e678), selecting the F2 progeny on ivermectin and scoring for markers in the survivors. avr-14 was three-point mapped by crossing DA1302 to CB4776 bli-3 unc-13 or DA568 dpy-5 eat-5 unc-29. F2 progeny were selected for ivermectin resistance, singled, and scored for the presence of markers in the progeny. Of 89 resistant progeny of the DA568 cross, we found three whose progeny contained Unc non-Eat non-Dpy animals and one with Dpy non-Eat non-Unc progeny. Of 98 ivermectin-resistant progeny of the CB4776 cross, there were 27 whose progeny contained Bli animals and seven with Unc progeny.
The alleles of Dyf genes were mapped to a chromosome by using the mapping strain DA438; ivermectin-resistant progeny were cloned from heterozygotes and markers were scored among the progeny. Noncomplementation tests then were performed between the mutants we isolated and candidate Dyf genes on that chromosome by scoring the Dyf phenotype.
Strains containing multiple mutations were constructed by using standard genetic techniques. avr-15 was scored by electropharyngeogram (6), glc-1 was scored by PCR (7), and avr-14 was scored by using test crosses to score ivermectin sensitivity in an avr-15 background (details available from the corresponding author on request).
Ivermectin Sensitivity Assay.
The whole-worm ivermectin sensitivity assay was performed as described (6). Survival at various concentrations was normalized to the average survival in the absence of ivermectin. For each curve two separate experiments were performed for a total of n = 6 at each point. To calculate the EC37, the dose-response curves were fit to the equation:
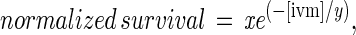 |
by using igor pro (WaveMetrics, Lake Oswego, OR) with x and y as free parameters. The y parameter is the EC37.
Ivermectin Binding.
Membranes (about 0.3–3 μg protein), prepared from C. elegans wild type (N2) and mutants by standard techniques, were incubated with [35S]sulfonamide-ivermectin (Drug Metabolism Group, Merck) from 60 fM to above 100 pM, for 1 day, by using techniques similar to those described (9). Bound radioactivity was separated from free by filtration; nonspecific retention of the ligand was measured by inclusion of 100 nM unlabeled ivermectin. Specific binding was modeled as binding to a single site or populations of sites with the same affinity for the ligand. The Kd was modeled at 3 pM for N2 and most mutants. The Bmax values for the different mutants are given relative to the N2 Bmax, which was 0.6 ± 0.08 pmol/mg protein (see Table 2).
Table 2.
High affinity ivermectin binding sites in worm membranes relative to wild type
| Wild-type GluCls Present | Bmax* | SEM | ||
|---|---|---|---|---|
| AVR-14 | AVR-15 | GLC-1 | 1.00 | |
| AVR-14 | GLC-1 | 0.37 | 0.15 | |
| AVR-14 | AVR-15 | 0.55 | 0.11 | |
| None | 0† | N.A. | ||
N.A., not available.
*Relative to N2, which had 0.6 pmol/mg pro.
† No high affinity binding site detected; if a site exists, its affinity is lower than 3 nM or constitutes less than 3% of the wild-type Bmax.
Cloning.
Cosmids were isolated and injected according to standard protocols (10). avr-14 was sequenced by designing sets of primers that spanned groups of exons and used to PCR from the mutant strains. After gel purification, the PCR products were sequenced by Applied Biosystems sequencers using the amplification primers for dye terminator reactions. avr-14/gbr-1 cDNAs were kindly provided by Adrian Wolstenholme (University of Bath, Bath, U.K.). These were cloned into the pT7 vector by using PCR to incorporate appropriate restriction sites. RNA transcription, oocyte manipulation, and electrophysiology have been described (6). PCR was used to amplify the 5′ end of avr-14 and add a PstI site in the sixth exon that could be used to clone into the green fluorescent protein (GFP) fusion vector, pPD95.77 (gift from A. Fire, J Ahn, G. Seydoux, and S. Xu, the Carnegie Institution of Washington, Washington, D.C.). This plasmid, pPD95.77.TM.avr-14.5′, was used to analyze GFP expression. Overlap extension PCR was used to generate a rescuing minigene construct, pBSaly.gbr3, consisting of the 5′ end of the avr-14 genomic fused in-frame at the introduced PstI site (which was removed in the process) to the avr-14 cDNA with the unc-54 3′ untranslated region from pPD30.69. Sequences of primers are available on request.
Laser Ablation.
I1 pharyngeal neurons were ablated as described (11). Operated worms were allowed to recover in the absence of ivermectin for 1 day. They then were transferred to plates containing 10 ng/ml of ivermectin and allowed to grow for 21 days. Statistical significance was tested by using a two-tailed Mann–Whitney U test.
Results
Isolation of Ivermectin-Resistant Mutants.
In a previously reported genetic screen for resistance to 10 ng/ml of ivermectin in an avr-15(ad1051) mutant background, we identified two classes of genes: alleles of four Dyf genes (osm-1, osm-5, dyf-11, and che-3), and two alleles of the GluCl subunit gene avr-14 (6). The Dyf class of genes includes more than 20 genes sharing defects in amphid sensory endings that result in the decreased ability of the sensory neurons to absorb dye from the environment. Analysis of the four Dyf mutants we isolated as well as a random sample of previously identified Dyf strains indicated that Dyf mutations generally confer low level (2- to 5-fold) resistance to ivermectin as single homozygous recessives, consistent with the reports of C. Johnson and Rand (12) and as cited in ref. 13; for example, see Table 4. In contrast, single homozygous avr-14 mutations confer no resistance to ivermectin in a wild-type background; resistance was synthetic, i.e., only a homozygous avr-14; avr-15 double mutant was resistant (6, 12, 14) (see Table 1).
Table 4.
EC37s for mutant strains without and with the Dyf mutation osm-1(ad1307)
| Wild-type GluCls present | Innexin present | Wild-type OSM-1
|
Mutant OSM-1
|
||||
|---|---|---|---|---|---|---|---|
| EC37, ng/ml | Error | EC37, ng/ml | Error | ||||
| AVR-14 | AVR-15 | GLC-1 | UNC-9 | 1.1 | 0.1 | 2.6 | 0.2 |
| AVR-15 | GLC-1 | UNC-9 | 0.6 | 0.1 | 2.5 | 0.1 | |
| AVR-14 | GLC-1 | UNC-9 | 1.0 | 0.1 | 8.2 | 0.4 | |
| AVR-14 | UNC-9 | 1.1 | 0.1 | 10.6 | 1.3 | ||
| GLC-1 | UNC-9 | 13.8 | 1.0 | 59 | 8.4 | ||
| AVR-14 | GLC-1 | 10.3 | 1.2 | 32.8 | 1.2 | ||
| UNC-9 | 4,264 | 1,750 | 5,490 | 2,270 | |||
Table 1.
Ivermectin EC37s for mutant strains
| Wild-type ivermectin receptors present | Wild-type innexins present | EC37, ng/ml | Error SEM | |||
|---|---|---|---|---|---|---|
| The GluCls AVR-14, AVR-15, and GLC-1 each independently make worms sensitive to ivermectin | ||||||
| AVR-14 | AVR-15 | GLC-1 | UNC-7 | UNC-9 | 1.1 | 0.1 |
| AVR-14 | UNC-7 | UNC-9 | 1.1 | 0.1 | ||
| AVR-15 | UNC-7 | UNC-9 | 1.4 | 0.3 | ||
| GLC-1 | UNC-7 | UNC-9 | 13.8 | 1.0 | ||
| Together, the three GluCls account for all sensitivity to >1,000 ng/ml | ||||||
| None | UNC-7 | UNC-9 | 4,264 | 1,750 | ||
| UNC-7 and UNC-9 gap junctions appear to act in parallel to the AVR-15 pathway | ||||||
| AVR-14 | AVR-15 | GLC-1 | UNC-7 | 0.6 | 0.1 | |
| AVR-14 | AVR-15 | GLC-1 | UNC-9 | 1.3 | 0.1 | |
| AVR-15 | GLC-1 | UNC-7 | 0.9 | 0.1 | ||
| AVR-15 | GLC-1 | UNC-9 | 0.5 | 0.1 | ||
| AVR-14 | GLC-1 | UNC-7 | 10.3 | 1.2 | ||
| AVR-14 | GLC-1 | UNC-9 | 733 | 136 | ||
| AVR-14 and UNC-9 appear to act via the same pathway | ||||||
| GLC-1 | UNC-7 | UNC-9 | 13.8 | 1.0 | ||
| AVR-14 | GLC-1 | UNC-7 | 10.3 | 1.2 | ||
| GLC-1 | UNC-7 | 14.0 | 0.5 | |||
| UNC-7 appears to contribute to ivermectin sensitivity via the AVR-14 and GLC-1 pathways | ||||||
| GLC-1 | UNC-7 | UNC-9 | 13.8 | 1.0 | ||
| AVR-14 | UNC-9 | 483 | 81.2 | |||
| AVR-14 | GLC-1 | UNC-9 | 733 | 136 | ||
| GLC-1 | UNC-9 | 2,110 | 595 | |||
| Evidence for a fourth ivermectin-sensitive channel that activates at very high concentrations and acts via UNC-7 | ||||||
| None | UNC-7 | UNC-9 | 4,264 | 1,750 | ||
| None | UNC-9 | >10,000 | NA | |||
NA, not applicable.
Molecular and Electrophysiological Characterization of avr-14.
To better understand the mechanism of synthetic ivermectin resistance, we cloned avr-14 by a combination of genetic mapping and identification of a candidate GluCl gene, gbr-2. gbr-2 encodes two alternatively spliced transcripts whose ORFs predict proteins that belong to the GluClα class of channel subunits (8). Two overlapping cosmids containing the gbr-2 gene, K07D2 and F56E2, as well as a minigene construct (see below) restored the ivermectin-sensitive phenotype when transformed into the avr-14; avr-15 double mutant (data not shown). We sequenced two alleles of avr-14 and found that one allele, ad1302, resulted from a missense V60E mutation in an exon common to both transcripts (Fig. 1A). This allele may be null for channel function; the mutated valine is conserved, even among vertebrate γ-aminobutyric acid type A and glycine receptors, and may play a role in subunit association (15). The second allele, ad1305, was a premature termination (nonsense) mutation that eliminates the last transmembrane domain of the channel encoded by transcript A and is likely to be null for the protein encoded by that transcript. We expressed RNAs corresponding to each of the two proteins encoded by gbr-2 in Xenopus oocytes. Oocytes injected with RNA encoding transcript A did not respond to ivermectin or glutamate. However, oocytes expressing transcript B gave a robust response to both ivermectin and glutamate (Fig. 1B) but did not respond to either of the chloride channel agonists γ-aminobutyric acid or glycine at 10 mM. As with the other characterized α-type subunits, the ivermectin response was only weakly desensitizing and was irreversible (5–7). We propose that the subunits encoded by avr-14/gbr-2 be called AVR-14A/GluClα3A and AVR-14B/GluClα3B.
Figure 1.

avr-14 encodes a subunit of an ivermectin-sensitive GluCl. (A) The mutations in alternatively spliced transcripts of avr-14. The exons encoding the extracellular domain (black) are shared by both transcripts. The exons encoding the transmembrane domains (stippled) are produced by alternatively spliced exons (white in AVR-14A and gray in AVR-14B). The region surrounding the conserved valine (bold) that is mutated to a glutamate in the ad1302 allele (resulting from a T to A mutation in the second base of the V60 codon) is shown lined up with the corresponding region in other GluCl subunits and in the rat glycine and γ-aminobutyric acid (GABA) type A-β channel subunits (29, 30). The ad1305 mutation results from an A to T change in the first base of the R387 codon. (B) Electrophysiology of oocytes expressing AVR-14B/GluClα3B. Oocytes were injected with RNA transcribed from AVR-14B/GluClα3B and were voltage clamped; 10 mM glutamate or 10 μM ivermectin (in 1% DMSO) were applied during the periods of time indicated by the bars. (C) Expression of an avr-14 reporter construct. A GFP reporter construct was generated by fusing GFP in-frame to the sixth exon of avr-14. GFP fluorescence of an L4 worm is shown. The identified neuronal cell bodies (arrows) are indicated by their three-letter code. Arrowheads indicate ventral cord neurons. Multiple fluorescent neurons in the ring ganglia are indicated by the bracket. (Scale bar indicates 50 μm).
avr-14 appears to act in the nervous system. We generated a reporter fusion between the avr-14 promoter and GFP that includes ≈3 kb of DNA upstream from the start ATG as well as the first six exons, which are common to both transcripts of avr-14. The avr-14∷GFP fusion was expressed exclusively in a subset of ≈40 extrapharyngeal neurons (Fig. 1C). Most of the neurons were in the ring ganglia of the head, but some motor neurons in the ventral cord and mechanosensory neurons also expressed GFP. Because GFP expression may not always accurately represent expression of the native protein, we wanted to confirm that the pattern of GFP we saw with our reporter construct could account for the ivermectin sensitivity conferred by avr-14. Therefore, we also constructed a minigene that consists of the same avr-14 promoter region fused to the avr-14B cDNA. When used to transform an avr-14; avr-15; gcl-1 triple mutant (see below), the combined minigene restored ivermectin sensitivity (data not shown).
Three GluCls Mediate Ivermectin Sensitivity.
Because simultaneous mutation of two GluCl subunits, avr-14 and avr-15, caused moderate ivermectin resistance (6, 14), we investigated the possible contribution of the third characterized GluCl subunit encoded by glc-1. By constructing double and triple GluClα-subunit mutants, we demonstrated that each of the GluCl genes represents a parallel genetic pathway that confers ivermectin sensitivity on C. elegans. Of the three double mutant strains in which only one of the three GluCls is wild type, only the strain expressing wild-type GLC-1 and lacking AVR-14 and AVR-15 was significantly resistant (Table 1). However, the avr-14; avr-15; glc-1 triple mutant, a strain in which all three GluClα subunit proteins are mutant, was ≈4,000-fold resistant (Table 1). In spite of having mutations in three GluClα subunits, these worms show surprisingly subtle behavioral changes including slightly inefficient eating and a tendency to reverse direction often (J.A.D., unpublished observation).
Innexins Contribute to Ivermectin Sensitivity by Altering the Structure of the Nervous System.
unc-1, unc-7, and unc-9 had been proposed to affect sensitivity to ivermectin and volatile anesthetics (16, 17). We did not find any resistance in the canonical loss-of-function unc-1(e719) allele either alone or in combination with avr-15 (data not shown). However, both unc-7 and unc-9, two genes encoding innexins (subunits of invertebrate gap junctions), contribute to ivermectin sensitivity (18–20). UNC-7 and UNC-9 act in parallel to AVR-15 because wild-type AVR-15 is sufficient to render worms sensitive to ivermectin regardless of the presence or absence of UNC-7 or UNC-9 (Table 1). However, strains with mutations in AVR-15 and UNC-7 or AVR-15 and UNC-9 showed increased resistance to ivermectin (Table 1). The UNC-9 gene product appears to be necessary for AVR-14-mediated sensitivity because: (i) when AVR-15 is mutated, mutation of either AVR-14 or UNC-9 resulted in strains with similar levels of resistance (Table 1), and (ii) when both AVR-14 and AVR-15 are mutated, the presence or absence of wild-type UNC-9 did not alter resistance substantially (Table 1), indicating that UNC-9 does not confer additional sensitivity in the absence of AVR-14. UNC-7 appears to mediate the effects of ivermectin acting via AVR-14 and GLC-1 and perhaps via an additional GluCl subunit. A strain in which UNC-7 and AVR-15 are mutated was more resistant than the strain with AVR-14 and AVR-15 mutated (Table 1), suggesting that mutating UNC-7 reduces sensitivity through a target in addition to AVR-14. However, mutating UNC-7 does not completely eliminate sensitivity mediated by AVR-14 because the strain with mutations in AVR-14, AVR-15, and UNC-7 was more resistant than the strain with mutated AVR-15 and UNC-7 (Table 1). On the other hand, the strain lacking all three GluCl subunits and UNC-7 was substantially more resistant than the strain lacking only the GluCl subunits (Table 1).
To determine whether the gap-junctional channels formed by UNC-7 and UNC-9 also might be targets of ivermectin, we performed ivermectin-binding studies on some of our mutant strains. As expected, membranes from strains lacking AVR-15 or GLC-1 had reduced ivermectin binding although they retain a fraction of the high affinity ivermectin binding found in wild type (Table 2). However, the strain lacking all three GluCl subunits had no detectable specific ivermectin binding. Lack of UNC-7 or UNC-9 had no significant effect on ivermectin binding (data not shown). Thus, the three genes encoding GluCl subunits appear to account for all high affinity ivermectin binding up to 100 pM (30 times the Kd of wild type). Although a small amount of ivermectin binding by the UNC-7 or UNC-9 proteins might have been missed, the amount they bind is not commensurate with their effects on ivermectin resistance. Rather than UNC-7 and UNC-9 being targets of ivermectin, their absence likely changes the connectivity of the nervous system to make it less susceptible to the effects of ivermectin on the GluCls.
One explanation for the effects of UNC-7- and UNC-9-containing gap junctions on ivermectin sensitivity is that they allow ivermectin-induced hyperpolarization of nonessential, GluCl-expressing cells to spread to other, essential excitable cells (19). Thus, in the absence of UNC-7 and UNC-9, ivermectin's effects would be restricted to cells expressing GluCls, inhibition of which does not cause severe toxicity. If so, what then might the essential cells be? One possibility is that the essential cells are in the pharynx. We observed that when the avr-15 mutant is exposed to ivermectin, it stops pumping. But when its pharynx is dissected away from the rest of the worm, pumping resumes. We explain these observations by supposing that ivermectin-induced hyperpolarization of extrapharyngeal neurons (through its action on AVR-14) flows via gap junctions back to the pharynx and inhibits pharyngeal function. If so, then ablating the neurons that connect the pharynx to the extrapharyngeal nervous system should break this link and decrease the ivermectin sensitivity of the avr-15 mutant. The pharynx is connected to the extrapharyngeal nervous system via gap junctions between the bilaterally symmetric I1 pharyngeal neurons and the extrapharyngeal RIP neurons. When we ablated the I1 neurons in an avr-15 glc-1 background, the worms were more resistant than mock-ablated worms (P = 0.0036; Table 3).
Table 3.
Effects of I1 ablation on sensitivity to 10 ng/ml of ivermectin
| Number of worms growing to each stage
|
|||
|---|---|---|---|
| Larva | Young adult | Fertile adult | |
| Control (avr-15 glc-1) | 18 | 1 | 0 |
| I1 ablated (avr-15 glc-1) | 9 | 3 | 7 |
Mutations in OSM-1 Act Additively with Other Genes to Increase Resistance.
OSM-1, the product of the Dyf gene osm-1, appears to act by a different mechanism than any of the other genes because worms lacking OSM-1 showed some resistance and all strains with a mutated OSM-1 were more resistant than strains of the same genetic background but with wild-type OSM-1 (with the possible exception of strains in which all three GluCls were lacking; Table 4). However, lack of OSM-1 seemed to preferentially affect the pathway defined by AVR-14 because worms lacking both OSM-1 and AVR-15 were more resistant than worms with only OSM-1 mutated whereas worms lacking AVR-14 and OSM-1 were just as sensitive as worms with only OSM-1 mutated.
Discussion
avr-14 Encodes a GluClα Subunit.
We have shown that the avr-14 ivermectin resistance locus corresponds to a GluCl gene cloned previously and known as gbr-2. Furthermore, we have demonstrated that this subunit forms a channel that is ivermectin- and glutamate-gated when expressed in Xenopus oocytes. However, we have only been able to observe a response to ligands in the avr-14B splice variant. This is odd because a mutation that specifically affects the avr-14A variant, avr-14(ad1305), confers ivermectin resistance to roughly the same extent as a mutation that affects both splice variants (J.A.D., unpublished observation). Furthermore, expressing only the avr-14B variant via our minigene construct is sufficient to confer ivermectin sensitivity on the triple GluCl mutant containing the avr-14(ad1302) allele. These results can be explained if these two splice variants associate in vivo. Although AVR-14B/GluClα3B appears sufficient to form a channel in oocytes, perhaps in vivo it must associate with the mutant ad1302 AVR-14A/GluClα3A subunits to form a channel.
Ivermectin Targets Three Members of the Family of Ivermectin-Sensitive GluCl Subunits.
Ivermectin has been shown to activate GluCls in several organisms including C. elegans. However, previous attempts to directly demonstrate the role of GluCls in ivermectin sensitivity by mutating individual GluCl genes were unsuccessful (6, 7), calling into question the role of GluCls in mediating physiologically relevant ivermectin toxicity. Our results clearly show that the GluCls are the physiologically relevant mediators of ivermectin toxicity and that the reason mutations in individual GluCls are not resistant is that ivermectin resistance is synthetic. Each of the three GluCls genes, avr-14, avr-15, and glc-1, constitutes a parallel genetic pathway that independently confers a high level of ivermectin sensitivity on worms. Consequently, mutations that reduce the sensitivity of any one pathway will not confer resistance (Table 1). A triple GluCl mutant, which reduces the sensitivity of all three pathways, is required before 4,000-fold resistance is achieved (Table 1).
The presence of a fourth ivermectin receptor is suggested by the increased resistance of the triple GluCl mutant when combined with an unc-7 mutation (Table 1). This observation could be explained if: (i) the mutations in AVR-14 and GCL-1 were not null for ivermectin sensitivity and mutating UNC-7 reduced the residual sensitivity of mutant AVR-14 or GCL-1 channels, or (ii) UNC-7 is necessary to confer sensitivity via yet another pathway, possibly one defined by one of the other GluCl-like subunits identified by the genome project. The first hypothesis predicts that mutating UNC-9 would have the same affect as mutating UNC-7, it would further reduce the sensitivity of worms with mutations in AVR-14 and AVR-15. This is not the case (Table 1) so we favor the second hypothesis. Because AVR-14 appears to contribute very little to ivermectin binding, it would not be surprising if a fourth, lower-affinity receptor were undetectable in our binding assay.
Ivermectin Can Inhibit Pharyngeal Pumping Via Two Pathways.
One way worms die from ivermectin is as a result of starvation caused by the inhibition of pharyngeal pumping (21–24). In light of this and previous studies it is clear that one of the genetic pathways leading to the inhibition of pharyngeal pumping is defined by the avr-15-encoded GluClα2 expressed in the pharyngeal muscle (6). A second pathway appears to consist of avr-14, unc-7, and unc-9 expressed in the extrapharyngeal nervous system. Three lines of evidence indicate that the extrapharyngeal nervous system is a separate target for ivermectin: (i) AVR-14 is not necessary for glutamatergic neurotransmission by the pharyngeal motor neuron M3, (ii) avr-14∷GFP is expressed only in extrapharyngeal neurons, and (iii) in the triple GluCl mutant, expression of AVR-14B in the nervous system is sufficient to confer ivermectin sensitivity. GluClα1, possibly in conjunction with UNC-7, appears to constitute a third genetic pathway in a set of as-yet-unidentified excitable cells.
However, ivermectin's interaction with any one of these three pathways is sufficient to inhibit pharyngeal pumping. How then does the interaction of ivermectin with GluCls in the extrapharyngeal nervous system inhibit pharyngeal function? Our results indicate that ivermectin-induced hyperpolarization spreads from avr-14-expressing extrapharyngeal neurons to the pharynx (Fig. 2). Breaking the connection between the pharynx and the extrapharyngeal nervous system of an avr-15 or an avr-15 glc-1 mutant either by: (i) dissecting the pharynx away from the rest of the worm, or (ii) laser ablation of the neurons that connect the pharynx to the extrapharyngeal nervous system, decreases ivermectin sensitivity.
Figure 2.

Preposed model to describe mechanisms of ivermectin sensitivity. Black arrows indicate the diffusion of ivermectin and the gray arrows indicate the flow of ivermectin-induced hyperpolarizing potential. Dyf gene mutations, which may reduce ivermectin permeability, act additively with all combinations of receptor mutants in the tier below. Ivermectin acts independently on each of the GluCl subunits to hyperpolarize cells: GluClα2 (AVR-15) acts in pharyngeal muscle, GluClα3 (AVR-14) acts in neurons. GluClα1 (GLC-1) is presumed to be neuronal but, because we do not know where gcl-1 is expressed, our model is not meant to imply that GluClα1 (GLC-1) and GluClα3 (AVR-14) are necessarily coexpressed or that they do or do not associate to form a channel. The effect of ivermectin on neurons expressing GluCls is not sufficient to kill the worms at low concentrations but requires that the ivermectin-induced hyperpolarization spread via gap junctions encoded by unc-7 and unc-9 to other excitable cells that are essential to the function of the worm. Our results indicate that the spread of hyperpolarization from the extrapharyngeal nervous system back to the pharynx is an important component of the gap-junction-mediated ivermectin sensitivity conferred by GluClα3 (AVR-14) and GluClα1 (GLC-1). The flow of hyperpolarizing potential from the extrapharyngeal neurons to the pharynx occurs via linking neurons such as I1 and RIP that may not themselves express GluCls.
Because unc-7 and unc-9 code for innexins (subunits of gap junctional channels), presumably making the avr-15; unc-7 or avr-15; unc-9 double mutant is yet another way of breaking the connection between the pharynx and the extrapharyngeal nervous system. This would explain why these double mutants, but not the avr-14; unc-7 or avr-14; unc-9 double mutants, show some resistance to ivermectin in our growth assay. We do not know where unc-7 and unc-9 are expressed and, in our model, they could be expressed anywhere in a chain of electrically coupled neurons linking the avr-14-expressing cells to the pharynx. Electron microscopic reconstruction of C. elegans indicates that there are gap junctions between the I1 and RIP neurons that form the connection between the pharyngeal and extrapharyngeal nervous systems (25). These gap junctional channels may require UNC-7 and UNC-9.
Dyf Mutations Appear to Affect Cuticle Permeability.
The Dyf mutants are so-named because of their inability to take up fluorescent dyes from the environment via the amphid sensory endings. It is therefore reasonable to think that the Dyfs confer resistance to ivermectin because they render the worm less permeable to the drug. Our results support this idea. All genetic backgrounds we tested (including wild type) have a higher EC37 when the Dyf gene osm-1 is mutated. This is the expected result if the equilibrium ivermectin concentration inside the worm as a fraction of the externally applied drug is reduced by the osm-1 mutation. Although not all Dyf mutants that are ivermectin resistant completely exclude dye [e.g., osm-5(ad1308)], it is possible that dye filling is reduced though not absent in these mutants or that the Dyf mutation differentially affects the permeabilities of the drug and the dye. Interestingly, the avr-15; osm-1 double mutant is significantly more resistant than the osm-1 single mutant whereas the avr-14; osm-1 double is not, suggesting that the osm-1 mutation preferentially reduces exposure of the extrapharyngeal nervous system to ivermectin. This makes sense in that the neurons with sensory endings at the amphid and that fill with dye are extrapharyngeal neurons.
What Can We Learn from the Genetics of Ivermectin Resistance in C. elegans?
The evolution of resistance in parasitic worms exposed to ivermectin is increasingly a problem. To better manage the use of ivermectin, it would be useful to be able to track this resistance by molecular means (26). Our study identifies several candidate genes that could be markers of resistance in parasites. Furthermore, the availability of clones from C. elegans will make it easier to identify homologs of these genes in parasitic worms. Interestingly, evidence suggests that ivermectin resistance in Haemonchus contortus is linked to GluCl genes (27). However, ivermectin-resistant strains of H. contortus with autosomal dominant resistance loci or wild-type ivermectin binding also have been observed (3).
Finally, as our ability to design drugs becomes more sophisticated and the pool of potential antibiotic drug targets grows, the choice of drug target becomes more important. Our results suggest principles to guide the design of antibiotic drugs. First, even proteins that have subtle loss-of-function phenotypes can be effective drug targets if the drug activates the target. Second, targeting a multigene family can help delay the evolution of resistance if it means that many drug targets must be mutated simultaneously for resistance to occur. If no single resistance allele can confer resistance on a strain, then the evolution of resistance requires the less probable simultaneous occurrence of two or more resistance alleles in different genes. This may account in part for the relative longevity of the avermectins and milbemycins as effective antiparasitic drugs (6, 28).
Acknowledgments
We thank Carl Johnson for sharing strains and unpublished data, Wes Shoop and Ann Smith for critical reading of the manuscript, Brande Thomas and Vivien Warren for assistance with the binding assay, Stan Zborovski for help with sequencing, and the Caenorhabditis Genetics Center for providing strains. L.A. was supported by an Established Investigator award from the American Heart Association. J.A.D. was supported by the American Cancer Society and the Haberecht Wild-Hare Idea program.
Abbreviations
- GluCl
glutamate-gated chloride channel
- GFP
green fluorescent protein
References
- 1.Campbell W C. Ivermectin and Abamectin. New York: Springer; 1989. [Google Scholar]
- 2.Prichard R. Vet Parasitol. 1994;54:259–268. doi: 10.1016/0304-4017(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 3.Sangster N. Parasitology. 1996;113:S201–S216. doi: 10.1017/s0031182000077982. [DOI] [PubMed] [Google Scholar]
- 4.Shoop W L. Parasitol Today. 1993;9:154–158. doi: 10.1016/0169-4758(93)90136-4. [DOI] [PubMed] [Google Scholar]
- 5.Cully D F, Vassilatis D K, Liu K K, Paress P S, Van der Ploeg L H, Schaeffer J M, Arena J P. Nature (London) 1994;371:707–711. doi: 10.1038/371707a0. [DOI] [PubMed] [Google Scholar]
- 6.Dent J A, Davis M W, Avery L. EMBO J. 1997;16:5867–5879. doi: 10.1093/emboj/16.19.5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vassilatis D K, Arena J, Plasterk R, Wilkinson H, Schaeffer J, Cully D, Van der Ploeg L. J Biol Chem. 1997;272:33167–33174. doi: 10.1074/jbc.272.52.33167. [DOI] [PubMed] [Google Scholar]
- 8.Laughton D L, Lunt G G, Wolstenholme A J. Gene. 1997;201:119–125. doi: 10.1016/s0378-1119(97)00436-8. [DOI] [PubMed] [Google Scholar]
- 9.Schaeffer J M, Haines H W. Biochem Pharmacol. 1989;38:2329–2338. doi: 10.1016/0006-2952(89)90473-5. [DOI] [PubMed] [Google Scholar]
- 10.Mello C C, Kramer J M, Stinchcomb D, Ambros V. EMBO J. 1992;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Avery L, Horvitz H R. Neuron. 1989;3:473–485. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- 12.Rand J B, Johnson C D. Methods Cell Biol. 1995;48:187–204. doi: 10.1016/s0091-679x(08)61388-6. [DOI] [PubMed] [Google Scholar]
- 13.Starich T A, Herman R K, Kari C K, Yeh W H, Schackwitz W S, Schuyler M W, Collet J, Thomas J H, Riddle D L. Genetics. 1995;139:171–188. doi: 10.1093/genetics/139.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson P. Methods Cell Biol. 1995;48:31–58. [PubMed] [Google Scholar]
- 15.Kuhse J, Laube B, Magalei D, Betz H. Neuron. 1993;11:1049–1056. doi: 10.1016/0896-6273(93)90218-g. [DOI] [PubMed] [Google Scholar]
- 16.Boswell M V, Morgan P G, Sedensky M M. FASEB J. 1990;4:2506–2510. doi: 10.1096/fasebj.4.8.2335273. [DOI] [PubMed] [Google Scholar]
- 17.Sedensky M M, Cascorbi H F, Meinwald J, Radford P, Morgan P G. Proc Natl Acad Sci USA. 1994;91:10054–10058. doi: 10.1073/pnas.91.21.10054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Starich T A, Lee R Y, Panzarella C, Avery L, Shaw J E. J Cell Biol. 1996;134:537–548. doi: 10.1083/jcb.134.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes T M, Hekimi S. J Neurochem. 1997;69:2251–2260. doi: 10.1046/j.1471-4159.1997.69062251.x. [DOI] [PubMed] [Google Scholar]
- 20.Phelan P, Stebbings L A, Baines R A, Bacon J P, Davies J A, Ford C. Nature (London) 1998;391:181–184. doi: 10.1038/34426. [DOI] [PubMed] [Google Scholar]
- 21.Arena J P, Liu K K, Paress P S, Frazier E G, Cully D F, Mrozik H, Schaeffer J M. J Parasitol. 1995;81:286–294. [PubMed] [Google Scholar]
- 22.Avery L, Horvitz H R. J Exp Zool. 1990;253:263–270. doi: 10.1002/jez.1402530305. [DOI] [PubMed] [Google Scholar]
- 23.Geary T G, Sims S M, Thomas E M, Vanover L, Davis J P, Winterrowd C A, Klein R D, Ho N F, Thompson D P. Exp Parasitol. 1993;77:88–96. doi: 10.1006/expr.1993.1064. [DOI] [PubMed] [Google Scholar]
- 24.Martin R J. Parasitology. 1996;112:247–252. doi: 10.1017/s0031182000084833. [DOI] [PubMed] [Google Scholar]
- 25.Albertson D G, Thomson J N. Philos Trans R Soc London Ser B. 1976;275:299–325. doi: 10.1098/rstb.1976.0085. [DOI] [PubMed] [Google Scholar]
- 26.Sangster N C. Int J Parasitol. 1999;29:115–124. doi: 10.1016/s0020-7519(98)00188-x. ; discussion 137–138. [DOI] [PubMed] [Google Scholar]
- 27.Blackhall W J, Pouliot J F, Prichard R K, Beech R N. Exp Parasitol. 1998;90:42–42. doi: 10.1006/expr.1998.4316. [DOI] [PubMed] [Google Scholar]
- 28.Martin R J, Murray I, Robertson A P, Bjorn H, Sangster N. Int J Parasitol. 1998;28:849–862. doi: 10.1016/s0020-7519(98)00048-4. [DOI] [PubMed] [Google Scholar]
- 29.Ymer S, Draguhn A, Kohler M, Schofield P R, Seeburg P H. FEBS Lett. 1989;258:119–122. doi: 10.1016/0014-5793(89)81630-8. [DOI] [PubMed] [Google Scholar]
- 30.Kuhse J, Schmieden V, Betz H. J Biol Chem. 1990;265:22317–22320. [PubMed] [Google Scholar]