

Abstract
Wolbachia are well known as bacterial symbionts of arthropods, where they are reproductive parasites, but have also been described from nematode hosts, where the symbiotic interaction has features of mutualism. The majority of arthropod Wolbachia belong to clades A and B, while nematode Wolbachia mostly belong to clades C and D, but these relationships have been based on analysis of a small number of genes. To investigate the evolution and relationships of Wolbachia symbionts we have sequenced over 70 kb of the genome of wOvo, a Wolbachia from the human-parasitic nematode Onchocerca volvulus, and compared the genes identified to orthologues in other sequenced Wolbachia genomes. In comparisons of conserved local synteny, we find that wBm, from the nematode Brugia malayi, and wMel, from Drosophila melanogaster, are more similar to each other than either is to wOvo. Phylogenetic analysis of the protein-coding and ribosomal RNA genes on the sequenced fragments supports reciprocal monophyly of nematode and arthropod Wolbachia. The nematode Wolbachia did not arise from within the A clade of arthropod Wolbachia, and the root of the Wolbachia clade lies between the nematode and arthropod symbionts. Using the wOvo sequence, we identified a lateral transfer event whereby segments of the Wolbachia genome were inserted into the Onchocerca nuclear genome. This event predated the separation of the human parasite O. volvulus from its cattle-parasitic sister species, O. ochengi. The long association between filarial nematodes and Wolbachia symbionts may permit more frequent genetic exchange between their genomes.
Synopsis
Filarial nematode worms cause hundreds of millions of cases of disease in humans worldwide. As part of efforts to identify new drug targets in these parasites, the Filarial Genome Project rediscovered that these worms carry within them a symbiotic bacterium, which may be a novel target. Fenn et al. investigated the relationships of these bacteria, from the genus Wolbachia, to those previously identified in arthropods using a new dataset of genome sequence data from the human parasite Onchocerca volvulus. O. volvulus causes river blindness in West Africa. The authors found that the Wolbachia strains found in nematodes are more closely related to each other than they are to the Wolbachia in insects, suggesting that the nematodes and their bacterial partners have been coevolving for some considerable evolutionary time and may indeed be good targets. In addition, the authors identified a fragment of Wolbachia DNA that was inserted in the genome of its nematode host and has subsequently degenerated. The insertion occurred before O. volvulus diverged from another nematode species, O. ochengi, found in cattle.
Introduction
Wolbachia are alphaproteobacteria that live intracellularly in a range of animal hosts [1]. Wolbachia belong to the Anaplasmataceae in the Rickettsiales, a diverse group of intracellular symbionts. In other Rickettsiales, the symbiosis is usually parasitic or pathogenic, and many of these bacteria cause significant human and veterinary disease problems. Rickettsiales have also been identified as symbionts of arthropods, and are implicated in causing reproductive manipulations in their hosts similar to those of Wolbachia. (See below; we note that our knowledge of these bacteria is likely to have a severe ascertainment bias, as disease-causing pathogens are obvious and important, whereas innocuous or even beneficial interactors, and free-living species, will be missed. In this context it is informative that unbiased surveys of ecosystems using PCR amplification of conserved genes are turning up rickettsia-like bacteria in many unexpected situations [2].)
In arthropods, where they were first discovered, Wolbachia are the causative agents of a number of fascinating reproductive manipulations [3]. These manipulations serve to promote the survival of infected female arthropods, which pass the Wolbachia vertically to their offspring. A range of phenotypes are caused by Wolbachia infection in arthropods, including killing or feminisation of genetic males, induction of parthenogenetic reproduction in haplo-diploid females, and induction of reproductive incompatibility between individuals that do not have the same infection status. The prevalence of Wolbachia in current arthropod faunas is very high [4,5]; this is due to rare but successful horizontal transfer of the infection between taxa, and is likely to play a role in speciation. Selective sweeps caused by introgression of new Wolbachia strains have strongly shaped mitochondrial population genetics [6], and genomic conflict between the bacterium and the nuclear genome may promote reproductive isolation [7]. There is limited congruence between host and bacterial phylogenies in the arthropod system.
Most arthropod Wolbachia derive from two relatively closely related clades, called A and B [1]. The only formally named Wolbachia is W. pipientis from the mosquito Culex pipiens, but divergence between the major clades is similar to that observed between species in other bacterial genera [8]. Variant arthropod Wolbachia have been described, from springtails, termites, and spiders, that define additional, more deeply separated clades (E, F, and G) [8,9]. Resolution of the relationships of these additional clades is currently poor. However, Wolbachia “infections” are not limited to the Arthropoda. Parasitic filarial nematodes of the Onchocercidae, including several major human pathogens, harbour intracellular Wolbachia [10–12]. No other nematodes are currently known to harbour Wolbachia [13], though other nematode–bacterial symbioses are common. In the onchocercids, the Wolbachia can be divided into two major clades, C and D [14], which, unlike the arthropod Wolbachia clades, show phylogenetic congruence with their hosts [15]. Thus, closely related filarial nematodes have closely related Wolbachia, and the association between nematode and bacterium appears to be one of long-term (>100 million years), stable, vertical transmission. The Wolbachia of one filarial species, Mansonella ozzardi, has been placed by analysis of a small number of genes in clade F with termite and weevil isolates.
Analysis of the relationship between the nematodes and their symbionts has revealed that they are likely to be mutualists [16]. Killing the bacteria with tetracycline affects nematode growth, moulting, fecundity, and lifespan [17,18]. In arthropods, in most cases, tetracycline treatment yields cured, healthy hosts, and related parasitic nematodes that do not harbour Wolbachia are unaffected by tetracycline treatment [18]. This feature of nematode–Wolbachia interaction has led to trialling of tetracycline antibiotics for treatment of human filariases, with very positive results [19–22].
In the Rickettsiales and Wolbachia, therefore, where the intracellular habit is ancestral, there has either been a loss of the parasitic or pathogenic phenotype in the nematode Wolbachia or evolution of novel parasitic mechanisms in the arthropod Wolbachia. Previous analyses of Wolbachia phylogeny, and of the relationships of the genus to other Rickettsiales, have been based on very few genes (the Wolbachia surface protein wsp, cell-division protein ftsZ, citrate synthase gltA, groEL chaperone, and small subunit ribosomal RNA [16S] genes) [1,14,15,23]. These analyses were equivocal concerning the deeper structure of the Wolbachia, and could not resolve the placement of the root of the genus; clades E, F, and G are significantly under-sampled. A major limiting factor has been the inferred length of the branches leading to the outgroup taxa. As the genes sequenced have generally been chosen for their ability to resolve within-clade, between-isolate relationships, they are not suited to robust resolution of the deeper relationships of Wolbachia. Studies on yeasts and other taxa have shown that extended, multigene datasets can often provide robust resolution when individual constituent genes cannot [24].
Given that clades A and B are very closely related, two possibilities seem most likely. The first is that the nematode symbionts and the arthropod parasites form two distinct radiations (i.e., the tree has the form [outgroup[[A,B],[C,D]]]; Tree 1 of Figure 1). The second is that one of the nematode symbiont clades (most probably clade C, found in Onchocerca species and close relatives) arises basal to the other clades (i.e., the tree has the form [outgroup[C[D[A,B]]]]; Tree 2). A final possibility is that nematode Wolbachia arose from within the arthropod-infecting clades (Tree 4). Trees 1 and 4 have been implicit in many discussions of Wolbachia evolution, possibly because of the historical accident that arthropod Wolbachia were the first to be identified, and are the more widely studied. We have generated genome sequence from a clade C Wolbachia, wOvo from the human parasite Onchocerca volvulus, and here analyse it along with genome sequence from the Wolbachia of Drosophila melanogaster (wMel) (clade A), Wolbachia from Brugia malayi (wBm) (nematode, clade D), and a series of anaplasmatacean outgroups to re-examine this question. We find that the root of Wolbachia is robustly placed between clades A and [C and D], and thus that the mutualist nematode symbionts likely arose from parasitic or pathogenic ancestors. The close coevolution of nematodes and their Wolbachia is underlined by the discovery of a segment of the Wolbachia genome translocated to the O. volvulus nuclear genome.
Figure 1. Hypotheses of Wolbachia Relationships.

While we analysed seven taxa, they can be treated as if they were four: outgroups (Anaplasma and Ehrlichia), clade A Wolbachia (wMel, wAna, and wSim), clade D Wolbachia (wBm), and clade C Wolbachia (wOvo). There are thus three possible placements of the root of Wolbachia: (1) Tree 1 [outgroups[A[C,D]]], (2) Tree 2 [outgroups[C[D,A]]], and (3) Tree 3 [outgroups[D[C,A]]]. As clade A included more than one taxon, trees with clade A paraphyletic are also possible. In practice only one such arrangement was found (Tree 4; [outgroups[A′[A′′[C,D]]]]), and may have arisen from analysis of paralogous genes.
Results
Five Segments of the Genome of Wolbachia from O. volvulus
Twenty-seven primer pairs derived from a range of putative genes from Wolbachia from O. volvulus (wOvo) were tested and yielded 11 probes (Table 1). Five of these identified positive clones in the O. volvulus genomic libraries, and the inserts of these clones were amplified by long-range PCR and sequenced (Table 2). The total unique sequence length of the segments is 70,830 bp, representing 6.5% of the estimated 1.1 Mb of the wOvo genome [25]. The proportion of the sequenced segments made up of guanine and cytosine bases (GC%) ranged from 31.8% to 35.38% with a mean value of 32.9%). The average GC% of wBm, wMel, and Rickettsia prowazekii is 34%, 35.2%, and 29.1%, respectively.
Table 1.
PCR Primers Used

Table 2.
Genome Sequence from wOvo
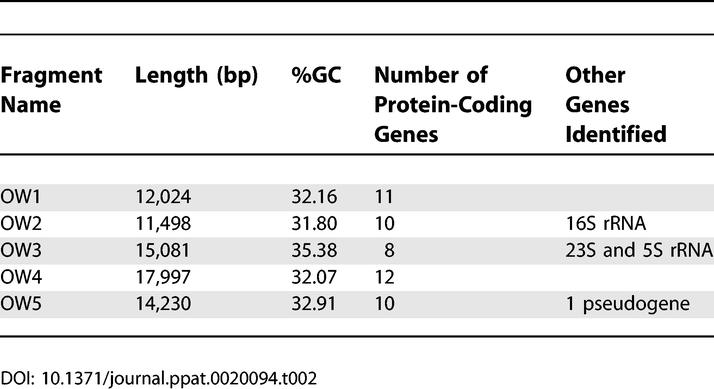
We identified 51 protein-coding genes and three ribosomal RNA genes (16S, 23S, and 5S) in the five segments (Table 3; Figure 2). Coding regions cover 76.6% of the total sequence, again within the expected range when compared to wBm, wMel, and R. prowazekii (67.4%, 81%, and 76%, respectively). This corresponds to a gene density of 0.72 protein-coding genes/kb, which is comparable to wBm and R. prowazekii (both 0.75 functional genes/kb) but much less than wMel (0.94 functional genes/kb). If the genome of wOvo is similar in size to those of wMel and wBm, it is estimated to have approximately 800 functional genes, like wBm (which has 806) [26], but many fewer than wMel (1,270) [27].
Table 3.
Genes Identified in Genomic Sequence from wOvo, and Their Support for Different Phylogenies

Figure 2. Synteny Comparisons between wOvo, wMel, and wBm.

Cartoons represent the conservation of local synteny between the sequenced wOvo fragments (OW1 to OW5) and the orthologous regions of the genomes of wBm and wMel. Genes are shown by small arrowed boxes, but are not drawn to scale. Double diagonal lines show breaks in synteny, and curled arrows show inversions of orientation.
Functional annotation was possible for the majority of the 51 protein-coding genes [26,27] (Table 3). Six are Wolbachia-specific, having no orthologue in any of the alphaproteobacterial genomes examined, or elsewhere. These include Wolbachia surface protein and five conserved hypothetical proteins. As these genes are present only in Wolbachia, they may encode proteins involved in the particular symbiotic biology of the bacteria. One gene, OW2-I, is wOvo-specific: no function can be ascribed by similarity. A partial pseudogene similar to an ATP-dependent caseinolytic protease ATP-binding subunit, ClpA, was identified (Figure 3). An orthologous ClpA gene is intact in wMel [27], is degraded in wBm [26], and is missing from R. prowazekii [28]. While it is possible that there is another copy of ClpA in the wOvo genome, this seems unlikely given the synteny of wOvo ClpA and flanking genes with wMel and wBm (see below). wBm ClpA is intact at the 3′/C-terminal end, but has a deletion of 21 bases and two in-frame stop codons compared to wMel ClpA. In the region that overlaps with the partial wOvo ClpA, the wBm representative is intact, while wOvo has 13 insertion/deletion (indel) changes, 12 of which cause frame shifts (Figure 3). ClpA acts as a molecular chaperone, and when in complex with the protease ClpP (ClpAP) it recognises and targets proteins for degradation. ClpX, another Clp regulator, is distinct from ClpA, and also forms complexes with ClpP (ClpXP) [29]. Both ClpP and ClpX are present in the genomes of wBm, wMel, and R. prowazekii. It has been reported that ClpAP and ClpXP have distinct substrate specificities in that ClpXP binds only substrate proteins that contain a recognition signal [30]. The benefits for mutualist Wolbachia of not having ClpA are unclear, as ClpA is the more generalist subunit, dealing with proteins damaged by heat shock and starvation.
Figure 3. A ClpA Pseudogene in wOvo Has Many Inactivating Mutations.

An alignment of the nucleotide sequences of ClpA from wMel (a functional gene) and wBm (a pseudogene, inactivated by mutations that generate two in-frame stop codons; otherwise intact), and the partial gene identified from wOvo from fragment OW5. The wOvo gene has multiple, independent, inactivating mutations in the 5′ region available for comparison. Yellow shading indicates in-frame indel events, red shading indicates frame-shifting indel events (observed only in wOvo OW5-K), and violet shading indicates the two in-frame stop codons in wBm ClpA.
A second wOvo serine protease subunit, identified as HtrA, was found in fragment OW4 (gene OW4-E). A HtrA from wOvo has been reported previously [31], but OW4-E differs from the published sequence, particularly in the 3′ half of the gene. Resequencing of wOvo HtrA from O. volvulus genomic DNA yielded the same sequence as OW4-E. No fragments or sequences corresponding to the published HtrA were recovered. Alignment of OW4-E and other alphaproteobacterial HtrA genes and the published sequence revealed many single base changes and several indel events that change the frame of the translated protein with respect to other HtrA sequences. The 3′ end of the published “wOvo” HtrA is, however, identical to wBm HtrA, while the 5′ end is nearly identical to OW4-E: it is likely to be an artefactual fusion between wOvo and wBm genes, with some indel sequencing errors also.
Synteny Comparisons between wOvo, wBm, and wMel
The arrangement of genes in the five fragments of the wOvo genome was compared to the sequenced genomes of other Wolbachia and Anaplasmataceae. None of the five wOvo fragments was fully syntenic with either fully sequenced Wolbachia (Figure 2). Fragment OW2 differed from wBm only in the presence of a wOvo-specific coding sequence (OW2-I). The other wOvo fragments had two or three rearrangements compared to wBm. Comparison to wMel identified between two and four rearrangements per fragment. Overall, wMel and wBm were more similar to each other in the compared regions, sharing many gene order structures compared to wOvo. Of the five instances where rearrangements compared to wOvo differ between wMel and wBm, wBm is more like wOvo in four (Figure 2). In the fifth (in OW4), gene OW4-L is inverted, but still linked, in wMel, while it is unlinked (but in the same transcriptional orientation) in wBm. None of the gene arrangements specific to wOvo, wBm, or wMel were found in the other Anaplasmataceae genomes surveyed (unpublished data).
Phylogenetic Analyses of Wolbachia Based on 46 Genes
We identified putative orthologues for the genes identified on the wOvo fragments from the complete and partial genomes of wBm, wMel, Wolbachia from D. ananassae (wAna), Wolbachia from D. simulans (wSim), Ehrlichia canis, E. ruminantium, and Anaplasma marginale. For each gene, we collected all homologues from all sequenced genes from alphaproteobacteria, constructed alignments, and analysed these phylogenetically using the neighbour joining (NJ) algorithm. For the set of target taxa (see Table 3) we selected those homologues that were robustly defined as orthologous to the wOvo genes. For two proteins (OW1-G and OW5-D) no orthologues were identified in Ehrlichia or Anaplasma, and for these we selected orthologues from R. typhi and R. prowazekii as outgroups. Calculation of the distance from each wOvo protein to that of E. canis, compared to its wMel or wBm orthologue, showed that there was no obvious long branch artefact that might artificially associate two of the three Wolbachia, and that the set of genes analysed embody a wide range of evolutionary rates (Figure 4). The gene set is thus suited to analysis of both local and deep phylogenetic problems [24].
Figure 4. The wOvo Genome Has Similar Evolutionary Rates to wMel and wBm.

The graph shows the relationship between evolutionary rates (mean difference) for all 46 protein-coding genes, calculated as distance to the outgroup E. ruminantium, between wOvo and wMel (red) and wOvo and wBm (blue). For both comparisons, the slope of the line is ≅1 (wOvo/wMel 0.977 ± 0.002; wOvo/wBm 0.981 ± 0.002), indicating that while wOvo has a lower rate than that of the other Wolbachia the difference is minor (~2% overall).
Each alignment of orthologues was then subjected to phylogenetic analysis using NJ, maximum likelihood (ML), and Bayesian ML models. The use of multiple methods of analysis is of utility in the identification of sequences or clades that behave differently or aberrantly under one method compared to others. The Bayesian ML analytical method is generally recognised to be very effective in dealing with biases in sequence alignments, though it is not foolproof [32]. NJ, as it effectively reduces all signal to a single pairwise difference, is most liable to systematic error. Under NJ, 28 of the 44 protein-coding genes yielded support (bootstrap values > 70%) for a close relationship between wMel and wBm to the exclusion of wOvo (i.e., Tree 2 of Figure 1; Table 4). Two genes supported Tree 1 and one Tree 3; the other genes did not yield phylogenies with >70% bootstrap support for any of the trees. Under Bayesian ML, only 15 of the individual proteins supported Tree 2 with significant posterior probability (>90%), while 11 supported Tree 1. Tree 3 was supported under Bayesian analysis of the same protein, OW1-G, that yielded Tree 3 in the NJ analysis. We note that we had to use Rickettsia outgroups for this gene as no orthologues were identified in Ehrlichia or Anaplasma genomes, and that this may have resulted in a long branch artefact. The ribosomal RNA genes yielded support for Tree 2 in NJ and Bayesian ML analyses, though the support was low. Surprisingly, despite the strong support for distinct trees by both methods for many genes, Shimodaira-Hasegawa (SH) tests found no cases in which there was a significant difference in support for Trees 1 or 2 (Table 3).
Table 4.
Summary of Support under Different Models of Phylogenetic Inference

Bayesian ML analyses were also carried out on a concatenated alignment of 42 protein-coding genes (excluding those lacking Anaplasma and Ehrlichia outgroups) using two models of protein evolution. The first used a single rate for all the sequences, while the second, more realistic model allowed each protein to evolve with its own rate multiplier. The second model was significantly better (harmonic mean LnL partitioned = −121,745.01; unpartitioned = −122,039.86; Bayes factor ≅ e294 ≅ 10127). Using a single rate yielded Tree 1, a result that might be expected considering the relative lengths of the proteins supporting Tree 1 versus Tree 2 (Table 3). A SH test showed highly significant support for Tree 1 (p = 0.003). Analysis using the partitioned model yielded Tree 1 with high posterior probabilities at all nodes (Figure 4). Although Bayesian ML analysis can overestimate support for trees, this result was found in multiple independent analyses.
Identification of a Lateral Gene Transfer Event from Wolbachia to the Nematode Nuclear Genome
Comparison of the sequenced wOvo genomic fragments to available O. volvulus DNA sequences identified a segment of O. volvulus genomic DNA that had significant nucleotide sequence identity to two distinct genes in wOvo (Figure 5). A 5,074-bp EcoRI fragment of O. volvulus genomic DNA had been isolated and sequenced because it contained a TATA box–binding protein gene (GenBank accession L13731) [33]. The TATA box–binding protein gene is located from residues ~2200 to 3500 of the fragment, but a full-length coding sequence was not predicted previously [33]. We resurveyed this sequence, identifying a likely 5′ trans-splice acceptor site at bases 2096 to 2101 and an initiation ATG at 2105 to 2107. The ~2 kb upstream of this trans-splice acceptor site are free of obvious coding features and have no BLASTx matches in public databases (unpublished data). We identified a region of 104 bases (from position 182 to 384 of L13731) that was 63% identical to wOvo OW4-C (Figure 6). There are three insertions (totalling four bases) and one deletion (of one base) in L13731 compared to wOvo OW4-C. Immediately following this section in L13731 is a stretch of 205 bases (385 to 589) that is 84% identical to wOvo OWJ-2 (with two insertions, of one base and 13 bases, and one deletion of one base). Neither of the wOvo-like segments in L13731 has a complete open reading frame because of the indel differences. Both of these wOvo genes have orthologues in wMel and wBm, but the region of wOvo OW4-C that is similar to L13731 is very divergent from the other Wolbachia genes (not shown). Alignment of the wMel and wBm orthologues of wOvo OWJ-2 (a predicted phosphomannomutase) to L13731 shows that the O. volvulus nuclear fragment is more similar to wOvo than it is to either of the other two Wolbachia (Figure 5A).
Figure 5. Wolbachia Relationships Inferred from 42 Protein-Coding Genes.

(A) Phylogram of consensus tree with inferred distances based on the ultimate model parameters inferred in MrBayes, and rooted by the Anaplasma and Ehrlichia outgroups.
(B) Cladogram showing Bayesian support for each node.
Figure 6. A Fragment of a Wolbachia Genome in the O. volvulus Nuclear Genome.

(A) Sequence alignment of a region upstream of the O. volvulus TATA box–binding protein gene from O. volvulus from Mali (residues 130 to 628 of GenBank accession L13731) and O. volvulus from Ghana (this work), and the orthologous insertion from O. ochengi. These nuclear sequences are aligned to fragments of two different genes from Wolbachia genomes: (1) wOvo OW4-C (residues 1290 to 1087 of the open reading frame) and the corresponding wOoc gene, and (2) wOvo OW2-J (a phosphomannomutase [pmm]; residues 252 to 443 of the open reading frame) and the corresponding fragments from the wOoc, wMel, and wBm orthologues. While wOvo OW4-C does have homologues in wMel and wBm, the region of the wOvo gene that aligns to the O. volvulus nuclear sequence is very poorly conserved. Inserted gaps are indicated by a dash. Residues identical in >50% of the aligned sequences are shaded.
(B and C) PCR verification of the presence of the Wolbachia-like gene fragments in O. volvulus and its close relative O. ochengi. Ethidium-bromide-stained gels are shown with lanes M, DNA size markers; 1, 2, 3, and 4, single-primer controls for the primers TATA_Phos, TATA_OW4C, Phos, and OW4C, respectively (see Table 1 for primer sequences); 5, PCR product of wOvo phosphomannomutase (primers TATA_Phos and Phos); 6, PCR product of wOvo OW4-C (primers TATA_OW4C and OW4C); and 7, PCR product of the Onchocerca genomic insertion (primers TATA_Phos and TATA_OW4C). In (B) the target was O. volvulus genomic DNA, while in (C) the target was O. ochengi genomic DNA.
The nematodes from which Li and Donelson [33] prepared their genomic DNA derived from Mali. As the fragment was sequenced from a genomic DNA clone it was possible that it was a cloning artefact. This possibility was excluded by firstly amplifying the putative insertion from our independent source of O. volvulus specimens (from Ghana), and secondly by identifying an orthologous insertion in the genome of the related cattle parasite O. ochengi. We carried out PCR assays using primers designed to be able to amplify either from the putative insertion in the nuclear genome, or from the copy resident in the wOvo genome. We were able to amplify, and confirm by sequencing (Figure 6A), the presence of the wOvo-like segments upstream of the O. volvulus TATA box–binding protein gene (Figure 6B). O. volvulus is one of a group of onchocercid species endemic in Africa. It is known to be close phylogenetically to O. ochengi, a cattle parasite that has a range overlapping that of O. volvulus, with which it shares some vector species [34]. We surveyed the genomes of O. ochengi for Wolbachia from O. ochengi (wOoc) genes and the putative nuclear insertion and confirmed their presence. Sequencing of the putative insertion fragment (Figure 6A) revealed five single base pair differences from O. volvulus. We were unable to confirm that the insertions were close to the O. ochengi TATA box–binding protein gene (unpublished data). By surveying the emerging genome sequence data for the filarial parasite B. malayi, we were able to identify a TATA box–binding protein gene, the orthologue of the O. volvulus gene, but did not find any significant sequence similarity to the wOvo gene fragments in the region upstream of this gene, and, indeed, did not identify any possible nuclear insertions of sequence similar to the five wOvo genome segments isolated in the B. malayi whole genome shotgun.
Discussion
The Genome of wOvo
The sequenced segments yielded 70 kb of genome sequence for wOvo. Additional rounds of screening failed to yield further wOvo fragments, and construction of Wolbachia-enriched genomic libraries was unsuccessful. It would be very informative to complete the wOvo genome and we are continuing to investigate routes to this end.
Relationships of Wolbachia Revealed by Sequence Phylogenetics and Synteny
We analysed the sequence of the genes encoded in the five wOvo fragments for phylogenetic signal, as for these we could identify credible orthologues in outgroup taxa. For individual genes, the signal was mixed, but biased towards Tree 2 of Figure 1. However, under ML models, none of the individual genes gave strong support to either of Trees 1 or 2. We identified no particular functional annotation to separate those genes supporting Tree 1 from those supporting Tree 2 (Table 3). As combining genes can yield resolution of phylogenetic problems, by summing the minor signal present in each gene such that it was detectable above the background noise of homoplasy [24], we generated a concatenated alignment of 42 of the wOvo proteins and their orthologues. Analysis of this concatenated alignment using unpartitioned or partitioned (more realistic, given the variation in inferred rates between genes; Figure 4) models yielded robust support for Tree 1, i.e., [outgroups[[wOvo, wBm],[wMel,wAna,wSim]]], equivalent to [outgroups[[C,D],[A]]] (Figure 5). Notably, the shortest inferred internal branch in the phylogeny was that linking the last common ancestor of all Wolbachia and the last common ancestor of the nematode (clade C and D) Wolbachia. The length of this branch compared to neighbouring ones in the phylogeny may explain the difficulty in robustly recovering a distinct phylogeny with more limited datasets. As genes from clade B Wolbachia are consistently very closely related to those from clade A rather than from clades C or D [1,35], we predict that inclusion of clade B in the analysis would yield a tree [outgroups[[C,D],[A,B]]].
Conserved gene arrangements (synteny) can be used to infer phylogenetic relationships between genomes. The wOvo fragments share some local synteny with both wMel and wBm. Where breakage of local synteny occurs, two features are apparent. Firstly, wBm and wMel are more similar to each other than either is to wOvo. Secondly, wBm is closer to wOvo than is wMel, as wMel has several unique rearrangements. Comparison to the outgroup genomes was uninformative because of the high levels of rearrangement that have taken place in Wolbachia genomes since they last shared a common ancestor with Anaplasmataceae [26,27]. Mapping of these changes in synteny onto the phylogeny derived from the sequence data suggests that the wOvo genome has undergone many more rearrangements since the last common ancestor of the three Wolbachia we have analysed than have either wBm or wMel.
We fully recognise that we have not been able to analyse with the larger dataset the more enigmatic and rarely described clades of Wolbachia, clades E, F, and G [8,9]. Current data suggest that clades E, F, and G arise basal to [A,B], but have not clearly resolved the pattern of branching compared to C and D [8,23]. We note that the standard three genes used for within-Wolbachia phylogenetics, wsp, ftsZ, and 16S ribosomal RNA, may not be the best set for analysing deeper relationships in the genus. Thus, wsp is essentially restricted to Wolbachia, while ftsZ has a high rate of evolutionary change, and is possibly subject to long branch artefacts. The ribosomal RNA genes yield Tree 1, though with relatively low NJ bootstrap support (66% for 23S and 5S, and 72% for 16S; Table 3). The addition of groEL and gltA genes to the analysis was unable to place the root with certainty [23].
Our sample of genes with a wide range of evolutionary rates has yielded strong support for one of the competing models. It will be very informative to utilise an expanded set of genes such as those sampled here to address the question of the relationships of the E, F, and G clades to the better known A, B, C, and D organisms.
The Evolution of Symbiotic Phenotypes in Wolbachia
As a whole, the Rickettsiales have lifestyles that involve intracellular replication in a eukaryotic host cell, and the outgroups analysed here have parasitic or pathogenic lifestyles. The support for Tree 1 suggests that the ancestor of all extant Wolbachia was probably an intracellular pathogen or parasite. Our analyses suggest that this intracellular pathogen was then tamed by, or evolved beneficial symbiotic relationships with, its nematode hosts, but evolved towards specific reproductive parasitism in the arthropod-infecting clade A (and B) strains. A single transfer of an ancestral Wolbachia to an onchocercid nematode host is most likely. The nematode Wolbachia have apparently coevolved with their hosts through strictly vertical descent, while the arthropod strains have undergone frequent (on an evolutionary timescale) horizontal transfers or host captures, while also maintaining themselves on a life-cycle timescale by vertical transmission. As arthropod Wolbachia are parasites, it is possible for individuals and populations to lose their infections. Importantly, it is also evident that nematodes can lose their Wolbachia, as Wolbachia-negative nematode species are nested within clades of infected taxa [16]. There is a correlation between the presence of WO phage in Wolbachia genomes [36] and the parasitic phenotype, and thus WO phage and/or genes transduced by WO phage may underpin parasitic manipulations [37]. There were no WO phage–like elements in the wOvo genome segments analysed.
Lateral Transfer of Wolbachia Genetic Material to the O. volvulus Nuclear Genome
Serendipitously, we identified two short fragments of Wolbachia genes in one of the few segments of the O. volvulus genome to have been sequenced. Transfer of Wolbachia genetic material into the host nuclear DNA has been noted previously, in the adzuki bean beetle, Callosobruchus chinensis, where a reasonably large segment of Wolbachia DNA has been inserted into the X chromosome [38]. The adzuki bean beetle insertion is not thought to be expressed.
The sequenced O. volvulus segment incorporates the gene for a TATA box–binding protein and a region 2 kb upstream. In this upstream region we detected two short segments that have significant pairwise identity to wOvo OW2-J and to wOvo OW4-C. We confirmed that the putative insertion was present in O. volvulus genomic DNA (and was not therefore a cloning artefact) by isolating it by specific PCR from an independent source of O. volvulus. Neither fragment is a complete gene, and both have been subject to mutational accumulation such that the open reading frames are no longer intact. The two genes do not lie beside each other in either the wOvo or wBm genomes. We suggest that an original insertion, perhaps of a relatively large portion of a Wolbachia genome, has been reduced by deletion, resulting in the close apposition of two fragmentary Wolbachia genes not found next door to each other in the bacterial chromosome. The insertional fragment is not unique to O. volvulus, as it is also present in the cattle onchocercid, O. ochengi. O. ochengi is very closely related to O. volvulus, and indeed O. volvulus in humans is thought to represent a recent host capture by, and vicariant speciation of, onchocercids of ungulates. No homologous insertion was detected in the partial genome sequence of B. malayi, but the orthologous TATA box–binding protein gene was identified. Examination of the region between the B. malayi TATA box–binding protein gene and the next gene upstream identified no sequences with significant similarity to the putative Wolbachia insertions (unpublished data). We also used PCR to screen for the insertion in the deer onchocercid O. flexuosa. O. flexuosa is interesting because it appears to lack Wolbachia entirely (as determined by PCR screens and electron microscopy) [39]. Identification of an insertional relic of Wolbachia would bolster suggestions that this species has lost its symbiont. However, we were unable to amplify any insertion fragments from O. flexuosa (unpublished data), leaving the question of symbiont loss unanswered. Nuclear integration of fragments of other cytoplasmic genomes, such as the mitochondrial and chloroplast genomes, is relatively common, but no plausible integrants of wBm were detected in the near-complete B. malayi genome [26]. Whether acquisition of Wolbachia genes by the host plays any part in host evolution remains conjectural. Similarly, the Wolbachia could capture host genes, but none of the sequenced genomes contain genes with signatures of animal, rather than alphaproteobacterial, origin.
Materials and Methods
Selection of wOvo probes and identification of wOvo genomic clones.
A series of probes were prepared from previously identified wOvo genes, including the 16S ribosomal RNA gene, wsp, ftsZ, hsp60, and others identified in the O. volvulus EST (expressed sequence tag) programme [40,41] (Table 1). Probes were labelled with alpha32P dCTP by oligo-primed synthesis. O. volvulus libraries in lambda phage, gifts of John Donelson [33] and Steve Williams, were plated on bacterial lawns, and the lifts were prepared for Southern hybridisation using standard methods. Initial hybridisations used a mix of probes from several genes. After autoradiography, positive plaques were identified by gene-specific PCR, and purified by dilution and reprobing. Inserts were isolated by long-range PCR using lambda-vector primers, and end sequenced. End-probes were generated and used to reprobe plaque lifts. Primer sequences are given in Table 1.
Sequencing and annotation.
Long-range PCR products were sequenced by standard shotgun methods at the Wellcome Trust Sanger Institute, and assembled using standard methods. The insert sequences were completed by a combination of directed sequencing of selected plasmid subclones, and primer walking. One clone insert proved to be a chimaera of human and Wolbachia DNA; the human segment was identified by its sequence identity to human genomic sequence, and was removed from the analysis. Genes were identified and annotated in the wOvo genome segments using Artemis [42]. The Artemis comparative tool, ACT, was used to display and investigate synteny relationships with the wBm [26] and wMel [27] genomes. A putative wOvo HtrA serine protease (GenBank accession AAP79877) similar to OW4-E had been published previously [31]. To test if wOvo has more than one HtrA gene or if the difference was due to technical error, primers (see Table 1) were designed within the OW4-E 5′ and 3′ extragenic regions. Multiple PCR and sequencing reactions were performed according to standard procedures using O. volvulus genomic DNA. The sequences were aligned and a consensus sequence was obtained. To assess the possible function of the wOvo-specific gene, OW2-I, SignalP v3.0 [43] and pSortb v2.0 [44] were used to identify a possible signal peptide and a probable cellular location.
Phylogenetic analysis.
For phylogenetic analysis, particularly since we desired to identify the root of the Wolbachia clade, it was essential to analyse alignments of orthologous sequences, and to exclude paralogues. Each protein-coding gene in wOvo was used to search (using BLAST [45]) a custom database of alphaproteobacterial proteins extracted from EMBL and GenBank to identify homologues. In addition, homologues were identified from the complete and partial genomes of wBm, wMel, wAna, wSim [46,47], A. marginale [48], E. ruminantium [49], and E. canis. For each wOvo protein, a multiple alignment was constructed using ClustalW [50] and subjected to NJ analysis in PHYLIP (using character difference) [51]. From the resulting phylograms we identified orthologous genes from the seven complete and partial genomes. Importantly, we excluded paralogues from genomes where an orthologue was absent. These paralogues were the best scoring match in the selected genome, but by phylogenetic analysis were clearly not orthologous to the wOvo query. The wAna and wSim genomes were assembled from whole genome shotgun reads “contaminating” those generated for the nuclear genome projects of their host species, and are incomplete. For wAna, we identified several genes that are present in one copy in other bacterial genomes but are duplicated (or partially duplicated) in the wAna assembly. We interpret these to be due to either misassemblies or the presence of two closely related Wolbachia genomes in D. ananassae. If one whole genome sequence shotgun survey includes DNA from two distinct Wolbachia, the genes we selected for subsequent analysis may be selected stochastically from two distinct genomes, but the close relationship implied by comparison of the “duplicated” segments in the assembly (>99% identity) means that they can effectively be considered a single taxon. Ehrlichia and Anaplasma orthologues of two genes were not found, and in these cases we identified orthologues in R. prowazekii and R. typhi to use as outgroups.
For the 44 proteins with matches, and the 16S and 23S/5S ribosomal RNA genes, we realigned each wOvo sequence with its orthologues. The alignments are available as Protocol S1 online. The protein alignments were combined and subjected to phylogenetic analysis using NJ and Bayesian ML methods. NJ was carried out in PAUP 3.6 [52] with mean character distances. Bootstrap support was estimated for NJ trees by 1,000 resamplings. Bayesian analyses of protein-coding genes were carried out in MrBayes 3.1 [53] under the fixed rate JTT model of protein evolution with gamma rate variation approximated by four rate categories and a proportion of invariant sites. For RNA genes, DNA alignments were analysed under the HKY model with gamma rate variation (four categories) and a proportion of invariant sites. For each gene, two independent runs were executed for 1,000,000 generations, and sampled every 1,000 generations, with default prior and Markov chain parameters. After visual confirmation of stationarity, the first 10% of saved trees were discarded as burn in. The significance of the difference in support for the two credible alternative hypotheses was tested for each gene using a likelihood ratio test. p-Values were calculated using the SH test as implemented in Tree-Puzzle 5.1 (http://www.tree-puzzle.de) using accurate (slow) parameter estimation. Since, for many genes, one of the trees was the one selected by ML, this test is more appropriate than the Kishino-Hasegawa likelihood ratio test, which requires that trees be specified a priori. For protein-coding genes, amino acid alignments were analysed under the JTT model with gamma rate variation (four categories) and a proportion of invariant sites.
Rokas et al. [24] have shown that the use of large datasets, employing many genes with varying rates, is effective in recovering “correct” topologies when single-gene analyses fail to do so. Bayesian analyses of the concatenated alignment of 42 protein-coding genes was carried out under two models. In the first model, all genes shared a fixed rate JTT model of protein evolution with gamma rate variation approximated by four rate categories and a proportion of invariant sites. In the second model, the Poisson model was used, along with a rate multiplier that allowed each gene to evolve at a different rate. In addition, independent gamma rate parameters and proportions of invariant sites were estimated for each gene. For the concatenated analyses, two independent runs were executed for 2,000,000 generations and sampled every 100 generations, with default prior and Markov chain parameters. After visual confirmation of stationarity, the first 10% of saved trees were discarded as burn in. To test whether the second, more complex model gave a significantly better fit to the data, harmonic mean likelihoods from runs using different models were used to calculate Bayes factors.
PCR testing of lateral gene transfer.
A potential lateral gene transfer event was detected through BLAST search of O. volvulus sequences in EMBL and GenBank using wOvo fragments and the wBm genome as queries. The Wolbachia genes and their putative nuclear homologues were aligned using ClustalW (Figure 6). To prove the existence of the insertion in the O. volvulus genome, a series of oligonucleotide primers was designed (Table 1) that would be useful in PCR amplification of the insertion event in the nuclear genome and the genes resident in the wOvo chromosome. O. volvulus and O. ochengi DNA was isolated from nematodes from nodules using standard procedures. PCRs were carried out using ~100 ng of O. volvulus or O. ochengi DNA, and analysed on 1% agarose gels. Positive PCR fragments were isolated and sequenced to confirm their identity.
Supporting Information
The data are in NEXUS format.
(414 KB TXT)
Accession Numbers
Sequence data reported in this paper have been deposited in EMBL (http://www.ebi.ac.uk/embl), GenBank (http://www.ncbi.nlm.nih.gov/Genbank/index.html), and DDBJ (http://www.ddbj.nig.ac.jp) with the accession numbers OW1 (CU062443), OW2 (CU062464), OW3 (CU062463), OW4 (CU062460), and OW5 (CU062461). The GenBank accession numbers for the O. volvulus (from Mali) TATA box–binding protein gene and a putative wOvo HtrA serine protease are L13731 and AAP79877, respectively. The GenBank accession number for the E. canis genome is CP000107.
Acknowledgments
We thank Achim Hoerauf for O. volvulus material, Sandy Trees for O. ochengi material, and Norbert Brattig for O. flexuosa material. O. volvulus genomic libraries were provided by John Donelson and Steve Williams. Graham Stone provided valuable comments on the manuscript. We acknowledge the assistance of the Wellcome Trust Sanger Institute core sequencing and informatics groups.
Abbreviations
- GC%
proportion of the sequenced segments made up of guanine and cytosine bases
- idel
insertion/deletion
- ML
maximum likelihood
- NJ
neighbour joining
- SH
Shimodaira-Hasegawa
- wAna
Wolbachia from Drosophila ananassae
- wBm
Wolbachia from Brugia malayi
- wMel
Wolbachia from Drosophila melanogaster
- wOoc
Wolbachia from Onchocerca ochengi
- wOvo
Wolbachia from Onchocerca volvulus
- wSim
Wolbachia from Drosophila simulans
Footnotes
Competing interests. The authors have declared that no competing interests exist.
Author contributions. KF, CC, MJ, MAQ, JP, and MB conceived and designed the experiments. KF, CC, MJ, MAQ, NEH, and JP performed the experiments. KF, CC, MJ, JP, and MB analyzed the data and wrote the paper.
Funding. This project was funded by the Wellcome Trust. Edinburgh computing facilities were funded by the Natural Environment Research Council.
References
- Werren JH, Zhang W, Guo LR. Evolution and phylogeny of Wolbachia: Reproductive parasites of arthropods. Proc Biol Sci. 1995;261:55–63. doi: 10.1098/rspb.1995.0117. [DOI] [PubMed] [Google Scholar]
- Vannini C, Petroni G, Verni F, Rosati G. A bacterium belonging to the Rickettsiaceae family inhabits the cytoplasm of the marine ciliate Diophrys appendiculata (Ciliophora, Hypotrichia) Microb Ecol. 2005;49:434–442. doi: 10.1007/s00248-004-0055-1. [DOI] [PubMed] [Google Scholar]
- Werren JH. Biology of Wolbachia. Annu Rev Entomol. 1997;42:587–609. doi: 10.1146/annurev.ento.42.1.587. [DOI] [PubMed] [Google Scholar]
- Werren JH, Guo L, Windsor DW. Distribution of Wolbachia among neotropical arthropods. Proc Biol Sci. 1995;262:197–204. [Google Scholar]
- Jiggins FM, Bentley JK, Majerus ME, Hurst GD. How many species are infected with Wolbachia? Cryptic sex ratio distorters revealed to be common by intensive sampling. Proc Biol Sci. 2001;268:1123–1126. doi: 10.1098/rspb.2001.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst GD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc Biol Sci. 2005;272:1525–1534. doi: 10.1098/rspb.2005.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh T, Noda H, Fujita T, Iwadate K, Higo Y, et al. Wolbachia and nuclear-nuclear interactions contribute to reproductive incompatibility in the spider mite Panonychus mori (Acari: Tetranychidae) Heredity. 2005;94:237–246. doi: 10.1038/sj.hdy.6800605. [DOI] [PubMed] [Google Scholar]
- Lo N, Casiraghi M, Salati E, Bazzocchi C, Bandi C. How many Wolbachia supergroups exist? Mol Biol Evol. 2002;19:341–346. doi: 10.1093/oxfordjournals.molbev.a004087. [DOI] [PubMed] [Google Scholar]
- Rowley SM, Raven RJ, McGraw EA. Wolbachia pipientis in Australian spiders. Curr Microbiol. 2004;49:208–214. doi: 10.1007/s00284-004-4346-z. [DOI] [PubMed] [Google Scholar]
- McLaren DJ, Worms MJ, Laurence BR, Simpson MG. Microorganisms in filarial larvae (Nematoda) Trans R Soc Trop Med Hyg. 1975;69:509–514. doi: 10.1016/0035-9203(75)90110-8. [DOI] [PubMed] [Google Scholar]
- Franz M, Buttner DW. The fine structure of Onchocerca volvulus: IV. The hypodermal chords of the female worm. Tropenmed Parasitol. 1983;34:122–128. [PubMed] [Google Scholar]
- Sironi M, Bandi C, Sacchi L, Di Sacco B, Damiani G, et al. Molecular evidence for a close relative of the arthropod endosymbiont Wolbachia in a filarial worm. Mol Biochem Parasitol. 1995;74:223–227. doi: 10.1016/0166-6851(95)02494-8. [DOI] [PubMed] [Google Scholar]
- Bordenstein SR, Fitch DHA, Werren JH. Absence of Wolbachia in nonfilariid nematodes. J Nematol. 2003;35:266–270. [PMC free article] [PubMed] [Google Scholar]
- Bandi C, Anderson TJ, Genchi C, Blaxter ML. Phylogeny of Wolbachia bacteria in filarial nematodes. Proc Biol Sci. 1998;265:2407–2413. doi: 10.1098/rspb.1998.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M, Anderson TJ, Bandi C, Bazzocchi C, Genchi C. A phylogenetic analysis of filarial nematodes: Comparison with the phylogeny of Wolbachia endosymbionts. Parasitology. 2001;122((Pt 1)):93–103. doi: 10.1017/s0031182000007149. [DOI] [PubMed] [Google Scholar]
- Fenn K, Blaxter M. Are filarial nematode Wolbachia obligate mutualist symbionts? Trends Ecol Evol. 2004;19:163–166. doi: 10.1016/j.tree.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Bandi C, McCall JW, Genchi C, Corona S, Venco L, et al. Effects of tetracycline on the filarial worms Brugia pahangi and Dirofilaria immitis and their bacterial endosymbionts, Wolbachia. Int J Parasitol. 1999;29:357–364. doi: 10.1016/s0020-7519(98)00200-8. [DOI] [PubMed] [Google Scholar]
- Hoerauf A, Nissen-Pahle K, Schmetz C, Henkle-Duhrsen K, Blaxter ML, et al. Tetracycline therapy targets intracellular bacteria in the filarial nematode Litomosoides sigmodontis and results in filarial infertility. J Clin Invest. 1999;103:11–18. doi: 10.1172/JCI4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoerauf A, Volkmann L, Hamelmann C, Adjei O, Autenrieth IB, et al. Endosymbiotic bacteria in worms as targets for a novel chemotherapy in filariasis. Lancet. 2000;355:1242–1243. doi: 10.1016/S0140-6736(00)02095-X. [DOI] [PubMed] [Google Scholar]
- Hoerauf A, Mand S, Adjei O, Fleischer B, Buttner DW. Depletion of Wolbachia endobacteria in Onchocerca volvulus by doxycycline and microfilaridermia after ivermectin treatment. Lancet. 2001;357:1415–1416. doi: 10.1016/S0140-6736(00)04581-5. [DOI] [PubMed] [Google Scholar]
- Taylor MJ, Makunde WH, McGarry HF, Turner JD, Mand S, et al. Macrofilaricidal activity after doxycycline treatment of Wuchereria bancrofti: A double-blind, randomised placebo-controlled trial. Lancet. 2005;365:2116–2121. doi: 10.1016/S0140-6736(05)66591-9. [DOI] [PubMed] [Google Scholar]
- Turner JD, Mand S, Debrah AY, Muehlfeld J, Pfarr K, et al. A randomized, double-blind clinical trial of a 3-week course of doxycycline plus albendazole and ivermectin for the treatment of Wuchereria bancrofti infection. Clin Infect Dis. 2006;42:1081–1089. doi: 10.1086/501351. [DOI] [PubMed] [Google Scholar]
- Casiraghi M, Bordenstein SR, Baldo L, Lo N, Beninati T, et al. Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: Clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology. 2005;151((Pt 12)):4015–4022. doi: 10.1099/mic.0.28313-0. [DOI] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804. doi: 10.1038/nature02053. [DOI] [PubMed] [Google Scholar]
- Sun LV, Foster JM, Tzertzinis G, Ono M, Bandi C, et al. Determination of Wolbachia genome size by pulsed-field gel electrophoresis. J Bacteriol. 2001;183:2219–2225. doi: 10.1128/JB.183.7.2219-2225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J, Ganatra M, Kamal I, Ware J, Makarova K, et al. The Wolbachia genome of Brugia malayi: Endosymbiont evolution within a human pathogenic nematode. PLoS Biol. 2005. e121. DOI: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed]
- Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: A streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004. e69. DOI: 10.1371/journal.pbio.0020069. [DOI] [PMC free article] [PubMed]
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Chandu D, Nandi D. Comparative genomics and functional roles of the ATP-dependent proteases Lon and Clp during cytosolic protein degradation. Res Microbiol. 2004;155:710–719. doi: 10.1016/j.resmic.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Sharma S, Hoskins JR, Wickner S. Binding and degradation of heterodimeric substrates by ClpAP and ClpXP. J Biol Chem. 2005;280:5449–5455. doi: 10.1074/jbc.M412411200. [DOI] [PubMed] [Google Scholar]
- Jolodar A, Fischer P, Buttner DW, Brattig NW. Wolbachia endosymbionts of Onchocerca volvulus express a putative periplasmic HtrA-type serine protease. Microbes Infect. 2004;6:141–149. doi: 10.1016/j.micinf.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Li S, Donelson JE. The gene for the TATA box-binding protein of Onchocerca volvulus. Mol Biochem Parasitol. 1993;61:321–324. doi: 10.1016/0166-6851(93)90078-c. [DOI] [PubMed] [Google Scholar]
- Xie H, Bain O, Williams SA. Molecular phylogenetic studies on filarial parasites based on 5S ribosomal spacer sequences. Parasite. 1994;1:141–151. doi: 10.1051/parasite/1994012141. [DOI] [PubMed] [Google Scholar]
- Zhou W, Rousset F, O'Neil S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc Biol Sci. 1998;265:509–515. doi: 10.1098/rspb.1998.0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui S, Kuroiwa H, Sasaki T, Inui M, Kuroiwa T, et al. Bacteriophage WO and virus-like particles in Wolbachia, an endosymbiont of arthropods. Biochem Biophys Res Commun. 2001;283:1099–1104. doi: 10.1006/bbrc.2001.4906. [DOI] [PubMed] [Google Scholar]
- Sinkins SP, Walker T, Lynd AR, Steven AR, Makepeace BL, et al. Wolbachia variability and host effects on crossing type in Culex mosquitoes. Nature. 2005;436:257–260. doi: 10.1038/nature03629. [DOI] [PubMed] [Google Scholar]
- Kondo N, Nikoh N, Ijichi N, Shimada M, Fukatsu T. Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect. Proc Natl Acad Sci U S A. 2002;99:14280–14285. doi: 10.1073/pnas.222228199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brattig NW, Buttner DW, Hoerauf A. Neutrophil accumulation around Onchocerca worms and chemotaxis of neutrophils are dependent on Wolbachia endobacteria. Microbes Infect. 2001;3:439–446. doi: 10.1016/s1286-4579(01)01399-5. [DOI] [PubMed] [Google Scholar]
- Lizotte-Waniewski M, Tawe W, Guiliano DB, Lu W, Liu J, et al. Identification of potential vaccine and drug target candidates by expressed sequence tag analysis and immunoscreening of Onchocerca volvulus larval cDNA libraries. Infect Immun. 2000;68:3491–3501. doi: 10.1128/iai.68.6.3491-3501.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnasch TR, Williams SA. The genomes of Onchocerca volvulus. Int J Parasitol. 2000;30:543–552. doi: 10.1016/s0020-7519(99)00184-8. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, et al. Artemis: Sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P. PSORT: A program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Dunning Hotopp JC, Delcher AL, Pop M, Smith DR, et al. Correction: Serendipitous discovery of Wolbachia genomes in multiple Drosophila species. Genome Biol. 2005;6:402. doi: 10.1186/gb-2005-6-7-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Hotopp JC, Delcher AL, Pop M, Smith DR, et al. Serendipitous discovery of Wolbachia genomes in multiple Drosophila species. Genome Biol. 2005;6:R23. doi: 10.1186/gb-2005-6-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayton KA, Kappmeyer LS, Herndon DR, Dark MJ, Tibbals DL, et al. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc Natl Acad Sci U S A. 2005;102:844–849. doi: 10.1073/pnas.0406656102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins NE, Liebenberg J, de Villiers EP, Brayton KA, Louw E, et al. The genome of the heartwater agent Ehrlichia ruminantium contains multiple tandem repeats of actively variable copy number. Proc Natl Acad Sci U S A. 2005;102:838–843. doi: 10.1073/pnas.0406633102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP: Phylogeny Inference Package, version 3.6 [computer program] Seattle: Department of Genome Sciences and Department of Biology, University of Washington; 2004. [Google Scholar]
- Swofford D. PAUP* 4b10: Phylogenetic Analysis using Parsimony * and other methods [computer program] Sunderland (Massachusetts): Sinauer Associates; 2001. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The data are in NEXUS format.
(414 KB TXT)