

Abstract
Changes in synaptic strength that underlie memory formation in the CNS are initiated by pulses of Ca2+ flowing through NMDA-type glutamate receptors into postsynaptic spines. Differences in the duration and size of the pulses determine whether a synapse is potentiated or depressed after repetitive synaptic activity. Calmodulin (CaM) is a major Ca2+ effector protein that binds up to four Ca2+ ions. CaM with bound Ca2+ can activate at least six signaling enzymes in the spine. In fluctuating cytosolic Ca2+, a large fraction of free CaM is bound to fewer than four Ca2+ ions. Binding to targets increases the affinity of CaM's remaining Ca2+-binding sites. Thus, initial binding of CaM to a target may depend on the target's affinity for CaM with only one or two bound Ca2+ ions. To study CaM-dependent signaling in the spine, we designed mutant CaMs that bind Ca2+ only at the two N-terminal or two C-terminal sites by using computationally designed mutations to stabilize the inactivated Ca2+-binding domains in the “closed” Ca2+-free conformation. We have measured their interactions with CaMKII, a major Ca2+/CaM target that mediates initiation of long-term potentiation. We show that CaM with two Ca2+ ions bound in its C-terminal lobe not only binds to CaMKII with low micromolar affinity but also partially activates kinase activity. Our results support the idea that competition for binding of CaM with two bound Ca2+ ions may influence significantly the outcome of local Ca2+ signaling in spines and, perhaps, in other signaling pathways.
Keywords: postsynaptic, protein design, synaptic plasticity, microdomains
Calmodulin (CaM) is a Ca2+ effector protein comprised of two Ca2+-binding lobes connected by a short linker region. Each lobe contains two EF hands that bind Ca2+ with low micromolar affinity. CaM mediates a host of regulatory effects of Ca2+ despite the fact that the Ca2+ concentration in the cytosol rarely reaches a level expected to saturate all four Ca2+-binding sites on free CaM (1). This sensitivity is possible because association of CaM with its targets stabilizes its Ca2+-bound conformation and, therefore, increases its affinity for Ca2+ (2–4). When CaM with two Ca2+ ions bound to either the N- or the C-terminal lobe associates with a target, the affinity of the remaining lobe for Ca2+ increases, often by at least one order of magnitude, causing high positive cooperativity of Ca2+ binding (5). Because of this property, we predict that when Ca2+ concentrations fluctuate in the cytosol, competition among targets for binding of CaM will depend on their affinities for CaM with one or two bound Ca2+ and also on the magnitude of cooperativity induced by binding to each target.
In the CNS, Ca2+ influx into postsynaptic spines of excitatory glutamatergic synapses is a key determinant of changes in synaptic strength that underlie memory formation. The direction of the change in strength is determined by the timing between release of glutamate at the synapse and depolarization of the postsynaptic membrane, caused, for example, by a back-propagating action potential. This timing controls the duration and extent of Ca2+ influx through NMDA-type glutamate receptors into the spine. Relatively large increases of a few micromolars in Ca2+ concentration occurring over 2 to 3 sec induce long-term potentiation of the synapse. In contrast, smaller changes of a few hundred nanomolars, lasting over several seconds to 1 min, induce long-term depression of the synapse (6–10).
How might these slight differences in Ca2+ influx produce such dramatic differences in regulation of the synapse? Ca2+ that flows into a spine through activated NMDA receptors binds to CaM and to several other Ca2+-binding proteins. Ca2+ binding to CaM enables its interaction with, and regulation of, several postsynaptic proteins. Two prominent targets of Ca2+/CaM are Ca2+/calmodulin-dependent protein kinase II (CaMKII) (11, 12) and calcineurin, a protein phosphatase (13, 14). The apparent affinities of these two proteins for Ca2+/CaM are quite different when measured at saturating Ca2+ concentrations (>200 μM). CaMKII is half maximally activated at ≈40–80 nM CaM (15–17), whereas calcineurin is half maximally activated at 1–10 nM CaM (18, 19). This severalfold difference in affinity for Ca2+/CaM has been invoked to explain the different synaptic effects of high and low Ca2+ fluxes (20). However, an accurate description of the critical initial steps of Ca2+ signaling in spines requires a precise understanding of how different CaM targets in the spine compete for Ca2+ under conditions in which quantities of Ca2+/CaM are limited and concentrations of Ca2+ are fluctuating.
In the work reported here, we focus on understanding interactions between CaM and CaMKII when the concentrations of Ca2+ or CaM are limiting. CaMKII is a dodecameric oligomer of catalytic subunits, each of which can bind one CaM (21–23). Binding of CaM to an individual subunit activates its kinase activity allowing the subunit to phosphorylate other proteins. In addition, binding of two CaM molecules to two adjacent subunits within a holoenzyme allows autophosphorylation of one or both of the subunits at Thr-286 (24). This autophosphorylation renders the subunit constitutively active and it remains so until the phosphate is removed by a protein phosphatase (25, 26). Activation and autophosphorylation of CaMKII has been shown to be crucial for induction of long-term potentiation (27).
To measure the affinity of CaMKII for CaM with less than four bound Ca2+ ions, we designed a mutant CaM that binds Ca2+ only at N-terminal sites (CaM-NWT) and a mutant CaM that binds Ca2+ only at C-terminal sites (CaM-CWT). To closely mimic conformations of CaM that would exist in the cytosol, we used computationally designed mutations to stabilize the inactivated Ca2+-binding domains in the “closed,” Ca2+-free conformation. We found that, when saturated with Ca2+, the mutant CaMs bind to CaMKII with low micromolar affinity and partially activate its kinase activity. Thus, the activity of CaMKII is more sensitive to small increases in cytosolic Ca2+ than previously believed. The mutant CaMs described here can be used to study the ability of CaM with two Ca2+ ions bound to the C- or N-terminal sites to associate with and activate other CaM targets. Such studies will enable more accurate understanding of the regulatory outcomes of local Ca2+ increases in many signaling pathways and cell types.
Results
Activation of CaMKII by Ca2+ and CaM Is Highly Cooperative.
Conditions usually used to study activation of CaM targets in vitro are necessarily different from those existing in cells. Most published assays of activation of proteins by CaM employ CaM concentrations in the range of 1 μM and concentrations of targets in the range of 20–50 nM. The actual concentrations of CaM and CaMKII in spines are estimated to be in the range of 10–30 μM (21, 28, 29), whereas fluctuations in the concentration of Ca2+ range over 0.1–30 μM near open NMDA-type receptor pores (30). To better approximate conditions under which competition for Ca2+/CaM might occur in a spine, we measured activation and autophosphorylation of CaMKII in reactions containing 0.28 μM CaMKII (molarity of catalytic subunits) and 12 μM CaM, concentrations that are 5- to 10-fold higher than those commonly used in enzymatic assays and yet are still practical. We observed a significant rate of autophosphorylation of CaMKII at as low as 350 nM Ca2+ (Fig. 1A), a concentration below the KD of even the highest-affinity Ca2+-binding site measured for free CaM (≈1 μM) (1, 31) and two orders of magnitude below the KD of the lowest affinity site (≈20–30 μM). These results suggest one or both of the following mechanisms. First, the cooperativity of binding among Ca2+, CaM, and CaMKII might be extremely high; that is, the presence of CaMKII might increase the apparent affinity of CaM for Ca2+ by more than one order of magnitude. Second, CaM might not require occupation of all its high-affinity Ca2+-binding sites to initiate autophosphorylation of CaMKII.
Fig. 1.

Cooperative binding of Ca2+ to CaM in the presence of CaMKII. (A) Autophosphorylation of CaMKII in the presence of WT CaM at varying Ca2+ concentrations. Reactions were performed for 1 min, as described in Methods, with 10 μM CaM, 0.28 μM CaMKII, and the indicated Ca2+ concentrations. Autophosphorylation was detected by immunoblotting with the phosphosite-specific antibody 22B1, as described in Methods. The position of the α-subunit of CaMKII is indicated. Note that the band shifts to slightly slower mobility as a second site, Thr-305, becomes autophosphorylated. (B) Increase in affinity of Ca2+ for CaM in the presence of CaMKII or CaMKII-cbp. Ca2+ binding to 5 μM CaM was measured in a competition assay with a fluorescent Ca2+-binding dye, Fluo4FF at 25°C. Ca2+ was titrated into solutions containing 5 μM Fluo4FF and WT CaM alone (▴), or in the presence of 5 μM CaMKII (■), or 5 μM CaMKII-cbp (♦). The data were fit with equations for Ca2+ binding at equilibrium to CaM alone or to CaM in the presence of CaMKII or CaMKII-cbp as described in Methods.
The magnitude of the increase in affinity of CaM for Ca2+ when CaM binds to CaMKII has not been measured previously under conditions approximating those in spines. We therefore compared the affinity of free CaM for Ca2+ to that of CaM in the presence of CaMKII and in the presence of a peptide having the sequence of the CaM-binding domain of CaMKII (CaMKII-cbp), at concentrations of 5 μM for each protein (Fig. 1B). We found that the concentrations of Ca2+ at which half of the Ca2+-binding sites on CaM are occupied are ≈15 μM for free CaM, ≈3–4 μM in the presence of CaMKII, and ≈1–2 μM in the presence of CaMKII-cbp. These data show that the presence of CaMKII increases the affinity of CaM for Ca2+ ≈5-fold under conditions approximating those in a postsynaptic spine. This effect undoubtedly contributes to the ability of Ca2+ concentrations <1 μM to activate CaMKII in our experiments; however, it is not high enough to account for it completely.
Computationally Designed Mutant CaMs That Bind Two Ca2+ ions at Either the C-Terminal or the N-Terminal Sites.
One traditional view held that occupation of all four Ca2+-binding sites on CaM is required to change the activity of most targets and, perhaps, also for high-affinity binding of CaM to targets. Discoveries of alternative modes of regulation by CaM, including some involving CaM with Ca2+ bound to only one of its two lobes, indicate that CaM is, in fact, more versatile in its regulatory mechanisms (32). CaM consists of N- and a C-terminal globular helical domains connected by a central flexible helix (Fig. 2A and B). Each globular domain contains two Ca2+-binding sites of the EF-hand variety. The C-terminal sites have ≈6-fold higher intrinsic affinity for Ca2+ than the N-terminal sites (31, 36); however, the on rate for Ca2+ binding to the N-terminal sites is ≈10-fold faster. In the absence of Ca2+, CaM adopts a closed conformation (33, 37, 38) in which it is not able to interact with most of its targets (Fig. 2A). Binding of Ca2+ to CaM causes a reorientation of helices inside the globular domains and a consequent exposure of patches of hydrophobic residues important for target recognition (34) (Fig. 2B). A shift toward this “open” CaM conformation is believed to be a prerequisite for interactions with many CaM-regulated proteins (1, 32, 39). Binding to most targets, including the CaM-binding domain of CaMKII (CaMKII-cbp), induces a conformation in which CaM wraps around the target domain, resulting in a coil-to-helix transition in the target (35, 40) (Fig. 2C).
Fig. 2.

Conformations of WT CaM and the designed mutant CaMs. (A) NMR structure of Ca2+-free CaM (PDB ID code 1CFD) (33). (B) X-ray crystal structure of Ca2+-bound CaM (PDB ID code 3CLN) (34). The hydrophobic patches important for target recognition are shown in red. Ca2+ atoms are shown as yellow spheres. (C) X-ray crystal structure of Ca2+/CaM bound to CaMKII-cbp, shown in pink (PDB ID code 1CM1) (35). (D and E) Models of the mutant CaMs designed to bind Ca2+ only in the C-terminal domain, CaM-CWT (D), and only in the N-terminal domain, CaM-NWT (E). These models were generated by combining the structure of the WT Ca2+/CaM domain with the structure of Ca2+-free CaM representing the mutated CaM domain. Mutant residues predicted by the computation to produce the best stabilized structure (Table 1) are shown in red. The figure was generated with PyMOL (DeLano Scientific, South San Francisco, CA).
To test whether binding of Ca2+ to both the C-terminal and the N-terminal lobes of CaM is necessary for activation of CaMKII under conditions that approximate those in the spine, we designed mutant CaMs that bind Ca2+ only in the C- or N-terminal lobes: CaM-CWT and CaM-NWT, respectively (Fig. 2 D and E and Table 1). Unlike previously reported mutant CaMs that are deficient in Ca2+ binding because of point mutations of Ca2+-ligating residues (36, 41, 42), our mutant CaMs were designed to mimic intermediate conformations that CaM might adopt in the cytosol by specifically stabilizing the closed conformation of the mutant Ca2+-binding loops. We used the protein design program ORBIT (43), which employs an empirical atom-based force field and a fast side-chain selection algorithm to search through different protein sequences to find the lowest energy sequence for a given protein structure. To abolish Ca2+ binding to a particular CaM domain, we redesigned the amino acid sequence of either the N-terminal or the C-terminal domain, leaving the other domain intact. For each calculation, a sequence of CaM residues forming the mutant Ca2+-binding site in the N- or C-terminal domain was optimized by using the Ca2+-free CaM structure as the template (ref. 33; Table 1). For each of these calculated sequences, we verified that the redesigned Ca2+ site had fewer residues with Ca2+ coordinating side chains.
Table 1.
Computationally designed N- and C-terminal mutant CaMs
| CaMs | Designed positions |
|||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site 1 |
Site 2 |
Site 3 |
Site 4 |
|||||||||||||||||
| 20 | 22 | 24 | 27 | 31 | 56 | 58 | 60 | 62 | 67 | 93 | 95 | 97 | 100 | 104 | 129 | 131 | 133 | 135 | 140 | |
| CaM WT | D | D | D | I | E | D | D | N | T | E | D | D | N | I | E | D | D | D | Q | E |
| CaM-CWT | T | K | Q | I | E | S | D | E | I | K | – | – | – | – | – | – | – | – | – | – |
| CaM-NWT | – | – | – | – | – | – | – | – | – | – | A | R | D | M | E | R | D | N | E | N |
All CaM residues that directly coordinate Ca2+ ions with their side chains (positions 20, 22, 24, 31, 56, 58, 60, and 67 for the N-terminal domain and 93, 95, 97, 104, 129, 131, 133, and 140 for the C-terminal domain) were included in the optimization. Residues 62 and 135 also were included because they coordinate Ca2+ ions with their backbone carbonyls. Residues 27 and 100, which do not coordinate Ca2+ ions, but are in close proximity to the ion and help define the Ca2+-binding loop, also were optimized. Bold lettering shows mutations with respect to the WT CaM sequence. –, positions were not included in the calculation for the mutant CaM indicated.
Assessment of Folding and Ca2+ Binding by Mutant CaMs.
The designed mutant CaMs were expressed in Escherichia coli and assessed by far-UV circular dichroism for proper folding and Ca2+ binding. In the absence of Ca2+, both mutant CaMs retain a predominantly α-helical structure essentially identical to that of WT CaM. Upon addition of Ca2+, both exhibit a small change in the circular dichroism signal, as does WT CaM, reflecting Ca2+-induced rearrangements of the EF-hand domains (Fig. 6, which is published as supporting information on the PNAS web site). Binding of Ca2+ to the C-terminal sites has been shown to be responsible for most of these changes in circular dichroism spectrum (44). Consistent with that finding, the structural transition of our N-terminal mutant (CaM-CWT) is nearly identical to that of WT CaM (Fig. 6 A and B).
We compared the Ca2+ binding stoichiometry of WT and mutant CaMs by electrospray mass spectrometry as described in ref. 45. In the presence of EGTA, the spectra of WT CaM and mutant CaMs contained a major peak corresponding to the molecular weight of the protein in the Ca2+-free state (data not shown). In the presence of saturating Ca2+, the WT CaM spectrum comprised a peak corresponding to Ca2+-free CaM and four additional peaks reflecting incremental additions of one Ca2+ ion. The major peak corresponds to four bound Ca2+ ions (Fig. 3A). For CaM-CWT and CaM-NWT, the major peaks correspond to one and two bound Ca2+ ions, as expected (Fig. 3 B and C).
Fig. 3.

High-affinity Ca2+-binding sites on WT and mutant CaMs. (A–C) Stoichiometry of Ca2+ binding to WT and mutant CaMs measured by electrospray mass spectrometry at 10 μM CaM and 200 μM Ca2+. (A) WT CaM. (B) CaM-CWT. (C) CaM-NWT. (D) Ca2+ binding to WT and mutant CaMs measured in a competition assay with the fluorescent Ca2+-binding dye, Fluo4FF. Ca2+ was titrated into a solution of Fluo4FF (10 μM) alone (●), Fluo4FF plus 10 μM CaM-CWT (▿),10 μM CaM-NWT (▴), or 10 μM WT CaM (■). The data were fit with a model assuming four high-affinity Ca2+-binding sites for WT CaM, two for CaM-CWT and CaM-NWT, and a single Ca2+-binding site for Fluo4FF (Table 2).
We measured the Ca2+-binding affinities of the mutant CaMs employing the fluorescent calcium sensor Fluo4FF. When CaM competes with Fluo4FF for available Ca2+, the fluorescence signal is reduced (Fig. 3D). The concentrations of Ca2+ and CaM over which the competition occurs reflect the affinity of CaM for Ca2+. The competition curves were fit with a four Ca2+ site model for WT CaM and a two Ca2+ site model for the mutant CaMs (46). Ca2+-binding affinities of WT and mutant CaMs range from 1 to 40 μM (Table 2) and are in approximate agreement with previously published values for WT CaM (1, 36). The data reveal positive cooperativity between the two neighboring Ca2+-binding sites in both mutant CaMs, as has been reported for WT CaM (1, 31). Thus, by several criteria, the two mutants behave as desired. In the presence of a saturating concentration of Ca2+, CaM-CWT binds two Ca2+ ions in the C-terminal lobe, whereas CaM-NWT binds two Ca2+ ions in the N-terminal lobe.
Table 2.
Kinetic properties of WT and mutant CaMs
| CaMs |
KD values for Ca2+, μM |
Kact for CaMKII, μM2 | kp of CaMKII, (sec−1)3 | |||
|---|---|---|---|---|---|---|
| KD1 | KD2 | KD3 | KD4 | |||
| WT CaM | 7.9 | 1.7 | 35 | 8.9 | 0.04 | 0.96 |
| CaM-CWT | 25 | 1.6 | – | – | 5 | 0.064 |
| CaM-NWT | 28 | 3.3 | – | – | 20 | 0.12 |
KD values for Ca2+ were calculated as described in Methods from the data in Fig. 3D with the CaLigator program (47) downloaded from www.bpc.lu.se/research/caligator. A model containing four Ca2+ sites was used for WT CaM, and one containing two Ca2+ sites was used for CaM-CWT and CaM-NWT. The concentration of WT CaM required for half-maximal rate of CaMKII autophosphorylation, Kact, is taken from Meyer et al. (16). Kact values for mutant CaMs were determined from the data in Fig. 4B. The intrinsic turnover rate of CaMKII autophosphorylation, kp, was determined from data in Fig. 5. Confidence intervals (95%) for kp values were 0.67–1.25 for WT CaM, 0.044–0.084 for CaM-CWT, and 0.079–0.16 for CaM-NWT.
We compared binding of WT and mutant CaMs to CaMKII-cbp at a saturating Ca2+ concentration by using circular dichroism to measure the coil-to-helix transition in the peptide upon binding, as previously described (ref. 48; Fig. 7, which is published as supporting information on the PNAS web site). Dissociation constants calculated by fitting the data to a 1:1 binding model show that WT CaM binds to CaMKII-cbp with a KD of 5 nM, similar to values reported for other CaM-binding peptide sequences (48). In contrast, CaM-CWT binds with a KD of 70 nM, and CaM-NWT binds with a much lower affinity KD of 6 μM.
Activation of CaMKII by Mutant CaMs.
We tested whether the mutant CaMs support activation of CaMKII by measuring their ability to activate autophosphorylation at Thr-286. Unexpectedly, we found that, at saturating Ca2+ concentration, either of the mutant CaMs (10 μM) supports substantial autophosphorylation of CaMKII in a 1-min reaction (Fig. 4A). This result means that CaM does not require Ca2+ bound to both of its lobes to activate CaMKII. The Kact values for CaM-CWT and CaM-NWT under these conditions were 5 and 20 μM, respectively (Fig. 4B), compared with a Kact value of ≈50 nM for WT CaM with four bound Ca2+ (Table 2). These results are consistent with the higher affinity of CaM-CWT for CaMKII-cbp and indicate that the binding of 2 Ca2+ ions to the C-terminal lobe of WT CaM results in more effective activation of CaMKII than binding of two Ca2+ ions to the N-terminal lobe.
Fig. 4.

Activation of autophosphorylation of CaMKII by WT and mutant CaMs. (A) Reactions were performed with 10 μM CaM and 0.28 μM CaMKII subunits for 1 min in the absence (−) or presence (+) of 300 μM Ca2+, as described in Methods. The positions of α and β subunits of CaMKII are indicated. (B) Dependence of autophosphorylation on the concentration of mutant CaMs. Autophosphorylation was performed as in A, except reactions were performed for 30 sec. To determine the Kact values reported in Table 2, the data were normalized to the level of autophosphorylation in saturating Ca2+/CaM for 1 min and fit with a model assuming binding of 1 CaM to 1 catalytic subunit. (C) Dependence of autophosphorylation on Ca2+ in the presence of WT and mutant CaMs. Reactions were performed as in A, except with 5 μM CaM, and quantified as in B. (D) Dependence of autophosphorylation on CaM concentration in the presence of WT and mutant CaMs. Reactions were performed as in A, except with 12 μM Ca2+, and quantified as in B. Mean ± SD of four experiments. WT (▾), CaM-CWT (●), and CaM-NWT (■).
To explore the ability of the mutant CaMs to activate CaMKII under conditions closer to those believed to exist in postsynaptic spines during synaptic activity, we measured autophosphorylation of CaMKII while varying Ca2+ concentration from 0 to 10 μM in the presence of 5 μM CaM. Autophosphorylation of CaMKII in the presence of CaM-CWT was first detectable at ≈1 μM Ca2+ and in the presence of CaM-NWT at ≈5 μM Ca2+ (Fig. 4C). The rate of autophosphorylation was half-maximal at 2 μM Ca2+ for WT CaM, at 6 μM Ca2+ for CaM-CWT, and at >32 μM Ca2+ for CaM-NWT. We then measured activation of CaMKII by each mutant CaM while varying their concentrations from 0.5 to 20 μM in 12 μM Ca2+. Under these conditions, WT CaM had a Kact of <0.4 μM, CaM-CWT ≈7 μM, and CaM-NWT >30 μM (Fig. 4D). These results show that, in the presence of Ca2+, both CaM-CWT, which mimics WT CaM with two Ca2+ ions bound at its C terminus (CaM2C), and CaM-NWT, which mimics WT CaM with two Ca2+ ions bound at the N terminus (CaM2N), can bind to and partially activate CaMKII in vitro under conditions, approximating those in stimulated postsynaptic spines. The results suggest that CaM2C is far more effective than CaM2N at binding and activating CaMKII in low concentrations of Ca2+.
We performed quench flow experiments to measure the turnover rates of autophosphorylation supported by WT and mutant CaMs at their saturating concentrations. The turnover rate supported by either mutant was ≈10% of that supported by WT CaM (Table 2 and Fig. 5), suggesting that binding of the mutant CaMs cannot produce the optimal conformational change required for a maximal rate of autophosphorylation. We conclude that both mutant CaMs support activation and autophosphorylation of CaMKII; however, their affinity for CaMKII is lower than that of WT CaM, and they support a lower enzymatic turnover number.
Fig. 5.

Determination of turnover number for autophosphorylation of CaMKII in the presence of WT and mutant CaMs. The time course of autophosphorylation of CaMKII was determined in saturating Ca2+/CaM. Data were gathered with a Quench Flow apparatus and normalized as described in Methods, then fit with the equation: A(1 − e−kp(t − t0)), where kp is the turnover rate (unknown), t0 (a negative term) is the time at which the extrapolated autophosphorylation curve crosses the x axis (Supporting Text), and A is a scaling factor. A, kp, and t0 were fit simultaneously by nonlinear regression performed with the Levenberg–Marquardt method in Mathematica. The kp values supported by each form of CaM are summarized in Table 2. Data were obtained from five experiments with WT CaM, five with CaM-CWT, and three with CaM-NWT. WT CaM (●), CaM-CWT (▿), and CaM-NWT (▴).
Discussion
We used a computational method to design mutant CaM proteins that bind two Ca2+ ions at either the N- or C terminus. At the inactivated terminus, we designed mutations that stabilize the closed structure of the Ca2+-binding loop favored when Ca2+ is not bound (33, 37, 38). We used these mutants to study how CaM with Ca2+ bound to only one of its two lobes (CaM2C or CaM2N) interacts with its target protein CaMKII. These interactions are of interest because the direction of the changes in synaptic strength that occur during storage of information in neural circuits is exquisitely sensitive to small changes in the concentration of postsynaptic Ca2+ that occur during repetitive electrical activity (6, 8). This sensitivity to Ca2+ occurs at concentrations at which the equilibrium concentration of CaM2C (reached in ≈50 msec) is the same or higher than the concentration of WT CaM with Ca2+ ions bound at all four sites (CaM4), as determined by simulations of Ca2+ binding to free CaM under conditions present in spines (ref. 49 and unpublished observations). Furthermore, because the N-terminal lobe of CaM binds Ca2+ with ≈10- to 100-fold higher kon and koff than the C-terminal lobe, as much as 20% of free CaM may be in the CaM2N conformation 5 msec after the onset of Ca2+ influx through NMDA receptors. Thus, competition for CaM2C or CaM2N within a synaptic spine may determine whether the synapse will be potentiated or depressed.
Using the values reported in this study we can estimate the relative concentrations of CaM2C bound to CaMKII (CaM2C·CaMKII), CaM2N·CaMKII, and CaM4·CaMKII when the Ca2+ concentration in the postsynaptic density is 0.5 μM or 2 μM Ca2+ (S.M. and M.B.K., unpublished data). We assumed concentrations of 30 μM CaMKII subunits and 10 μM free CaM and a KD of 50 nM for binding of CaM4 to CaMKII. At 0.5 μM Ca2+, the concentration of CaM2C·CaMKII is ≈0.8 μM, which is ≈10 times more than the concentration of CaM4·CaMKII. Because of the low affinity of CaM2N for CaMKII, its concentration is ≈80 times lower than the concentration of CaM2C·CaMKII. At 2 μM Ca2+, the concentrations of CaM2C·CaMKII (2.3 μM) and CaM4·CaMKII (3.2 μM) are roughly equal; both still exceed CaM2N·CaMKII by a factor of 80. These numbers show that the enhanced affinity for the third and fourth Ca2+ ions bound to CaM when CaMKII is present results in formation of a significant amount of CaM4·CaMKII at 2 μM Ca2+. However, this mechanism doesn't appear to account quantitatively for the substantial activation of autophosphorylation at concentrations of Ca2+ as low as 0.35 μM (Fig. 1). We conclude that activation of CaMKII by binding of CaM2C alone likely contributes to activation of CaMKII during small increases in Ca2+ in the spine above the basal concentration of 80 nM such as might occur during low-frequency stimulation. This finding is consistent with the recently reported structural arrangement of the holoenzyme of CaMKII (22) (see Supporting Text and Fig. 8, which are published as supporting information on the PNAS web site). Our findings demonstrate the importance of understanding the kinetics of interactions of CaM2C and CaM2N with their various targets to correctly simulate the orchestration of biochemical responses to Ca2+ influx into the spine.
Previous studies have shown that the individual N- or C-terminal lobes of CaM with bound Ca2+ can activate certain effector proteins, phosphorylase kinase (50), myosin light chain kinase, neuronal nitric oxide synthase (51), and adenylate cyclase (52). These studies relied either on isolated N-terminal and C-terminal fragments of CaM generated by proteolysis (50, 51) or on point mutants of CaM that have lost one or more Ca2+-binding sites (52). More recent studies have made use of CaM with single-point mutations in each of the two N-terminal sites (CaM12) or the two C-terminal sites (CaM34) to show that the individual lobes of CaM have distinct functional effects on voltage-activated Ca2+ channels (53, 54). Although these mutants have been useful for understanding the qualitative ability of individual CaM domains to interact with target proteins, they are not ideal for determining the actual binding constants of such interactions as they occur in vivo. The reason that single-point mutations or proteolytic fragmentation may introduce structural changes in CaM that do not represent its intermediate conformations in the cytosol. These changes may influence their rates of binding to targets (55). By stabilizing the mutated Ca2+-binding loops of CaM in the structure assumed by apo-CaM, we have attempted to mimic as closely as possible the transient forms of CaM loaded with Ca2+ ions at either the N or C terminus. These mutant CaMs will be useful for understanding how Ca2+ controls mechanisms of synaptic plasticity that underlie learning and memory. They also may be useful for studying Ca2+-signaling events in other cell types.
Methods
Computational Methods.
The sequences for the mutant CaMs were designed by using the protein design program ORBIT (ref. 43; see Supporting Text). The sequences of the N- and C-terminal CaM domains were optimized with the Ca2+-free CaM structure as a template (33) to produce CaM-CWT and CaM-NWT, respectively.
Expression of CaM Proteins.
cDNAs encoding CaM-CWT and CaM-NWT mutants were constructed with an inverse PCR procedure beginning with the WT CaM cDNA cloned into pET-15 (Novagen, San Diego, CA). WT and mutant CaMs were expressed and purified as described in ref. 56.
Peptide Synthesis.
The CaMKII-cbp peptide corresponding to the CaM-binding domain of CaMKII (sequence -AKSKWKQAFNATAVVRHMRKLQ-) was synthesized and purified in the Caltech Peptide Synthesis Facility as described in ref. 56.
Electrospray Mass Spectrometry.
Electrospray ionization mass spectrometry was performed on an API365 LC/MS/MS system (PE SCIEX) with a nanospray ion source (Protana, Toronto, ON, Canada) operated at an infusion mode of 0.2 μl/min and a spray voltage of −1.4 kV. The data were collected in the negative ion mode. The Ca2+-bound samples (10 μM CaM) were diluted into 4 mM NH4HCO3/200 μM CaCH3CO2/15% CH3OH, pH 8.0. For the Ca2+-free samples, CaCH3CO2 was replaced with 500 μM EGTA.
Ca2+-Binding Affinities.
Ca2+ binding to WT and mutant CaMs was measured by competition with the Ca2+-binding fluorescent dye Fluo4FF (Molecular Probes, Eugene, OR). We determined the intrinsic binding affinity of Fluo4FF for Ca2+ (KD ≈ 13 μM) in the absence of CaM by titrating concentrated CaCl2 into a 10 μM solution of Fluo4FF in 50 mM Tris/100 mM NaCl/1 mM MgCl2, pH 7.2 at 25°C and recording fluorescence after addition of each aliquot of Ca2+ on a Hitachi (Tokyo, Japan) F-4500 fluorescence spectrophotometer (excitation at 488 nm and emission recorded at 516 nm). The measured fluorescence signal F was used to calculate the free Ca2+ concentration [Ca2+]free, the concentration of Ca2+ bound to the dye [Ca2+·Dye], and the affinity of CaM for Ca2+ in the presence of targets as described in Supporting Text.
CaMKII Autophosphorylation Assay.
Reactions were initiated by addition of purified CaMKII (0.28 μM) to a final volume of 100 μl of 50 mM Tris/0.1 M NaCl2/0.9 mg/ml BSA/50 μM ATP/5 mM DTE, pH 7.2, the desired concentration of WT or mutant CaM, and desired Ca2+ concentration determined by mixing appropriate ratios of 100 mM CaCl2 and 100 mM EGTA as described in instructions supplied with the Ca2+ buffer kit (Molecular Probes, Carlsbad, CA). The buffer was warmed to 30°C for 1 min, reactions were initiated, proceeded for 1 min, and were stopped by the addition of 3% SDS/2% 2-mercaptoethanol. Samples containing 0.2 μg of CaMKII were fractionated by SDS/PAGE, transferred by electrophoresis onto a nitrocellulose membrane for 1 h at 350 mA in 25 mM Tris base/0.2 M glycine/20% methanol. Membranes were immunoblotted with 7.5 μg antibody 22B1 in 15 ml of 50 mM Tris, pH 7.4/0.5 M NaCl/0.05% Tween 20 (TTBS) plus 2% normal goat serum for 3 h at 4°C. Monoclonal antibody 22B1 (anti-phospho-CaMKII; Affinity Bioreagents, Golden, CO) is specific for α and β subunits of CaMKII only when they are phosphorylated at Thr-286/287 (in β). Bound antibodies were visualized by measuring fluorescence of a secondary antibody, (IRDye800-Anti-mouse; Rockland, Gilbertsville, PA; 1.5 μg in 30 ml of TTBS) with an Odyssey Imaging System (Li-Cor Biosciences, Lincoln, NE). Conditions were adjusted so that fluorescence intensity was linear over the range of CaMKII measured. The amount of autophosphorylated CaMKII was calculated by quantifying the intensity of each band with the Odyssey software and normalizing it to the intensity of a band corresponding to standard fully autophosphorylated CaMKII prepared by incubating in the assay solution for 1 min with 300 μM Ca2+ and 10 μM WT CaM.
Measurement of Turnover Numbers for Autophosphorylation of CaMKII.
Initial rates of autophosphorylation were measured at saturating levels of Ca2+/CaM and ATP with the use of a temperature-controlled Kintek Model RQF-3 Quench Flow apparatus (Kintek Corp., Austin, TX). Reactions were initiated at 100 ms, 300 ms, 1, 3, 10, and 30 sec by rapid mixing of solution 1 (25 mM Tris·HCl, pH 7.2/0.1 M NaCl/1 mM MgCl2/2 mM Ca2+/0.9 mg/ml BSA/5 mM DTE/1.4 μM CaMKII catalytic subunits, and either 12 μM WT CaM, 30 μM CaM-CWT, or 60 μM CaM-NWT) and solution 2 (identical to solution 1 except that CaMKII and CaM were replaced by 200 μM ATP). The two solutions were kept at 4°C until they were transferred to the quench flow apparatus and warmed for 1 min to 30°C. Autophosphorylation was initiated by rapid mixing of 16 μl of each solution and terminated by rapid addition of a final concentration of 1% SDS, 3.3 mM glycine·HCl, pH 2.9 (see Supporting Text) After quenching, all reactions were brought to a final concentration of 3% SDS, 66 mM Tris·HCl (pH 7.2)/2% 2-mercaptoethaol/5% glycerol/40 μg/ml Bromophenol Blue and subjected to SDS/PAGE followed by immunoblotting with antibody 22B1 as described for the autophosphorylation assay. Intensities of autophosphorylated bands were quantified with the Odyssey Imaging System. The levels of autophosphorylation were normalized to that of fully autophosphorylated CaMKII incubated in the assay for 30 sec (WT CaM) or 100 sec (mutant CaMs). The amount of autophosphorylated CaMKII in each sample lane was obtained from a standard curve built for each set of experiments by measuring the intensity of 0.06, 0.12, and 0.23 μg of fully autophosphorylated CaMKII and fitting the standard data with a power function.
Supplementary Material
Acknowledgments
We thank K. Beckingham (Rice University, Houston, TX) for providing a plasmid encoding calmodulin, E. Winfree and laboratory for the use of their spectrofluorometer, J. Oh and E. Marcora for CaMKII, A. Ingemar for help with CaLigator, and J. Zhou for help with mass spectrometry. This work was supported by the Howard Hughes Medical Institute, the Ralph M. Parsons Foundation, and an IBM Shared University research grant (to S.L.M.), the Sloan-Schwartz Foundation (to M.H.C), and U.S. Public Health Service Grants NS047300 (to M.H.C) and NS44306 (to M.B.K.).
Abbreviations
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- CaM
calmodulin
- CaM-NWT
mutant CaM that binds Ca2+ only at N-terminal sites
- CaM-CWT
mutant CaM that binds Ca2+ only at C-terminal sites
- CaMKII-cbp
peptide having the sequence of the CaM-binding domain of CaMKII
- CaM2C
WT CaM with two Ca2+ ions bound at its C terminus
- CaM2N
WT CaM with two Ca2+ ions bound at its N terminus
- CaM4
WT CaM with Ca2+ ions bound at all four sites.
Footnotes
The authors declare no conflict of interest.
References
- 1.Klee CB. In: Calmodulin. Cohen P, Klee CB, editors. Vol 5. Amsterdam, The Netherlands: Elsevier; 1988. pp. 35–56. [Google Scholar]
- 2.Wang JH, Sharma RK, Huang CY, Chau V, Chock PB. Ann NY Acad Sci. 1980;356:190–204. doi: 10.1111/j.1749-6632.1980.tb29611.x. [DOI] [PubMed] [Google Scholar]
- 3.Burger D, Stein EA, Cox JA. J Biol Chem. 1983;258:14733–14739. [PubMed] [Google Scholar]
- 4.Olwin BB, Edelman AM, Krebs EG, Storm DR. J Biol Chem. 1984;259:10949–10955. [PubMed] [Google Scholar]
- 5.Xia Z, Storm DR. Nat Rev Neurosci. 2005;6:267–76. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- 6.Yang SN, Tang YG, Zucker RS. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- 7.Abbott LF, Nelson SB. Nat Neurosci. 2000;3(Suppl):1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- 8.Franks KM, Sejnowski TJ. BioEssays. 2002;24:1130–1144. doi: 10.1002/bies.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi GQ, Rubin J. Trends Neurosci. 2005;28:222–228. doi: 10.1016/j.tins.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Rubin JE, Gerkin RC, Bi GQ, Chow CC. J Neurophysiol. 2005;93:2600–2613. doi: 10.1152/jn.00803.2004. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- 12.Lisman J, Schulman H, Cline H. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 13.Quinlan EM, Halpain S. Neuron. 1996;16:357–368. doi: 10.1016/s0896-6273(00)80053-7. [DOI] [PubMed] [Google Scholar]
- 14.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 15.Miller SG, Kennedy MB. J Biol Chem. 1985;260:9039–9046. [PubMed] [Google Scholar]
- 16.Meyer T, Hanson PI, Stryer L, Schulman H. Science. 1992;256:1199–1202. doi: 10.1126/science.256.5060.1199. [DOI] [PubMed] [Google Scholar]
- 17.Hudmon A, Schulman H. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 18.Hubbard MJ, Klee CB. J Biol Chem. 1987;262:15062–15070. [PubMed] [Google Scholar]
- 19.Klee CB, Cohen P. In: Calmodulin. Cohen P, Klee CB, editors. Amsterdam, The Netherlands: Elsevier; 1988. pp. 225–248. [Google Scholar]
- 20.Lisman J. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bennett MK, Erondu NE, Kennedy MB. J Biol Chem. 1983;258:12735–12744. [PubMed] [Google Scholar]
- 22.Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg OS, Deindl S, Comolli LR, Hoelz A, Downing KH, Nairn AC, Kuriyan J. FEBS J. 2006;273:682–694. doi: 10.1111/j.1742-4658.2005.05088.x. [DOI] [PubMed] [Google Scholar]
- 24.Hanson PI, Meyer T, Stryer L, Schulman H. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 25.Miller SG, Kennedy MB. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 26.Miller SG, Patton BL, Kennedy MB. Neuron. 1988;1:593–604. doi: 10.1016/0896-6273(88)90109-2. [DOI] [PubMed] [Google Scholar]
- 27.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 28.Watterson DM, Harrelson WG, Jr., Keller PM, Sharief F, Vanaman TC. J Biol Chem. 1976;251:4501–4513. [PubMed] [Google Scholar]
- 29.Erondu NE, Kennedy MB. J Neurosci. 1985;5:3270–3277. doi: 10.1523/JNEUROSCI.05-12-03270.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabatini BL, Oertner TG, Svoboda K. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- 31.Linse S, Helmersson A, Forsen S. J Biol Chem. 1991;266:8050–8054. [PubMed] [Google Scholar]
- 32.Yamniuk AP, Vogel HJ. Mol Biotechnol. 2004;27:33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- 33.Kuboniwa H, Tjandra N, Grzesiek S, Ren H, Klee CB, Bax A. Nat Struct Biol. 1995;2:768–776. doi: 10.1038/nsb0995-768. [DOI] [PubMed] [Google Scholar]
- 34.Babu YS, Bugg CE, Cook WJ. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 35.Wall ME, Clarage JB, Phillips GN. Structure (London) 1997;5:1599–612. doi: 10.1016/s0969-2126(97)00308-0. [DOI] [PubMed] [Google Scholar]
- 36.Maune JF, Klee CB, Beckingham K. J Biol Chem. 1992;267:5286–5295. [PubMed] [Google Scholar]
- 37.Zhang M, Tanaka T, Ikura M. Nat Struct Biol. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- 38.Finn BE, Evenas J, Drakenberg T, Waltho JP, Thulin E, Forsen S. Nat Struct Biol. 1995;2:777–783. doi: 10.1038/nsb0995-777. [DOI] [PubMed] [Google Scholar]
- 39.Chin D, Means AR. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 40.Meador WE, Means AR, Quiocho FA. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 41.Krueger JK, Bishop NA, Blumenthal DK, Zhi G, Beckingham K, Stull JT, Trewhella J. Biochemistry. 1998;37:17810–17817. doi: 10.1021/bi981656w. [DOI] [PubMed] [Google Scholar]
- 42.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 43.Dahiyat BI, Mayo SL. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 44.Maune JF, Beckingham K, Martin SR, Bayley PM. Biochemistry. 1992;31:7779–7786. doi: 10.1021/bi00149a006. [DOI] [PubMed] [Google Scholar]
- 45.Veenstra TD, Johnson KL, Tomlinson AJ, Naylor S, Kumar R. Biochemistry. 1997;36:3535–3542. doi: 10.1021/bi9628329. [DOI] [PubMed] [Google Scholar]
- 46.Wyman J, Gill SJ. Binding and Linkage: Functional Chemistry of Biological Molecules. Mill Valley, CA: University Science Books; 1990. [Google Scholar]
- 47.Andre I, Linse S. Anal Biochem. 2002;305:195–205. doi: 10.1006/abio.2002.5661. [DOI] [PubMed] [Google Scholar]
- 48.Shifman JM, Mayo SL. Proc Natl Acad Sci USA. 2003;100:13274–13279. doi: 10.1073/pnas.2234277100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franks KM, Bartol TM, Sejnowski TJ. Neurocomputing. 2001;38–40:9–16. [Google Scholar]
- 50.Kuznicki J, Grabarek Z, Brzeska H, Drabikowski W, Cohen P. FEBS Lett. 1981;130:141–145. doi: 10.1016/0014-5793(81)80683-7. [DOI] [PubMed] [Google Scholar]
- 51.Persechini A, McMillan K, Leakey P. J Biol Chem. 1994;269:16148–16154. [PubMed] [Google Scholar]
- 52.Gao ZH, Krebs J, VanBerkum MF, Tang WJ, Maune JF, Means AR, Stull JT, Beckingham K. J Biol Chem. 1993;268:20096–20104. [PubMed] [Google Scholar]
- 53.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 54.Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 55.Mukherjea P, Beckingham K. Biochem Mol Biol Int. 1993;29:555–563. [PubMed] [Google Scholar]
- 56.Shifman JM, Mayo SL. J Mol Biol. 2002;323:417–423. doi: 10.1016/s0022-2836(02)00881-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
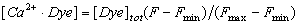{kind=link}