

Abstract
Angiogenesis is a crucial step in many pathologies, including tumor growth and metastasis. Here, we show that tilted peptides exert antiangiogenic activity. Tilted (or oblique-oriented) peptides are short peptides known to destabilize membranes and lipid cores and characterized by an asymmetric distribution of hydrophobic residues along the axis when helical. We have previously shown that 16-kDa fragments of the human prolactin/growth hormone (PRL/GH) family members are potent angiogenesis inhibitors. Here, we demonstrate that all these fragments possess a 14-aa sequence having the characteristics of a tilted peptide. The tilted peptides of human prolactin and human growth hormone induce endothelial cell apoptosis, inhibit endothelial cell proliferation, and inhibit capillary formation both in vitro and in vivo. These antiangiogenic effects are abolished when the peptides' hydrophobicity gradient is altered by mutation. We further demonstrate that the well known tilted peptides of simian immunodeficiency virus gp32 and Alzheimer's β-amyloid peptide are also angiogenesis inhibitors. Taken together, these results point to a potential new role for tilted peptides in regulating angiogenesis.
Keywords: 16-kDa N-terminal fragment of prolactin
Angiogenesis, the formation of new blood vessels from preexisting ones, is crucial in both health and disease. Its involvement in tumor growth and metastasis (1) makes it an important potential target for anticancer therapy. Many angiogenesis inhibitors are cryptic fragments of endogenous molecules displaying no angiogenesis-related activity [angiostatin, endostatin, platelet factor-4 (PF-4), tumstatin, and thrombospondin (2)]. From some of these inhibitors, shorter peptides retaining antiangiogenic activity have been isolated, such as the PF-4 peptide 47-70 (3), endostatin fragments 2 and 5 (amino acids 60–70 and amino acids 171–183) (4), and tumstatin peptides T3 and T7 (amino acids 69–88 and amino acids 74–98) (5). This observation suggests that the antiangiogenic regions of these peptides may be exposed in the isolated fragments but buried in the full-length proteins.
The 16-kDa N-terminal fragment of prolactin (16K PRL) is antiangiogenic in vitro (6–11) and in vivo. Generation of the 16K fragment from PRL has been attributed to cathepsin D (12). Its ability to prevent angiogenesis in tumor and retinopathy mouse models has raised interest in its potential therapeutic use (13–15). We have sought to identify in human 16K PRL (16K hPRL) a peptide that might be responsible for its antiangiogenic activity. Although the 16K fragments of the other three human PRL/GH-family members are also potently antiangiogenic (16), the sequence similarity of these fragments is low (≈35% similarity between all mammalian PRL/GH sequences). This consideration led us to seek a peculiar common structural feature rather than a similar sequence.
Tilted (or oblique-oriented) peptides are short helical peptides (11 to 20 aa long) characterized by a peculiar distribution of hydrophobic residues: they are amphipathic and their net hydrophobicity increases from one end of the helix to the other. A characteristic of tilted peptides family is that they do not share high similarity in the primary sequence. Molecular modeling therefore predicts that they will adopt a tilted position at a lipid/water interface and that this orientation should disturb the parallelism of lipid acyl chains. Tilted peptides have been detected in various proteins with different functions. They were first identified in viral fusion proteins, protein signal sequences, neurotoxic proteins, and proteins involved in lipid metabolism (17). Little is known of the roles played by tilted peptides, with the exception of viral peptides clearly involved in virus-induced fusion events (18). A common feature of tilted peptides is their ability to induce liposome fusion in vitro (19).
Here, we have identified, in all four 16K fragments, a domain showing the characteristic structural features of tilted peptides. We show that the 14-aa tilted peptide sequences of 16K hPRL and the 16-kDa fragment of the human growth hormone (16K hGH) are sufficient to exert the antiangiogenic activity of their parent molecules both in vitro and in vivo. We also show that two tilted peptides from proteins unrelated to angiogenesis have similar antiangiogenic properties. Our findings may have important implications for the study of antiangiogenic mechanisms.
Results
Identification of Tilted Peptide Sequences in 16K Fragments of the Human PRL/GH Family.
Structural analysis (see Materials and Methods) revealed in each N-terminal 16-kDa fragment of the human PRL/GH family a region with tilted peptide-like properties. The peptides were 3D constructed as an α-helix, and their insertion into a modeled membrane was simulated. As expected, they were found to adopt an oblique orientation in the membrane (see Fig. 1A for the 16K hPRL tilted peptide).
Fig. 1.

Structural modeling, location, and analysis of the liposome fusion property of the prolactin tilted peptide. (A) Insertion of the tilted peptide of hPRL into a modeled membrane. The most stable position in the membrane as predicted by IMPALA for the tilted peptide of hPRL is represented. Mid-plane (yellow grid) = bilayer center (z = 0). Upward and downward from the mid-plane: first upper (bottom) plane (pink grid) = lipid acyl chain/polar head groups interface at 13.5 Å from the center; second upper (bottom) plane (mauve grid) = lipid/water interface (z = 18 Å). The N terminus and C terminus of the peptide are indicated. (B and C) Location of the tilted peptide and cathepsin D cleavage sites in the hPRL structure. Shown is the solution structure of hPRL (1R5W), according to ref. 20. The tilted peptide is highlighted in green and is only partially accessible to the solvent. Three of the cathepsin D cleavage sites identified by Piwnica et al. (12), in red, are located on the same surface in the region of the tilted peptide (12). (B and C) Ribbon representation. (B) hPRL is seen from the side with helices 2 and 3 to the front. (C) hPRL is tilted 90°C backward and seen from the bottom. (D) The tilted peptide of hPRL induces liposome fusion. Shown is the time course of lipid mixing induced by the hPRL tilted peptide. The peptide, dissolved in DMSO, was added at different concentrations to a mixture (1:4 wt/wt) of R18-labeled and unlabeled large unilamellar vesicles (LUVs) (PC/PE/SM/Chol). The peptide-to-lipid (P/L) molar ratio ranged from 1/20 to 1/5. The increase in the relative R18 fluorescence because of probe dilution was monitored at room temperature. ♦, DMSO (blank); ×, 1/20 P/L ratio; ▵, 1/10 P/L ratio; ■, 1/5 P/L ratio.
The sequences and properties of these tilted peptides are summarized in Table 1. Mutants of the PRL and GH tilted peptides (respectively, POPRLmut and POGHmut) were designed so as to abolish the hydrophobicity gradient. POPRLmut has the same amino acid composition as its wild-type counterpart, but Leu-2 is permuted with Asn-13 and Val-6 with Ser-11. In POGHmut, Leu-2, Leu-6, and Leu-7 are replaced with Ser, Arg-3 with Gln, and Ser-5 with Leu (Table 1).
Table 1.
Characteristics of the peptides used in this study
| Peptide | Sequence | α, ° | 〈H〉 |
|---|---|---|---|
| POPRL | FLSLIVSILRSWNE | 50 | 0.33 |
| POPRLmut | FNSLISSILRVWLE | 10 | 0.33 |
| POGH | LLRISLLLIQSWLE | 55 | 0.41 |
| POGHmut | LSQILSSLIQSWLE | 5 | 0.35 |
| POSIV | GVFVLGFLGFLA | 50 | 0.93 |
| POBamyl | GAIIGLMVGGVVIA | 50 | 0.78 |
α, Angle of insertion (between the peptide helix axis and a vector perpendicular to the hydrophobic/hydrophilic interface); 〈H〉, hydrophobicity calculated using the consensus scale of Eisenberg (39).
All PRL/GH-family members share a 3D fold characterized by a 4-helix bundle (Fig. 1 B and C) (20). The four α-helices wind around each other and are connected by loops in an up-up-down-down topology. A long loop connects helices 1 and 2, and another connects helices 3 and 4. The tilted peptide is located in the N-terminal region of the second helix and is quite buried in the structure, being surrounded by the other three helices and the third loop.
The Tilted Peptide of hPRL Induces Liposome Fusion.
We first synthesized the tilted peptide of hPRL (POPRL) and tested it for the ability to induce lipid fusion. R18-labeled (R18 is a fluorescent lipophilic probe) and R18-free liposomes [large unilamellar vesicles (LUVs)] were mixed, and the increase in fluorescence intensity because of dequenching of the probe was measured as a function of time. The tilted peptide of hPRL was found to induce lipid mixing in a dose-dependent fashion (Fig. 1D).
Recombinant Protein Production.
Being hydrophobic, tilted peptides are usually quite insoluble and hard to produce in vitro. We therefore produced them in fusion with the maltose-binding protein (MBP), known to improve the solubility of peptides fused to it. The MBP-fused tilted peptides of hPRL and hGH (respectively named poPRL and poGH) and their mutant counterparts (m-poPRL and m-poGH) were produced as soluble protein in Escherichia coli and purified by maltose affinity chromatography. As a control, we engineered an MBP protein (named MBP*) with the C terminus of the fusion proteins but devoid of the tilted peptide sequence.
The Tilted Peptides of hPRL and hGH Inhibit Proliferation of Endothelial Cells and Induce Their Apoptosis.
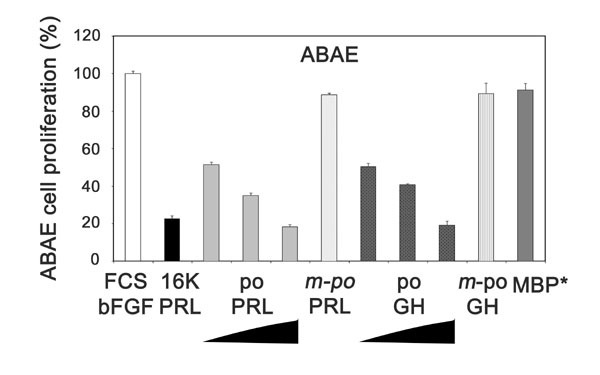
16K hPRL inhibits the proliferation of endothelial cells and induces their apoptosis (9, 11). Likewise, both poPRL and poGH were found to inhibit adult bovine aortic endothelial (ABAE) cell proliferation in a concentration-dependent manner (Fig. 6, which is published as supporting information on the PNAS web site).
Flow cytometry applied to bovine adrenal cortex capillary endothelial (BACE) cells treated with 10 nM 16K hPRL revealed, as expected, the presence of subdiploid cells (the sub-G1 peak) typical of a population undergoing apoptosis. Respectively, 33% or 51% of the cells were found in the sub-G1 phase after treatment with 40 nM poPRL or 320 nM poGH (Fig. 2A and B. Results for ABAE cells were similar (Fig. 2B).
Fig. 2.

The tilted peptides of hPRL and hGH induce apoptosis of BACE and ABAE cells by activating caspase-3. (A and B) poPRL and poGH induce endothelial cell apoptosis. FL3, red fluorescence intensity. (B) Percentage of cells entering apoptosis (sub-G1 population). BACE (A and B) or ABAE (B) cells were treated with 10 nM 16K hPRL or with a fusion protein for 18 h. Cell cycle progression was monitored by measuring cell DNA content by flow cytometry analysis. (C) poPRL and poGH activate caspase-3 in endothelial cells. poPRL (10, 20, and 40 nM), poGH (80, 160, and 320 nM), or 10 nM 16K hPRL induced caspase-3 activation in endothelial cells as compared with untreated cells (Un). Control proteins, i.e., 80 nM mutated poPRL (m-poPRL), 640 nM mutated poGH (m-poGH), and 640 nM MBP* did not. BACE cells were treated for 18 h with the indicated proteins. Each result is expressed as an enhancement factor (treated vs. untreated cells). Each bar represents the mean ± SD; n = 3.
To confirm these observations, we tested the ability of poPRL and poGH to induce caspase-3 activation, one of the most common events in the apoptosis signaling pathway. Both showed a concentration-dependent activating effect (BACE cells; Fig. 2C). At 40 nM and 320 nM, respectively, poPRL caused 30-fold and poGH caused 35-fold activation, similar to that obtained with 10 nM 16K hPRL. Results for ABAE cells were similar (data not shown). The MBP*, m-poPRL, and m-poGH controls failed to inhibit proliferation and to induce apoptosis, even at concentrations twice as high. The experiments were repeated three times with similar results.
Because the peptides were produced in E. coli, it was important to demonstrate that the activity of the preparations used was not caused by bacterial contaminants such as endotoxin. Levels of endotoxin in each peptide preparation are not related to the activity of tilted peptides. Furthermore, using the endotoxin blocker polymyxin-B, we showed that poPRL and poGH actions are independent of endotoxin contamination (Fig. 7, which is published as supporting information on the PNAS web site).
The tilted Peptides of hPRL and hGH Inhibit Capillary Formation both in Vitro and in Vivo.
BACE cells plated between two collagen gels develop into a network of new capillary-like vessels and thus provide an in vitro model of capillary formation. Both poPRL and poGH prevented network formation, whereas m-poPRL and m-poGH had no effect (Fig. 3A and B). The results were similar in three independent experiments.
Fig. 3.

The tilted peptides of hPRL and hGH inhibit capillary network formation in vitro and in vivo. BACE cells were plated between two collagen gels and treated with the indicated protein for 16 h. Living cells were labeled with calcein-AM. (A) Photographs were taken under a fluorescence microscope. (B) Quantitative analysis of network structure performed by measuring tube lengths. Each bar represents the mean ± SD, n = 3. (C) Representative examples of CAMs taken from a typical experiment. CAMs were treated with 40 μg of 16K hPRL, m-poPRL, poPRL, m-poGH, or poGH. (D) Quantification performed by measuring the area devoid of capillaries in the region surrounding the disk. Values are means ± SD, n (BSA) = 8, n (16K hPRL) = 7, n (poPRL) = 6, n (m-poPRL) = 7, n (poGH) = 8, and n (m-poGH) = 12. ∗, P < 10−5 vs. BSA; ∗∗, P < 10−4 vs. m-poPRL; ∗∗∗, P < 10−6 vs. m-poGH.
We next studied the effects of these proteins on in vivo neovascularization in the early-stage chick chorioallantoic membrane (CAM) assay (Fig. 3 C and D). Avascular areas appeared around methylcellulose disks containing 40 μg of poPRL or poGH, whereas m-poPRL and m-poGH had no significant effect.
16K hPRL Mutated so as to Abolish the Hydrophobicity Gradient of Its Tilted Peptide Shows Reduced Activity.
The poPRL and poGH fusion proteins are thus potent angiogenesis inhibitors. To ascertain that the tilted peptide of 16K hPRL is responsible for its antiangiogenic action, we engineered into the 16K hPRL sequence the same mutations as in POPRLmut. Both the mutant and wild-type fragments were produced as MBP fusion proteins (respectively, MBP-16KhPRLmut and MBP-16KhPRL). In a caspase-3 assay performed on ABAE cells, caspase-3 activation was 40–50% lower in MBP-16KhPRLmut-treated than in MBP-16KhPRL-treated cells (Fig. 4). The results are representative of three similar experiments.
Fig. 4.

16K hPRL mutated so as to abolish the hydrophobicity gradient of its tilted peptide shows reduced activity. MBP-16KhPRLmut (■) and MBP-16KhPRL (♦). ABAE cells were treated for 18 h with the indicated protein. Each result is expressed as an enhancement factor (treated vs. untreated cells). Data are means ± SD, n = 3.
The Tilted Peptides of SIV (Simian Immunodeficiency Virus) gp32 and Alzheimer's β-Amyloid Peptide Inhibit Angiogenesis.
The tilted properties of peptides of SIV gp32 and Alzheimer's β-amyloid peptide have been extensively studied elsewhere. Their characteristics are shown in Table 1. Although the parent molecules display no angiogenesis-related function, we produced these tilted peptides as MBP fusion proteins (poSIV and poβamyl, respectively) and tested them for antiangiogenic activity.
Both poSIV and poβamyl were found to exert concentration-dependent inhibition of endothelial cell proliferation (Fig. 5A) and to activate caspase-3 in endothelial cells (Fig. 5B). At the highest concentration tested, proliferation decreased by 30 to 35%, and caspase-3 activation reached 30-fold with poSIV and 40-fold with poβamyl. The results are representative of three similar experiments. Both poSIV and poβamyl prevented BACE cells plated between two collagen gels from forming a capillary network (Fig. 5 C and D), and both were found to inhibit blood vessel formation significantly in a bioassay where CAM was treated for 48 h with 40 μg of fusion proteins and MBP* (Fig. 5 E and F). Invariably in these assays, MBP* had no effect.
Fig. 5.

The tilted peptides of SIV pg32 and the β-amyloid peptide inhibit angiogenesis. (A) poSIV and poBamyl inhibit ABAE cell proliferation. Proliferation was assessed by measuring [3H]thymidine uptake. Cells were treated with 1% FCS and 1 ng/ml basic FGF (bFGF) and increasing concentrations of fusion proteins (80, 160, or 320 nM poSIV or poBamyl, 640 nM MBP*) or with 10 nM 16K hPRL (16K PRL). The data are expressed as percentages of the incorporation measured with 1% FCS and 1 ng/ml bFGF alone. (A and B) Each bar represents a mean ± SD, n = 3. (B) The poSIV and poBamyl fusion proteins activate caspase-3 in BACE cells. BACE cells were treated for 18 h with the indicated protein (40, 80, or 160 nM poSIV or poBamyl). We used 10 nM 16K hPRL (16K PRL) as a positive control and 640 nM MBP* as a negative control. Each result is expressed as an enhancement factor [treated vs. untreated (Un) cells]. (C and D) poSIV and poBamyl inhibit capillary network organization in vitro. BACE cells were plated between two collagen gels and treated with the indicated protein for 16 h. Living cells were labeled with calcein-AM. (C) Photographs were taken under a fluorescence microscope. (D) Quantitative analysis of network structure performed by measuring tube lengths. Each bar represents a mean ± SD, n = 3. (E and F) poSIV and poBamyl inhibit capillary formation in vivo. (E) Representative examples of CAM are taken from a typical experiment. (F) Quantification was done by measuring the area devoid of capillaries in the region surrounding the disk. Values are means ± SD, n (poSIV) = 6, n (poBamyl) = 8, and n (MBP) = 7. ∗, P < 10−3 vs. MBP*; ∗∗, P < 10−2 vs. MBP*.
Discussion
This study demonstrates the antiangiogenic activity of tilted peptides. Strikingly, not only do the tilted peptides of two known angiogenesis inhibitors (16K hPRL and 16K hGH) display such activity, but also so do those of two proteins unrelated to angiogenesis.
Antiangiogenic effects of 16K GH and 16K PRL have been demonstrated at three levels: inhibition of endothelial cell proliferation (7), apoptosis induction (9), and inhibition of capillary formation (16, 21). Here, we show that the MBP-fused isolated tilted peptides of these molecules exert all these effects, unlike MBP-fused variants of these peptides, mutated so as to disrupt the hydrophobicity gradient.
The same mutational strategy has been used successfully to suppress the ability of tilted peptides to induce liposome fusion in vitro (19) and, when applied to the tilted peptides of the SIV and bovine leukemia virus (BLV) envelope glycoproteins, to reduce strongly or even abolish virus fusion processes (22, 23). Here, likewise we show that an MBP-fused variant of 16K hPRL, characterized by mutations that respect the amino acid composition but disrupt the hydrophobicity gradient in the tilted peptide region, is less antiangiogenic than MBP-fused wild-type 16K hPRL. The antiangiogenic activity of 16K hPRL is thus at least partially due to its tilted peptide.
It remains necessary to explain why the tilted peptides of 16K fragments fail to confer antiangiogenic properties to the full-length hormones. The 3D structure of hPRL has been solved recently, more precisely, by NMR spectroscopy (20). In it (Fig. 1 B and C), the tilted peptide is located in the N-terminal region of the second helix and is quite buried by the three other helices and the third loop. Although the structure of 16K hPRL is not yet known, we might speculate that protease cleavage yields a fragment where the tilted peptide is more exposed. Piwnica et al. (12) have recently shown that cleavage of PRL by cathepsin D generates three fragments corresponding to amino acids 1–132, 1–147, and 1–150, characterized by molecular masses of 15, 16.5, and 17 kDa, respectively (12). All of them show antiangiogenic activity. The identified cleavage sites are all located in the same region of the folded protein, very close to loop 3 and to the C-terminal end of the third helix hiding the tilted peptide. This location suggests that the tilted peptide could indeed be more exposed in the fragment than in the parent molecule.
Although poPRL and poGH are very antiangiogenic, they remain, respectively, 4 and 32 times less potent than 16K hPRL in vitro. An unknown in these experiments is how fusion with MBP [used as a molecular chaperone to promote peptide solubility and stability (24)] might affect the presentation of the tilted peptide. MBP is a large protein with two distinct globular domains separated by a deep groove, and at its surface lie several clusters of hydrophobic residues (25). Given the hydrophobicity profile of tilted peptides, MBP might interact with them in such a way as to hinder, to some extent, their activity.
Alternatively, the antiangiogenic action of 16K hPRL and 16K hGH might be mediated by several regions within these fragments. Isolated peptides corresponding to several distinct regions of either endostatin or tumstatin have indeed been shown to exert antiangiogenic effects (5, 26). In keeping with this hypothesis, mutations that fully abolish the activity of poPRL reduce only by ≈50% the activity of MBP-16K hPRL.
We show here that the tilted peptides of SIV gp32 and Alzheimer's β-amyloid peptide are also antiangiogenic. When fused to MBP, both peptides inhibit endothelial cell proliferation, trigger endothelial cell apoptosis by activating caspase-3, and inhibit capillary formation in vitro and in vivo. Neither parent protein seems related to angiogenesis. SIV gp32 is an envelope glycoprotein playing an important role in viral fusion; Alzheimer's β-amyloid peptide 1-42 is involved in neurotoxicity. The “oblique orientation” properties of these tilted peptides have been described at length (19, 22), but to our knowledge, data on relationships between endothelial cells and SIV gp32 or β-amyloid peptides are scant: β-amyloid peptides have been shown to cause endothelial cell death (27, 28).
Much remains to be learned about the mechanisms through which tilted peptides inhibit angiogenesis. Do these mechanisms involve protein–membrane interactions as in the case of tilted peptides in viral fusion proteins? In virus fusion proteins, the tilted peptide is involved in virus entry into the host cell. Its predicted oblique insertion across the lipid–water interface is proposed to trigger local phospholipid disorganization and to generate a new lipid phase favoring fusion (17). Modification of the insertion angle of the bovine leukemia virus (BLV) and SIV peptides is apparently sufficient to abolish fusion (22, 23).
Another possibility is that antiangiogenic activity is initiated by a protein–protein interaction, as suggested for the β-amyloid peptide and Apo proteins. Lins et al. (29) suggest that specific and complementary interactions may exist between apolipoproteins and tilted peptides (29), because the β-amyloid tilted peptide interacts with ApoE2 and ApoE3 but not with ApoA1, and the opposite is true of the SIV tilted peptide (30). This observation suggests that here is evidence of some specificity in the action of tilted peptides. Our results on nonendothelial tumor cells (B16F10 and MDA-MB-231, data not shown) suggest that the antiangiogenic action of poPRL and poGH targets endothelial cells specifically, as previously shown for 16K hPRL (9). Keeping in mind this specificity of action, we favor the view that an initial event in signaling should involve a protein–protein interaction rather than a protein–membrane interaction.
If a protein–protein interaction is necessary to confer activity to the tilted peptide, do the peptides act via receptors? Although Clapp et al. (31) identified a saturable high-affinity specific binding site for 16K PRL on endothelial cell membranes, and despite numerous efforts by different laboratories to find a receptor, until now no receptor has been found. Among angiogenesis inhibitors, 16K hPRL is not the only one whose specific receptor has not yet been identified. For example, the molecular mechanism underlying the antiangiogenic activity of endostatin, a much more extensively studied antiangiogenic factor, is not yet fully elucidated. Endostatin has been shown to bind to α5β1 integrin (32), or with low affinity to glycopican-1 and glycopican-4, or to as yet unidentified receptors (33). So far, none of these binding sites can fully explain the antiangiogenic action of endostatin.
In conclusion, our results open new avenues for investigating the antiangiogenic properties of the 16K fragments of PRL/GH family members. They highlight a potential role of tilted peptides in angiogenesis. These results could have important implications for the development of novel therapies.
Materials and Methods
Molecular Modeling: Sequence Analysis.
Tilted peptides were detected in the hPRL and hGH protein sequences by using different methods to analyze the distribution of the mean hydrophobicity along the sequence (34). These methods are described in Supporting Text, which is published as supporting information on the PNAS web site.
Membrane Insertion.
We used IMPALA (Integral Membrane Protein and Lipid Association), developed in ref. 35, to insert peptides 3D constructed as an α-helix into an implicit bilayer. IMPALA simulates the insertion of any molecule (protein, peptide, or drug) into a bilayer by adding energy restraint functions to the usual energy description of molecules (35, 36).
The position of the structure with the lowest restraint values is considered the most stable in the bilayer. This method is discussed in Supporting Text.
Production of Recombinant Proteins.
Recombinant 16K hPRL was produced in E. coli as described (10). Oligonucleotides encoding tilted and mutated peptides were inserted into the pMal-C2x plasmid at the 3′ end of the MBP coding sequence (New England Biolabs, Hitchin, U.K.). A modified MBP, named MBP*, was engineered to have a C terminus identical to that of the fusion protein but no tilted peptide sequence. MBP fusion proteins were also made with the full-length 16K hPRL sequence, mutated or not in its tilted peptide region. All recombinant fusion proteins were produced in E. coli as soluble proteins. The cells were disrupted, and soluble proteins were recovered by centrifugation. The proteins were first purified by affinity chromatography on amylose resin (New England Biolabs) and then by anion exchange chromatography (Hitrap Q; Amersham Pharmacia Biotech, Arlington Heights, IL) according to the manufacturer's instructions. In each protein preparation, the endotoxin level was lowered to <175 endotoxin units/mg (as quantified by the “endotoxin testing service” at Cambrex Biosciences, Verviers, Belgium) by means of an endotoxin affinity chromatography on EndoTrap resin (Profos, Regensburg, Germany).
Cell Cultures.
Synthesis of the PRL Tilted Peptide (POPRL).
The tilted peptide of 16K hPRL was chemically synthesized by Eurogentec S.A. (Seraing, Belgium). Its sequence is FLSLIVSILRSWNE. The peptide is N-acetylated and C-amidated.
Liposome Fusion Experiments.
Large unilamellar vesicles (LUVs) were prepared and the lipid phase fusion assay was performed as described in ref. 18 and in Supporting Text.
Endothelial Cell Proliferation Assay.
ABAE cells were growth-arrested by contact inhibition for 48 h and plated at a density of 2 × 104 cells per well (in 24-well plates) in 0.5 ml of 1% FCS/DMEM. They were treated with 1 ng/ml basic FGF (bFGF) and the specified recombinant protein at the indicated dose for 16 h. Then, they were incubated with [methyl-3H]thymidine 5′ triphosphate (Amersham Biosciences, Buckinghamshire, U.K.), and measurement of [3H]thymidine incorporation was performed as described in ref. 11.
DNA Fragmentation Assay.
BACE or ABAE cells were plated at a density of 3.5 × 105 cells per 5-cm plate in 5 ml of 10% FCS/DMEM. They were treated or not with increasing concentrations of a recombinant protein for 18 h, then harvested by trypsinization, washed with ice-cold PBS, and fixed for 3 h at 4°C with 80% ethanol in PBS. After centrifugation followed by a 15-min incubation at 37°C in PBS containing 50 μg/ml propidium iodide and 300 ng/ml RNase (Boehringer Mannheim, Ingelheim, Germany), the cells were analyzed with a Coulter EPICS XL flow cytometer equipped with an argon laser emitting at 488 nm (Coulter, Hialeah, FL). Graphical and population analyses were performed with FlowJo 6.01 software (Treestar, San Carlos, CA).
Caspase-3 Assay.
BACE or ABAE cells were plated at a density of 2 × 104 cells per well (in 24-well plates) in 0.5 ml of 10% FCS/DMEM. After 24 h, they were treated for 18 h with the specified protein at the indicated dose. Caspase-3 activity was measured with the CaspACE Assay System, Fluorimetric (Promega, Madison, WI) according to the manufacturer's instructions.
In Vitro Capillary Formation.
A collagen gel assay for angiogenesis was performed as described (38) with some modifications. Briefly, 5 vol of rat tail collagen (4 mg/ml; Serva, Heidelberg, Germany), 10× M199 medium (1 vol), and 500 mM NaHCO3 (1 vol), PBS (4 vol) were mixed on ice. Two hundred microliters of this mixture was poured per well (of 24-well plates) and allowed to gel at 37°C for 2 h. BACE cells (200 cells per well) were plated, overlaid with a second gel layer, and incubated with 500 μl of 10% FCS/DMEM for 16 h with the specified recombinant protein. The cells were incubated with calcein-AM (2 μM). Quantitative analysis of the network structure was performed by measuring tube lengths on a PC computer with Scion Image Software (Scion, Frederick, MD). Each sample was analyzed in triplicate, and a representative field of each well was examined at ×200 magnification. Pictures were made with an Olympus fluorescence microscope and camera linked to the Analysis software (Soft Imaging System GmbH, Münster, Germany).
In Vivo Early-Stage Chick CAM Bioassay.
On day 3 of development, fertilized chick embryos were removed from their shells, placed in Petri dishes, and incubated at 37°C. On the 7th day, disks (5 mm) of methylcellulose (0.5%, Sigma) containing 40 μg of recombinant protein and 4 μg of BSA were placed on the chick CAM. After 48 h, white India ink was injected into the chorioallantoic sac, and the avascular area was determined with Analysis software (Soft Imaging System GmbH) and by phase analysis allowing automatic quantitative evaluation of the area.
Supplementary Material
Acknowledgments
This work was supported by grants from the Fonds pour la Recherche Industrielle et Agricole (FRIA) and Télévie (to N.-Q.-N.N. and S.P.T.) and from the Fonds National pour la Recherche Scientifique (FNRS), the Fédération Belge Contre le Cancer, and the Université de Liège (Fonds Spéciaux). L.L. and R.B. are, respectively, Research Associate and Research Director at the FNRS of Belgium.
Abbreviations
- 16K PRL
16-kDa N-terminal fragment of prolactin
- 16K hPRL
human 16K PRL
- 16K hGH
16-kDa N-terminal fragment of human growth hormone
- MBP
maltose-binding protein
- ABAE
adult bovine aortic endothelial
- BACE
bovine adrenal cortex capillary endothelial
- CAM
chorioallantoic membrane
- SIV
simian immunodeficiency virus.
Footnotes
The authors declare no conflict of interest.
References
- 1.Carmeliet P. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 2.Cao Y. Int J Biochem Cell Biol. 2001;33:357–369. doi: 10.1016/s1357-2725(01)00023-1. [DOI] [PubMed] [Google Scholar]
- 3.Jouan V, Canron X, Alemany M, Caen JP, Quentin G, Plouet J, Bikfalvi A. Blood. 1999;94:984–993. [PubMed] [Google Scholar]
- 4.Wickstrom SA, Alitalo K, Keski-Oja J. J Biol Chem. 2004;279:20178–20185. doi: 10.1074/jbc.M312921200. [DOI] [PubMed] [Google Scholar]
- 5.Maeshima Y, Yerramalla UL, Dhanabal M, Holthaus KA, Barbashov S, Kharbanda S, Reimer C, Manfredi M, Dickerson WM, Kalluri R. J Biol Chem. 2001;276:31959–31968. doi: 10.1074/jbc.M103024200. [DOI] [PubMed] [Google Scholar]
- 6.D'Angelo G, Struman I, Martial JA, Weiner RI. Proc Natl Acad Sci USA. 1995;92:6374–6378. doi: 10.1073/pnas.92.14.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Angelo G, Martini JF, Iiri T, Fantl WJ, Martial JA, Weiner RI. Mol Endocrinol. 1999;13:692–704. doi: 10.1210/mend.13.5.0280. [DOI] [PubMed] [Google Scholar]
- 8.Lee H, Struman I, Clapp C, Martial JA, Weiner RI. Endocrinology. 1998;139:3696–3703. doi: 10.1210/endo.139.9.6194. [DOI] [PubMed] [Google Scholar]
- 9.Martini JF, Piot C, Humeau LM, Struman I, Martial JA, Weiner RI. Mol Endocrinol. 2000;14:1536–1549. doi: 10.1210/mend.14.10.0543. [DOI] [PubMed] [Google Scholar]
- 10.Tabruyn SP, Sorlet CM, Rentier-Delrue F, Bours V, Weiner RI, Martial JA, Struman I. Mol Endocrinol. 2003;17:1815–1823. doi: 10.1210/me.2003-0132. [DOI] [PubMed] [Google Scholar]
- 11.Tabruyn SP, Nguyen NQ, Cornet AM, Martial JA, Struman I. Mol Endocrinol. 2005;19:1932–1942. doi: 10.1210/me.2004-0515. [DOI] [PubMed] [Google Scholar]
- 12.Piwnica D, Touraine P, Struman I, Tabruyn S, Bolbach G, Clapp C, Martial JA, Kelly PA, Goffin V. Mol Endocrinol. 2004;18:2522–2542. doi: 10.1210/me.2004-0200. [DOI] [PubMed] [Google Scholar]
- 13.Bentzien F, Struman I, Martini JF, Martial JA, Weiner RI. Cancer Res. 2001;61:7356–7362. [PubMed] [Google Scholar]
- 14.Kim J, Luo W, Chen DT, Earley K, Tunstead J, Yu-Lee LY, Lin SH. Cancer Res. 2003;63:386–393. [PubMed] [Google Scholar]
- 15.Pan H, Nguyen NQ, Yoshida H, Bentzien F, Shaw LC, Rentier-Delrue F, Martial JA, Weiner RI, Struman I, Grant MB. Invest Ophthalmol Vis Sci. 2004;45:2413–2419. doi: 10.1167/iovs.03-1001. [DOI] [PubMed] [Google Scholar]
- 16.Struman I, Bentzien F, Lee H, Mainfroid V, D'Angelo G, Goffin V, Weiner RI, Martial JA. Proc Natl Acad Sci USA. 1999;96:1246–1251. doi: 10.1073/pnas.96.4.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brasseur R, Pillot T, Lins L, Vandekerckhove J, Rosseneu M. Trends Biochem Sci. 1997;22:167–171. doi: 10.1016/s0968-0004(97)01047-5. [DOI] [PubMed] [Google Scholar]
- 18.Lins L, Charloteaux B, Thomas A, Brasseur R. Proteins. 2001;44:435–447. doi: 10.1002/prot.1109. [DOI] [PubMed] [Google Scholar]
- 19.Pillot T, Goethals M, Vanloo B, Talussot C, Brasseur R, Vandekerckhove J, Rosseneu M, Lins L. J Biol Chem. 1996;271:28757–28765. doi: 10.1074/jbc.271.46.28757. [DOI] [PubMed] [Google Scholar]
- 20.Teilum K, Hoch JC, Goffin V, Kinet S, Martial JA, Kragelund BB. J Mol Biol. 2005;351:810–823. doi: 10.1016/j.jmb.2005.06.042. [DOI] [PubMed] [Google Scholar]
- 21.Clapp C, Martial JA, Guzman RC, Rentier-Delrue F, Weiner RI. Endocrinology. 1993;133:1292–1299. doi: 10.1210/endo.133.3.7689950. [DOI] [PubMed] [Google Scholar]
- 22.Horth M, Lambrecht B, Khim MC, Bex F, Thiriart C, Ruysschaert JM, Burny A, Brasseur R. EMBO J. 1991;10:2747–2755. doi: 10.1002/j.1460-2075.1991.tb07823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voneche V, Callebaut I, Kettmann R, Brasseur R, Burny A, Portetelle D. J Biol Chem. 1992;267:15193–15197. [PubMed] [Google Scholar]
- 24.Bach H, Mazor Y, Shaky S, Shoham-Lev A, Berdichevsky Y, Gutnick DL, Benhar I. J Mol Biol. 2001;312:79–93. doi: 10.1006/jmbi.2001.4914. [DOI] [PubMed] [Google Scholar]
- 25.Spurlino JC, Lu GY, Quiocho FA. J Biol Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 26.Chillemi F, Francescato P, Ragg E, Cattaneo MG, Pola S, Vicentini L. J Med Chem. 2003;46:4165–4172. doi: 10.1021/jm0308287. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Chen S, Ku G, Ahmed SH, Xu J, Chen H, Hsu CY. J Cereb Blood Flow Metab. 2001;21:702–710. doi: 10.1097/00004647-200106000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H. Neurobiol Aging. 2003;24:437–451. doi: 10.1016/s0197-4580(02)00135-5. [DOI] [PubMed] [Google Scholar]
- 29.Lins L, Thomas-Soumarmon A, Pillot T, Vandekerchkhove J, Rosseneu M, Brasseur R. J Neurochem. 1999;73:758–769. doi: 10.1046/j.1471-4159.1999.0730758.x. [DOI] [PubMed] [Google Scholar]
- 30.Pillot T, Goethals M, Vanloo B, Lins L, Brasseur R, Vandekerckhove J, Rosseneu M. Eur J Biochem. 1997;243:650–659. doi: 10.1111/j.1432-1033.1997.00650.x. [DOI] [PubMed] [Google Scholar]
- 31.Clapp C, Weiner RI. Endocrinology. 1992;130:1380–1386. doi: 10.1210/endo.130.3.1311239. [DOI] [PubMed] [Google Scholar]
- 32.Wickstrom SA, Alitalo K, Keski-Oja J. Cancer Res. 2002;62:5580–5589. [PubMed] [Google Scholar]
- 33.Karumanchi SA, Jha V, Ramchandran R, Karihaloo A, Tsiokas L, Chan B, Dhanabal M, Hanai JI, Venkataraman G, Shriver Z, et al. Mol Cell. 2001;7:811–822. doi: 10.1016/s1097-2765(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 34.Brasseur R. Mol Membr Biol. 2000;17:31–40. doi: 10.1080/096876800294461. [DOI] [PubMed] [Google Scholar]
- 35.Ducarme P, Rahman M, Brasseur R. Proteins. 1998;30:357–371. [PubMed] [Google Scholar]
- 36.Lins L, Charloteaux B, Heinen C, Thomas A, Brasseur R. Biophys J. 2006;90:470–479. doi: 10.1529/biophysj.105.068213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gospodarowicz D, Massoglia S, Cheng J, Fujii DK. J Cell Physiol. 1986;127:121–136. doi: 10.1002/jcp.1041270116. [DOI] [PubMed] [Google Scholar]
- 38.Cavallaro U, Tenan M, Castelli V, Perilli A, Maggiano N, Van Meir EG, Montesano R, Soria MR, Pepper MS. J Cell Biochem. 2001;82:619–633. doi: 10.1002/jcb.1190. [DOI] [PubMed] [Google Scholar]
- 39.Eisenberg D, Weiss RM, Terwilliger TC. Nature. 1982;299:371–374. doi: 10.1038/299371a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}