

Abstract
Inhibition of erythrocyte (RBC) promotion of platelet reactivity could improve the antiplatelet effect of aspirin (ASA). We tested different ASA regimens for optimal inhibition of platelets and the effects of RBC in patients with a history of vascular diseases. Collagen-induced platelet activation (14C-5HT, TXA2 release) and platelet recruitment (proaggregatory activity of cell-free releasates from activated platelets) were measured in PRP, platelet-RBC (Hct 40%) and whole blood (WB) in 206 patients initially on 200–300 mg ASA/day. Their regimen was modified to bi-weekly 500 mg (loading dose, L) plus daily or twice-daily low-dose ASA (50 or 100 mg). TXA2 was inhibited with all regimens. Percentage of patients with suboptimal inhibition of platelet recruitment in WB was: 200–300 ASA/day (41%), L-50/day (87%), L-100/day (58%), L-50/twice-daily (39%) and L-100/twice-daily (20%; p<0.05 vs. other regimens). 14C-5HT release was inhibited to the greatest extent with L-100/twice-daily in PRP+RBC or WB (p<0.05 vs. other regimens) due to greater inhibition of the RBC prothrombotic effect. Compared to other ASA regimens, L-100 twice-daily (equivalent to 221 mg ASA/day in the 14 day cycle), reduced by >50% the proportion of patients with suboptimal inhibition of platelet recruitment in WB and inhibited 14C-5HT release to the greatest extent.
Keywords: aspirin, blood cells, platelets, erythrocytes, cardiovascular diseases, cerebrovascular disorders, thrombosis
Abbreviations: ADP: adenosine 5′-diphosphate, ASA: acetylsalicylic acid, BID: twice-daily, 14C-5HT: 14C-serotonin, COX-1: cyclooxygenase-1, L: loading dose, OD: daily, PRP: platelet-rich plasma, RBC: erythrocyte, TXA2: thromboxane A2, WB: whole blood
INTRODUCTION
Increased platelet reactivity promotes thrombotic and ischemic events in patients with occlusive vascular diseases. Thus, long term aspirin therapy is utilized for secondary prevention of platelet-driven cardiovascular and cerebrovascular diseases. Aspirin prophylaxis prevents new events in 25% of patients with occlusive vascular disorders1. Nevertheless, many patients experience an additional event despite aspirin treatment.
Several factors contribute to this shortcoming of aspirin therapy. ASA-treated platelets can be activated by cyclooxygenase-1 (COX-1)-independent mechanisms with high doses of physiological agonists such as thrombin, collagen, or ADP2–4. Lack of complete inhibition of thromboxane A2 (TXA2) production may also influence the clinical outcome in patients who received aspirin5. Moreover, cell-cell interactions between different cell types in the circulation as well as the vessel wall constitute additional factors which could modify the response of ASA-treated platelets to agonists. For example, platelet-neutrophil interactions can down-regulate the early stages of platelet reactivity, thus enhancing the antithrombotic effect of aspirin6,7. Also, close apposition of activated platelets with intact erythrocytes (RBC) increases platelet activation and the proaggregatory capacity of cell-free releasates (recruitment)3,8,9. The mechanisms involved are not fully characterized, but include an increase of platelet arachidonate release and eicosanoid synthesis3,8 (even in the presence of aspirin)3 and a 6.9- and 4.9-fold increase in ADP and ATP release, respectively (in the absence of RBC lysis) 8. In addition, platelet-erythrocyte interactions increase αIIbβ3 receptor activation and P-selectin exposure on recruiting platelets9. These prothrombotic effects of RBC result in a reduction of the inhibitory effect of aspirin on platelet reactivity3,10,11.
Previously, we observed that in patients with vascular disease, daily treatment with moderate doses of ASA (200 to 300 mg) was insufficient to block platelet recruitment in 45% of patients due to the prothrombotic effect of RBC on platelet reactivity, even though TXA2 synthesis was inhibited11. A major component of the aspirin-sensitive prothrombotic effect of RBC on platelets can be blocked with a single dose of 500 mg8,10. However, with time RBC recover from this ASA-blockade. To maintain inhibition of the RBC prothrombotic effect, normal subjects require an additional dose of 500 mg ASA every 2 weeks, which supplements the daily dose of 50 mg ASA10. No data is currently available concerning these effects of aspirin in patients with vascular disease.
Erythrocyte-enhanced platelet reactivity could contribute to occurrence of occlusive events in patients with vascular diseases. Therefore, the purpose of this study was to establish a novel ASA regimen which would simultaneously inhibit platelet reactivity plus the aspirin-sensitive prothrombotic effect of erythrocytes in patients with vascular disease, which would down-regulate platelet reactivity in whole blood. We found that the number of patients with incomplete inhibition of platelet recruitment in the presence of RBC or WB was reduced by half if they ingested an intermittent dose of 500 mg ASA at 2-week intervals plus 100 mg twice daily (equivalent to 221 mg/day in the 14 day-cycle), when compared to a regimen of 200 or 300 mg ASA daily. Our modified ASA regimen also significantly down-regulated platelet 14C-5HT release.
METHODS
Patients and Controls
Ninety-eight patients with a history of ischemic heart disease (mean age=63.79±10.00 years; range: 36–81, men/women 80/18) and 108 patients with ischemic cerebrovascular disease (mean age=63.92±10.34 years; range: 39–82, men/women 79/29) (Table 1), treated with 200 mg ASA/day (Adiro, Bayer, Spain) or 300 mg ASA/day (Tromalyt, Madaus Cerafarma, Spain) for at least three months, were consecutively included in the study. The study conforms to the ethical guidelines for human research and was approved by the Institutional Review Board of the Hospital Universitario La Fe, and patients gave informed consent. Included patients were not taking other platelet inhibitory or anticoagulant medications. Compliance with ASA treatment was ascertained by personal interview prior to venipuncture and TXA2 inhibition >80%.
Table 1.
PATIENT DIAGNOSES AND MEDICATIONS
| Ischemic Heart Disease (n=98) | n | % |
|---|---|---|
| Myocardial infarction | 25 | 25 |
| Unstable angina | 34 | 35 |
| Stable angina | 31 | 32 |
| Congestive heart failure | 8 | 8 |
| Medications | ||
| Vasodilators | 52 | 53 |
| Calcium channel blockers | 43 | 44 |
| β-Adrenergic blockers | 36 | 37 |
| Antihypertensives | 38 | 39 |
| Hypoglycemics | 23 | 23 |
| Lipid-lowering agents | 30 | 31 |
| Sedatives or hypnotics | 17 | 17 |
| Cerebrovascular Disease (n=108) | ||
| Lacunar infarct | 48 | 44 |
| Atherothrombotic infarct | 39 | 36 |
| Transient ischemic attack | 16 | 15 |
| Stroke of undetermined etiology | 5 | 5 |
| Medications | ||
| Vasodilators | 9 | 8 |
| Calcium channel blockers | 33 | 31 |
| Antihypertensives | 46 | 43 |
| Hypoglycemics | 31 | 29 |
| Lipid-lowering agents | 17 | 16 |
| Sedatives or hypnotics | 11 | 10 |
After evaluation of platelet reactivity when the patients were treated with 200/300 mg ASA daily, their ASA regimen was altered to a loading dose (L) of 500 mg ASA (Bayer, Spain) every two weeks (to down-regulate the prothrombotic effect of erythrocytes10) supplemented with daily (OD) or twice-daily (BID) ASA (mg): L+50−OD (n=31), L+100−OD (n=33), L−50−BID (n=78) or L+100−BID (n=95). This daily dosing prolongs the effect of the intermittent dose on RBC-dependent prothrombotic effects10 and inhibits the reactivity of newly formed platelets in the circulation. The regimens were continued for at least two months, followed by re-evaluation of platelet reactivity. During this period other concomitant medications were not changed. Some patients followed more than one regimen to optimize the ASA effect.
Platelet recruitment, 14C-5HT release and TXA2 synthesis were also evaluated in normal control subjects not taking aspirin, as indicated in Results.
Blood Cell Collection and Processing
Citrate-anticoagulated venous blood (129 mmol/L; 9:1, vol:vol) was collected from patients and controls into siliconized glass tubes (Vacutainer; Becton Dickinson, USA) after an overnight fast. Patient samples were obtained before daily ASA ingestion, and, when the intermittent dose was prescribed, 11–14 days after the last 500 mg dose. Since platelet life span is 7–10 days2, the effect of the intermittent dose would primarily affect RBC at the time of analysis.
Platelet-rich plasma (PRP) and platelet-poor plasma (PPP) were prepared by differential centrifugation (200g and 2,500g, respectively, 10 min, 22 °C). Following removal of PRP, PPP and buffy coat, 1 ml of erythrocytes, taken from the center of the erythrocyte column, was removed for subsequent testing.
Collagen (1 μg/mL) (Horm, Nycomed, Linz, Austria)-induced serotonin release was the marker for the platelet release reaction to monitor platelet activation. Platelets were radiolabeled with serotonin (14C-5HT, Amersham International, Buckinghamshire, England) as described3. Supernatants of blood to which only agonist buffer had been added served as controls, and their radioactivity content was subtracted from sample values3.
TXA2 was measured in cell-free supernatants of collagen (1 μg/ml)-stimulated whole blood by radioimmunoassay of the stable TXA2 metabolite TXB28. Agonist buffer alone served as control.
Measurement of Platelet Activation and Recruitment
Platelet activation and recruitment were independently evaluated using the activation-recruitment system, a two-stage in vitro cell-cell interaction procedure as previously described3,8,9. Platelets (1.8×108/ml, PRP), platelets plus erythrocytes (PRP+RBC, hematocrit 40%), or whole blood (WB) were incubated (10 min, 37°). Collagen (1 μg/mL) was added as platelet agonist and the tube contents mixed by inversion (10 sec). A cell-free releasate was obtained within 1 min by centrifugation (13,000×g, 50 sec). This was employed for biochemical studies, to assess platelet activation (14C-5HT, TXA2), or as an inducer of platelet aggregation in autologous PRP (recruitment), expressed as maximal height (in mm) of the aggregation response3,8. Erythrocyte promotion of platelet reactivity only takes place with metabolically-intact erythrocytes in contact with activated platelets in the absence of lysis3,8.
The therapeutic effect of ASA was considered optimal when platelet recruitment induced by 1 μg/mL collagen was inhibited by at least 80% vs. the average of ASA-free population of normal subjects. This derives from our observations10,11, that in normal subjects, 2 hr following ingestion of a single dose of 500 mg ASA, the level of inhibition is close to 100%, with a lower limit of 80%.
Statistical Analyses
Data are expressed as mean ± SEM or percentage. For quantitative data analysis one way ANOVA plus Duncan’s tests were used. For comparisons of qualitative data, Chi-squared tests were used. Statistical significance was established at p<.05.
RESULTS
1. Effect of Different Therapeutic Aspirin Regimens on Inhibition of Platelet Recruitment in Patients with Vascular Disease
To discern participation of different cell types in the ASA effect, studies were performed in parallel in PRP, PRP+RBC (hematocrit 40%) and WB. Of the patients treated with 200/300 mg ASA/day upon entering the study, 20% did not reach the optimal degree of inhibition of platelet recruitment in PRP (Figure 1). Importantly, in PRP+RBC or WB, this proportion increased to 51% and 41% (Figure 1). This took place in a setting of blocked TXA2 synthesis (Table 2), in agreement with a previous report11.
Figure 1. Percentage of Patients with Insufficient Inhibition of Platelet Recruitment.
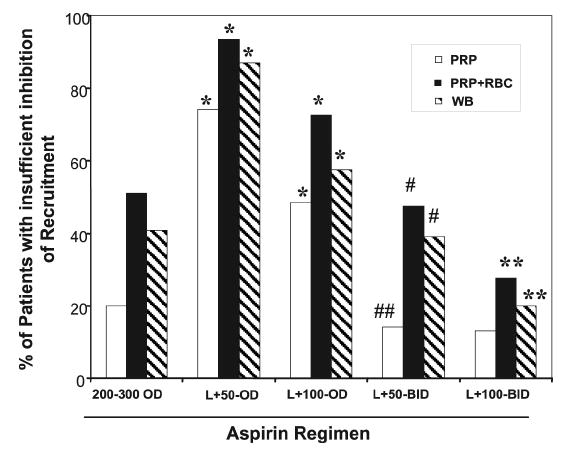
Platelet recruitment was evaluated in PRP plus RBC (Hct 40%) and WB (Methods) in patients treated with 200/300 mg ASA OD (n=206) and after changing the regimen to intermittent 500 mg at 2-week intervals (L) supplemented with daily (OD) or every 12 hours (BID) doses (mg): L+50−OD (n=31), L+100−OD (n=33), L+50−BID (n=78) or L+100−BID (n=95). The proportion of patients with insufficient inhibition markedly increased with L+50−OD and L+100−OD vs. 200/300 mg OD (*p< .05). The L+50−BID regimen had a similar proportion of patients with insufficient inhibition of recruitment as the 200/300 mg OD, in PRP, PRP+RBC and WB. The L+100−BID regimen yielded the same results in PRP as 200/300 mg OD. However, a 50% reduction in the proportion of patients with insufficient inhibition was observed in PRP+RBC or WB with L+100−BID (**p< 0.005). The comparison of L+50−BID with L+100−OD indicated that a twice-a-day ASA regimen significantly reduced the percentage of patients with insufficient inhibition in PRP (##p< 0.01), in the presence or RBC or WB (#p< 0.05). The chi-squared test was used for statistical analysis.
Table 2. Thromboxane B2 Production (ng/mL).
TXB2 formed in collagen-stimulated WB was assayed by radioimmunoassay (Methods) in ASA-treated patients and ASA-free controls. Assays were performed at days 11–14 in samples from patients on regimens involving administration of 500 mg ASA at 2-week intervals (i.e. just prior to the next 500 mg dose, and past the life-span of the platelets treated by the previous 500 mg dose). Significant inhibition of TXB2 synthesis was observed with all ASA regimens vs. controls (p< .05). Although TXB2 was drastically inhibited, the L+50−OD regimen showed the least inhibition of the ASA regimens (p< .05). ND= not detectable
| Thromboxane B2(ng/mL). | |||
|---|---|---|---|
| Aspirin Regimen | mean ± SEM | Range | n |
| 200 or 300 mg/day | 0.59 ± 0.01 | ND- 9 | 206 |
| 500 mg 2-week intervals plus: | |||
| 50 mg/day (L+50-OD) | 4.16 ± 0.67 | ND - 9 | 31 |
| 100 mg/day (L+100-OD) | 1.23 ± 0.22 | ND - 5 | 33 |
| 50 mg/BID (L+50-BID) | 0.82 ± 0.12 | ND - 6 | 78 |
| 100 mg/BID (L+100-BID) | 0.51 ± 0.15 | ND - 6 | 95 |
| ASA-free controls | 66.06±2.48 | 22–123 | 73 |
Subsequently, the ASA regimen was changed in order to optimize the ASA effect on platelet reactivity. The first group of patients received a loading dose (500 mg) at 2-week intervals combined with 50 mg ASA/day (L+50−OD). This dose effectively blocked both platelet reactivity and the aspirin-sensitive prothrombotic effect of erythrocytes in normal subjects as previously reported10. For patients with a previous vascular event, this regimen greatly increased the number of patients with insufficient inhibition of platelet recruitment in platelets alone, in the presence of RBC and in WB when compared to 200/300 mg ASA/day (Figure 1). This occurred in a setting of blocked TXA2 formation (93% vs. aspirin-free controls) (Table 2). However, our subsequent ASA regimens led to greater TXA2 inhibition than the L+50−OD (Table 2).
Hence, the daily ASA dose was increased to 100 mg/day (L+100−OD). This regimen reduced the percentage of patients with insufficient blockade of platelet recruitment (vs. the L+50−OD regimen). Nevertheless, inhibition remained less than optimal in 48, 72 and 58% of patients as measured in PRP, PRP+RBC or WB, respectively (Figure 1).
Since high platelet turnover may occur in some patients, and to minimize the daily dose of aspirin, the aspirin dose was altered to a loading dose plus 50 mg twice-daily (L+50−BID). Importantly, L+50−BID when compared to the L+100−OD regimen (same dose but administered once daily), significantly reduced the proportion of patients with insufficient inhibition in PRP (14% vs. 48%, 3.4-fold) and 1.5-fold in PRP+RBC (47% vs. 72%) or in WB (39% vs. 58%) (Figure 1). Furthermore, L+50−BID produced optimal down-regulation of platelet recruitment in a similar percentage of patients as single, but higher doses of aspirin (200/300 mg OD) (Figure 1).
A regime of L+100−BID yielded similar results in PRP alone as L+50−BID and 200/300 mg OD, suggesting that the effect of ASA was maximal in platelets alone. However, it reduced by half the proportion of patients with insufficient inhibition in WB, to 20%. With 200/300 mg OD the proportion was 41% and with L+50−BID 39% (Figure 1). The improved effects of L+100−BID, therefore, is attributable to greater down-regulation of the aspirin-sensitive prothrombotic effect of erythrocytes with that regimen. In addition, a parallel 40% reduction was found in the proportion of patients with partial inhibition of platelet recruitment in platelet-erythrocyte suspensions (Figure 1). Thus, RBC are the component of whole blood responsible for the prothrombotic effects observed in WB as well as its significant reduction in patients treated with this aspirin regimen.
2. Patients with Insufficient Inhibition of Platelet Recruitment
The maximal intensity of the proaggregatory activity of cell-free releasates from collagen-stimulated whole blood (recruiting activity) from aspirin-treated patients was compared with that of controls. With all ASA regimens the means of recruitment values in whole blood were significantly inhibited as compared to ASA-free controls. However, the proportion of patients with sufficient inhibition depended upon the aspirin regimen utilized (Figure 2). Interestingly, individual patients who did not achieve optimal aspirin-suppression of platelet recruitment demonstrated platelet responses in the range of the aspirin-free control group of normal subjects (Figure 2). Thus, in a large number of patients aspirin protection was insufficient, rendering them more susceptible to a new event. Therefore, the increased proportion of patients with optimal inhibition achieved by the L−100−BID regimen (Figures 1, 2) may experience a more favorable clinical result.
Figure 2. Reduction of platelet recruitment in WB as induced by ASA.
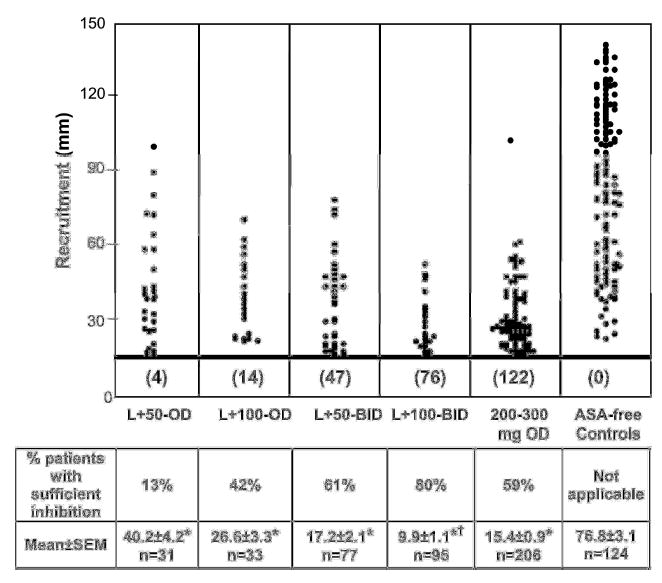
Platelet recruitment was evaluated in WB of patients treated with the different ASA regimens and compared with aspirin-free normal subjects (Methods). The horizontal heavy line designates the demarcation between patient data that are considered to represent optimal and suboptimal inhibition by ASA. Below the line inhibition was considered optimal (Methods), and the number of patients falling in this region is indicated. Above the line are data points of patients with insufficient inhibition. In many patients platelet reactivity remained within the range of ASA-free controls, despite successful ASA-treatment, as shown by blockade of TX formation. Importantly, the proportion of patients with sufficient inhibition correlated with the ASA regimen, indicated as “% of patients with sufficient inhibition”. When the mean recruitment of all patients was calculated for each ASA regimen, significant inhibition was detected in all instances when compared to ASA-free controls (* p< 0.001). Importantly, the L+100−BID treatment was the most favorable, and resulted in significantly lower recruitment († p< 0.05) than any other regimen. Mean and SEM of the recruitment activity for each group is indicated; n=total number of patients in each group. ANOVA plus Duncan’s test was used for these statistical analyses.
3. Serotonin Release from Platelets of Aspirin-treated Patients with Vascular Disease. Effects of Different Aspirin Regimens
Serotonin (14C-5HT) release is a biochemical marker for platelet activation. All aspirin regimens reduced serotonin release in PRP, PRP+RBC and WB, as compared to the aspirin-free control group, although the extent of inhibition varied (Figure 3).
Figure 3. Effect of different ASA regimens on platelet 5HT release.
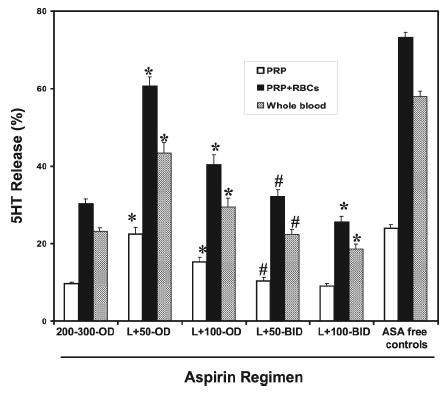
14C-5HT radiolabeled PRP, PRP plus RBC (Hct 40%) or WB was stimulated with collagen (1 μg/mL) and 5HT release was evaluated in cell-free supernatants (Methods). Patients were treated with different ASA regimens: L+50−OD (n=31); L+100−OD (n=33); L+50−BID (n=77); L+100−BID (n=95); 200/300 mg OD (n=206); and compared to ASA-free normal controls (n=84). ASA reduced 5HT release in PRP, PRP+RBC and WB vs. ASA-free (p< 0.05) in all instances. Twice-daily ASA produced greater inhibition (L+50−BID vs. L+100−OD; # p< 0.05). While comparable inhibition was observed with the L+50−BID, L+100−BID and 200/300 mg OD regimens in PRP, inhibition in PRP+RBC and WB was significantly greater with L+100−BID (*p< 0.05) due to a reduction in the ASA-sensitive prothrombotic effect of RBC. ANOVA plus Duncan’s test was used for the statistical analyses.
In platelet-erythrocyte suspensions or WB, administration of the L+50−OD and L+100−OD regimens was less inhibitory for 14C-5HT release than 200/300 mg OD. Interestingly, L+50−BID administration equaled the effect of 200/300 mg OD. Serotonin release was significantly lowered by the same aspirin dose administered twice daily (L+50−BID vs. L+100−OD), and was even lower with L+100−BID (Figure 3). Importantly, while 14C-5HT release in platelets alone was similar in L+50−BID, L+100−BID and 200/300 mg OD, it was significantly lower with L+100−BID in the presence of erythrocytes (PRP+RBC or WB, Figure 3), implying greater down-regulation by ASA of the prothrombotic effect of RBC. In all instances, 14C-5HT release in WB was lower than in PRP+RBC, in all probability due to the inhibitory effect of neutrophils6.
DISCUSSION
The present data indicate that administration of 200/300 mg ASA OD is insufficient for optimal inhibition of platelet recruitment in the presence of RBC or WB in a high proportion of patients with vascular disease (51 and 41% respectively - Figure 1). This is in agreement with our previous research 11. The extent of recruitment is strongly modulated by platelet-erythrocyte interactions3,8, in a manner which is partially inhibitable by ASA10,11. Therefore, we studied ASA regimens that would inhibit platelet COX-1-dependent responses as well as the ASA-inhibitable RBC effect in patients with vascular diseases. This would enhance down-regulation of platelet reactivity in the physiological milieu of WB. This had previously been achieved in normal subjects with an intermittent loading dose (500 mg) at 2-week intervals supplemented with 50 mg ASA OD10. In sharp contrast to the reduction in platelet responsiveness observed in normal subjects10, the L+50−OD or L+100−OD regimens resulted in a significant increase in the number of patients with insufficient inhibition of platelet recruitment. The latter was evaluated in PRP alone, in PRP plus RBC, and in whole blood, and compared to 200/300 mg OD (Figure 1). We observed that 87% (L+50−OD) and 58% (L+100−OD) of patients had insufficient inhibition of platelet recruitment in WB (Figure 1). This paralleled results with combined PRP plus RBC (Figure 1) and increased 14C-5HT release (Figure 3). These less than optimal ex vivo results may be clinically relevant because 81–100 mg ASA OD is widely used for secondary prevention of occlusive vascular diseases. These patients may have had a greater proportion of activated platelets in their circulation, and may represent a group of patients at increased risk for a vascular event.
The difference in optimal dosage with normal subjects10 in all probability reflects greater platelet reactivity in patients. This could be a consequence of underlying vascular disease or increased platelet turnover12, which results in a greater number of ASA-free platelets into the circulation. The latter is certainly a contributory factor as indicated by the strong reduction in percentage of patients with insufficient inhibition with L+50−BID compared to L+100−OD, especially in PRP alone (14.1% vs. 48.5%), but also in the presence of erythrocytes: PRP+RBC (47.4 vs. 72%) or WB (39 vs. 57.6%) (Figure 1). This confirms our previous experimental data in which 10% of ASA-free platelets (the estimated daily platelet turnover) mixed with autologous ASA-treated platelets and RBC induced a marked increase in platelet recruitment8. A similar phenomenon was demonstrated during induction of experimental thrombosis in animals in vivo13. Furthermore, newly formed platelets with an active cyclooxygenase are detectable as early as 4 hours after ASA ingestion14. Therefore, ensuring that circulating platelets are fully aspirin-treated is critical for optimal control of platelet reactivity.
Increasing the daily dose from L+50−BID to L+100−BID did not modify the percentage of patients with insufficient inhibition of recruitment evaluated in PRP alone (13–14%, Figure 1) or 14C-5HT release (Figure 3). Since TXA2 synthesis was blocked (Table 2), these platelet responses are COX-independent. As previously shown, such responses can be increased by either ASA-free or ASA-treated RBC8. Importantly, the L+100-BID regimen drastically reduced by 50% the percentage of patients with insufficient inhibition when recruitment was evaluated in PRP plus RBC or WB as compared to other regimens (Figure 1). This demonstrates increased blockade of the ASA-sensitive prothrombotic effect of RBC in the patients by this ASA regimen, which is also shown by a reduction in 14C-5HT release. The enhancing effect of erythrocytes on platelet reactivity may be a contributory factor to the phenomenon known as “aspirin resistance”, defined as a higher degree of platelet reactivity than expected in ASA-treated patients. As demonstrated herein, this could be markedly reduced by altering the ASA regimen. With the L+100−BID treatment, 80% of patients had optimal inhibition in WB. Interestingly, this is achieved with a change in timing of ASA administration, rather than in the total amount of ASA ingested. If we calculate the loading dose (500 mg) plus the daily dose (100 mg BID for 14 days), the ASA ingested is equivalent to 221 mg/day in each cycle, within the range (75–325 mg) of ASA recommended for treatment15–17. Remarkably, with the L+50−BID regimen (equivalent to an average of 129 mg/day in the 14-day cycle), the proportion of patients with optimal inhibition was similar to the 200/300 mg OD regimen, with only about half the amount of ASA administered.
We did not further increase the aspirin dose in the remaining 20% of patients who did not achieve optimal inhibition with the L+100−BID regimen in WB (Figure 1). Of those patients, optimal control was not achieved in 13% due to responsiveness in platelets alone (Figure 1). In the remaining 7%, platelets were inhibited, but blockade of the ASA-sensitive prothrombotic effect of RBC was not fully achieved. This resulted in an increased response in WB (Figure 1). Since TXA2 generation was blocked (Table 2), the platelet responses were elicited by mechanisms independent of COX-1 and/or COX-2. These would include formation of aspirin-insensitive eicosanoids such as 8-iso-PGF2α18, protein tyrosine phosphorylation4, other signal transduction mechanisms or genetic variations in platelet proteins19.
Platelet activation as evaluated by 14C-5HT secretion from dense granules was increased by RBC and reduced by ASA (Figure 3), as previously reported3,10,11. The reduction was greatest in patients treated with the L+100−BID regimen and as a result, 5HT release was lowest in the presence of RBC or in WB in this group of patients. Reduction of released 5HT from activated platelets (a vasoconstrictor and platelet agonist20) reflects a reduction in the detrimental effects of continuously elevated platelet reactivity.
We observed an individual variability of aspirin effects in patients depending on the specific dose-regime and the prothrombotic effect of RBC. However, in our patient population, all ASA regimes significantly reduce platelet activation (5HT, TXA2) and recruitment. The latter is in accordance with the beneficial effect of aspirin at any dose found in a recent meta-analysis of ASA clinical trials1, resulting from the fact that ASA always reduces platelet reactivity to some extent. However, composite data from such studies may tend to mask individual variability in responsiveness to the drug23.
A correlation between the effect of aspirin on platelet reactivity ex vivo and clinical outcomes was reported by others5,21,22. Thus, the parallel blockade of platelet reactivity and the aspirin-sensitive component of the prothrombotic effect of erythrocytes in a large proportion of patients with vascular disease may enhance the clinical benefit of aspirin treatment.
In conclusion, the administration of an intermittent biweekly dose of 500 mg ASA along with 100 mg ASA every 12 hours provides optimal inhibition of platelet recruitment in WB in 80% of patients with previous cardiovascular or cerebrovascular events. This reduced by 50% the proportion of patients who did not achieve optimal inhibition in WB from 41% on a regimen of 200 or 300 mg ASA OD to 20% on our L+100−BID regimen. Patients not achieving optimal inhibition may require additional platelet inhibitory therapy, alone or in combination with ASA. Nevertheless, further studies would be necessary concerning the association between ex vivo platelet reactivity and clinical events.
Acknowledgments
We thank Dr V. Martínez-Sales for TXA2 determinations. The technical assistance of M.C. Insa, C. Olmeda and C. Peña is gratefully acknowledged.
Footnotes
MTS and JV contributed equally to this study
Supported in part by grants from the Spanish Fondo de Investigaciones Sanitarias FIS01/1208 (MTS, JV, JA, ES), FIS03/0270 (JV, MTS, AL) FISC03/06, Agencia Valenciana de Ciencia y Tecnologia AVCiT-G03/005 (MTS, JV, AL, JC), NIH grants HL 47073, HL 46403, and NS 41462 (AJM, MJB.), and Merit Review grants from the Department of Veterans Affairs (AJM, MJB).
References
- 1.Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomized trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br Med J. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcus AJ, Broekman MJ, Drosopoulos JHF, Islam N, Pinsky DJ, Sesti C, et al. Heterologous cell-cell interactions. thromboregulation, cerebroprotection and cardioprotection by CD39 (NTPDase-1) J Thromb Haemost. 2003;1:2497–2509. doi: 10.1111/j.1538-7836.2003.00479.x. [DOI] [PubMed] [Google Scholar]
- 3.Santos MT, Valles J, Marcus AJ, Safier LB, Broekman MJ, Islam N, et al. Enhancement of platelet reactivity and modulation of eicosanoid production by intact erythrocytes. J Clin Invest. 1991;87:571–80. doi: 10.1172/JCI115032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santos MT, Moscardo A, Valles J, Martinez M, Piñon M, Aznar J, et al. Participation of tyrosine phosphorylation in cytoskeletal reorganization,αIIbβ3 integrin receptor activation and aspirin-insensitive mechanisms of thrombin-stimulated human platelets. Circulation. 2000;102:1924–30. doi: 10.1161/01.cir.102.16.1924. [DOI] [PubMed] [Google Scholar]
- 5.Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–5. doi: 10.1161/01.cir.0000013777.21160.07. [DOI] [PubMed] [Google Scholar]
- 6.Valles J, Santos MT, Marcus AJ, Safier LB, Broekman MJ, Islam N, et al. Down-regulation of human platelet reactivity by neutrophils. Participation of lipoxygenase derivatives and adhesive proteins. J Clin Invest. 1993;92:1357–65. doi: 10.1172/JCI116709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Farre A, Caramelo C, Esteban A, Alberola ML, Millas I, Monton M, et al. Effects of aspirin on platelet-neutrophil interactions: Role of nitric oxide and endothelin-1. Circulation. 1995;91:2080–8. doi: 10.1161/01.cir.91.7.2080. [DOI] [PubMed] [Google Scholar]
- 8.Valles J, Santos MT, Aznar J, Marcus AJ, Martinez-Sales V, Portoles M, et al. Erythrocytes metabolically enhance collagen-induced platelet responsiveness via increased thromboxane production, ADP release, and recruitment. Blood. 1991;78:154–62. [PubMed] [Google Scholar]
- 9.Valles J, Santos MT, Aznar J, Martinez M, Moscardo A, Pinon M, et al. Platelet-erythrocyte interactions enhance αIIbβ3 integrin receptor activation and P-selectin expression during platelet recruitment: down-regulation by aspirin ex vivo. Blood. 2002;99:3978–84. doi: 10.1182/blood.v99.11.3978. [DOI] [PubMed] [Google Scholar]
- 10.Santos MT, Valles J, Aznar J, Marcus AJ, Broekman MJ, Safier LB. Prothrombotic effects of erythrocytes on platelet reactivity: reduction by aspirin. Circulation. 1997;95:63–8. doi: 10.1161/01.cir.95.1.63. [DOI] [PubMed] [Google Scholar]
- 11.Valles J, Santos MT, Aznar J, Osa A, Lago A, Cosin J, et al. Erythrocyte promotion of platelet reactivity decreases the effectiveness of aspirin as an antithrombotic therapeutic modality: the effect of low-dose aspirin is less than optimal in patients with vascular disease due to prothrombotic effects of erythrocytes on platelet reactivity. Circulation. 1998;97:350–5. doi: 10.1161/01.cir.97.4.350. [DOI] [PubMed] [Google Scholar]
- 12.Dale GL. Platelet kinetics. Curr Opin Hematol. 1997;4:330–4. doi: 10.1097/00062752-199704050-00006. [DOI] [PubMed] [Google Scholar]
- 13.Cerskus AL, Ali M, Davies BJ, McDonald JVD. Possible significance of small numbers of functional platelets in a population of aspirin-treated platelets in vitro and in vivo. Thromb Res. 1980;18:389–97. doi: 10.1016/0049-3848(80)90334-5. [DOI] [PubMed] [Google Scholar]
- 14.Di Minno G, Silver MJ, Murphy S. Monitoring the entry of new platelets into the circulation after ingestion of aspirin. Blood. 1983;61:1081–5. [PubMed] [Google Scholar]
- 15.Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101:1206–18. doi: 10.1161/01.cir.101.10.1206. [DOI] [PubMed] [Google Scholar]
- 16.Smith SC, Jr, Blair SN, Bonow RO, Brass LM, Cerqueira MD, Dracup K, et al. AHA/ACC Scientific Statement: AHA/ACC guidelines for preventing heart attack and death in patients with atherosclerotic cardiovascular disease: 2001 update: A statement for healthcare professionals from the American Heart Association and the American College of Cardiology. Circulation. 2001;104:1577–9. doi: 10.1161/hc3801.097475. [DOI] [PubMed] [Google Scholar]
- 17.Coull BM, Williams LS, Goldstein LB, Meschia JF, Heitzman D, Chaturvedi S, et al. Anticoagulants and antiplatelet agents in acute ischemic stroke: report of the Joint Stroke Guideline Development Committee of the American Academy of Neurology and the American Stroke Association (a division of the American Heart Association) Stroke. 2002;33:1934–42. doi: 10.1161/01.str.0000028456.18614.93. [DOI] [PubMed] [Google Scholar]
- 18.Patrono C, Fitzgerald GA. Isoprostanes: potential markers of oxidant stress in atherothrombotic disease. Arterioscler Thromb Vasc Biol. 1997;17:2309–15. doi: 10.1161/01.atv.17.11.2309. [DOI] [PubMed] [Google Scholar]
- 19.Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nat Rev Drug Discov. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 20.Crowley ST, Dempsey EC, Horwitz KB, Horwitz LD. Platelet-induced vascular smooth muscle cell proliferation is modulated by the growth amplification factors serotonin and adenosine diphosphate. Circulation. 1994;90:1908–18. doi: 10.1161/01.cir.90.4.1908. [DOI] [PubMed] [Google Scholar]
- 21.Gum PA, Kottke-Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–5. doi: 10.1016/s0735-1097(02)03014-0. [DOI] [PubMed] [Google Scholar]
- 22.Grotemeyer KH, Scharafinski HW, Husstedt IW. Two-year follow-up of aspirin responder and aspirin non responder. A pilot-study including 180 post-stroke patients. Thromb Res. 1993;71:397–403. doi: 10.1016/0049-3848(93)90164-j. [DOI] [PubMed] [Google Scholar]
- 23.Schafer AI. Genetic and acquired determinants of individual variability of response to antiplatelet drugs. Circulation. 2003;108:910–11. doi: 10.1161/01.CIR.0000088843.52678.8A. [DOI] [PubMed] [Google Scholar]