

Abstract
The mechanisms leading to prostate cancer metastasis are not understood completely. Although there is evidence that the CXC chemokine receptor (CXCR) 4 and its ligand CXCL12 may regulate tumor dissemination, their role in prostate cancer is controversial. We examined CXCR4 expression and functionality, and explored CXCL12-triggered adhesion of prostate tumor cells to human endothelium or to extracellular matrix proteins laminin, collagen, and fibronectin. Although little CXCR4 was expressed on LNCaP and DU-145 prostate tumor cells, CXCR4 was still active, enabling the cells to migrate toward a CXCL12 gradient. CXCL12 induced elevated adhesion to the endothelial cell monolayer and to immobilized fibronectin, laminin, and collagen. Anti-CXCR4 antibodies or CXCR4 knock out significantly impaired CXCL12-triggered tumor cell binding. The effects observed did not depend on CXCR4 surface expression level. Rather, CXCR4-mediated adhesion was established by α5 and β3 integrin subunits and took place in the presence of reduced p38 and p38 phosphorylation. These data show that chemoattractive mechanisms are involved in adhesion processes of prostate cancer cells, and that binding of CXCL12 to its receptor leads to enhanced expression of α5 and β3 integrins. The findings provide a link between chemokine receptor expression and integrin-triggered tumor dissemination.
Keywords: Adhesion, CXCR4, CXCL12, integrins, prostate carcinoma cells
Introduction
Chemokines are a family of low-molecular-weight (8–10 kDa) proinflammatory cytokines that bind to G-protein-coupled receptors. Their primary functions are chemoattraction and activation of specific leukocytes in diverse immunoinflammatory responses. However, increasing evidence suggests that they also play key roles in neoplastic transformation and passage of tumor cells through the endothelial vessel wall and extracellular matrix. Among the chemokines and chemokine receptors identified to date, the membranous CXC chemokine receptor 4 (CXCR4) and its ligand stromal-derived factor-1 (SDF-1, synonymous to CXCL12) are thought to play a central role in regulating the metastasis of many solid tumors, including those of the lung, breast, and kidney.
The extent to which the CXCR4-CXCL12 axis is involved in prostate cancer, the most common nondermatologic malignancy worldwide, is still not clear. There is evidence that high expression levels of CXCR4 and positive staining for its ligand CXCL12 might correlate with the presence of metastatic disease in prostate cancer patients [1,2]. The binding of secreted CXCL12 to CXCR4 at the tumor cell surface is assumed to activate the cellular motor machinery and to trigger tumor migration from the blood vessel into the target tissue. Nevertheless, the hypothesis of CXCR4-driven prostate tumor cell adhesion has not yet been proven. Whether the CXCR4 surface level in fact correlates with tumor aggressiveness has not been established. Tumor cells isolated from patients with prostate carcinoma compared to benign prostatic hyperplasia have shown different migratory capacities in an invasion chamber model, although the amount of CXCR4-expressing cells did not differ between the groups [3]. In a similar model, the same number of PC3 and LNCaP prostate carcinoma cells moved toward a chemotactic CXCL12 gradient, although fluorescence-activated cell sorter (FACS) analysis revealed significantly higher levels of cell surface CXCR4 on LNCaP when compared to PC3 [2,4]. In contrast to this, Darash-Yahana et al. [5] detected no or very low CXCR4 surface expression on PC3 and LNCaP cell lines. Both cell types did not respond to CXCL12. Immunohistochemical evaluation of PC-3 cells demonstrated uniform cytoplasmic—but no surface—CXCR4 staining [6], and the analysis of prostate specimen from primary prostate cancer sections and prostate bone metastases has not revealed CXCR4 surface localization [3,7].
These findings might conflict with the idea that prostate tumor cell adhesion and migration are mediated by CXCL12-CXCR4 interaction. If the concept of CXCR4-mediated tumor invasion is valid, we should expect functionally active CXCR4 receptors along the tumor cell membrane. In this study, we investigated whether the membrane of prostate tumor cells is occupied by CXCR4. In the next step, the involvement of CXCR4 in tumor cell adhesion and migration processes was evaluated.
Using DU-145 and LNCaP prostate tumor cells as culture model, we have demonstrated that CXCR4 was expressed on the cell surface, although to a very low extent. The receptor amount was sufficient to respond to a CXCL12 stimulus, indicating that migratory activity does not depend on receptor quantity. We demonstrate, for the first time, that CXCR4 engagement triggers tumor cell adhesion to endothelial cells as well as to extracellular matrix proteins. CXCR4 does not act as an anchoring molecule that allows firm cellular attachment, but rather as a signaling receptor that activates α5 and β3 integrin subunits. Moreover, we demonstrate that the process of CXCR4 stimulation by CXCL12 is accompanied by the downregulation of p38 MAPK and p38 MAPK phosphorylation.
Materials and Methods
Chemokines and Antibodies
Human CXCL12 was purchased from Strathmann (Amsterdam, The Netherlands), whereas phycoerythrin (PE)-conjugated monoclonal antibodies against CXCR4 (IgG2a; clone 12G5) and CXCR3 (IgG1; clone 49801.111) were purchased from R&D Systems (Wiesbaden, Germany). Anti-ERK1 (clone MK12), phospho-specific anti-ERK1/2 (pT202/pY204; clone 20A), anti-JNK (clone 37), phospho-specific anti-JNK (pT183/pY185; clone 41), anti-p38 (clone 27), and phospho-specific anti-p38 (pT180/pY182; clone 30) monoclonal antibodies were obtained from BD Biosciences (Heidelberg, Germany). Integrin-linked kinase (ILK; clone 3), focal adhesion kinase (FAK; clone 77), and phospho-specific FAK (pY397; clone 18) were purchased from BD Biosciences. Anti-β-actin monoclonal antibody was obtained from Sigma (Taufenkirchen, Germany).
Cell Cultures
DU-145 and LNCaP prostate carcinoma cells were purchased from DSMZ (Braunschweig, Germany). Tumor cells were grown and subcultured in RPMI 1640 medium (Seromed, Berlin, Germany) supplemented with 10% fetal calf serum (FCS), 100 IU/ml penicillin, and 100 µg/ml streptomycin at 37°C in a humidified 5% CO2 incubator.
Human umbilical vein endothelial cells (HUVEC) were isolated and harvested by enzymatic treatment with chymotrypsin. HUVEC were grown in Medium 199 (Biozol, Munich, Germany), 10% FCS (Gibco, Karlsruhe, Germany), 10% pooled human serum (Blood Bank of The German Red Cross, Frankfurt am Main, Germany), 20 µg/ml endothelial cell growth factor (Boehringer Mannheim, Mannheim, Germany), 0.1% heparin (Roche, Basel, Switzerland), 100 ng/ml gentamycin (Gibco), and 2% 1 M HEPES buffer (Seromed). To control the purity of HUVEC cultures, cells were stained with fluorescein isothiocyanate-labeled monoclonal antibody against factor VIII-associated antigen (von Willebrand factor; clone F8/86; Dako, Hamburg, Germany) and analyzed microscopically or by FACScan [FL-1H (log) channel histogram analysis, 1 x 104 cells/scan; BD Biosciences]. Cell cultures with purity >95% were serially passaged. Subcultures from passages 2 to 4 were selected for experimental use.
Transfection of Tumor Cells with Small Interfering RNA (siRNA)
siRNA was constructed and directed against CXCR4 [gene accession no. NM_003467; sense: r(GCA GUC CAU GUC AUC UAC A)dTdT; antisense: r(UGU AGA UGA CAU GGA CUG C)dCdT]. LNCaP or DU-145 cells were transfected at 70% confluence with 8 nM siRNA using RNAiFect transfection reagent (Qiagen, Hilden, Germany). Optimum transfection was achieved in RPMI 1640 medium supplemented with 5% FCS and a 1:6 siRNA/RNAiFect ratio. The viability of tumor cells was assessed by propidium iodide double-stranded DNA intercalation or quantitative fluorescence analysis of enzyme-catalyzed fluorescein-diacetate metabolism.
Tumor Cell Adhesion and Migration
HUVEC were transferred to six-well multiplates (Falcon Primaria; BD Biosciences) in complete HUVEC medium. When confluency had been reached, DU-145 or LNCaP cells were detached from culture flasks by accutase treatment (PAA Laboratories, Cölbe, Germany), and 0.5 x 106 cells were then added to the HUVEC monolayer for 60 minutes. Subsequently, nonadherent tumor cells were washed off using warmed (37°C) Medium 199. The remaining cells were fixed with 1% glutaraldehyde.
Cell migration toward CXCL12 was examined using six-well Transwell chambers (Greiner, Frickenhausen, Germany) with 8-mm pores. DU-145 or LNCaP cells were removed from culture flasks and resuspended at 0.5 x 106 cells/ml in a serum-free culture medium. CXCL12 (0–500 ng/ml) was placed in lower wells. Test cells were then placed in the upper chamber for 60 minutes. After incubation, the upper surface of the Transwell membrane was wiped gently with a cotton swab to remove nonmigrating cells. Cells that migrated to the lower surface of the membrane were stained using hematoxylin.
In each experimental setting, adherent or migrated tumor cells were counted in five different fields of a defined size (5 x 0.25 mm2) using a phase-contrast microscope, and the mean cellular adhesion/migration rate was calculated. For neutralization studies, cells were pretreated with 20 µg/ml anti-human CXCR4 or anti-human CXCR3 monoclonal antibodies for 60 minutes, or tumor cells were transfected with CXCR4 siRNA and collected 48 hours later. Cells were then applied for adhesion and migration experiments.
Attachment to Extracellular Matrix Components
Six-well plates were coated with collagen [diluted to 100 µg/ml in phosphate-buffered saline (PBS); Seromed], laminin (diluted to 50 µg/ml in PBS; BD Biosciences), or fibronectin (diluted to 50 µg/ml in PBS; BD Biosciences) overnight. Plastic dishes served as background control. Plates were washed with 1 % bovine serum albumin (BSA) in PBS to block nonspecific cell adhesion. Thereafter, 0.5 x 106 tumor cells/well were added for 60 minutes. Subsequently, nonadherent tumor cells were washed off, and the remaining adherent cells were fixed with 1% glutaraldehyde and counted microscopically. The mean cellular adhesion rate (adherent cellscoated well - adherent cellsbackground) was calculated from five observation fields.
Tumor Cell Binding to Immobilized Receptor Protein Chimeras
Chimeric receptor globulins were constructed as described previously [8]. Proteins containing the extracellular domain of E-selectin, ICAM-1, VCAM-1, or P-selectin were expressed in COS-7 cells. COS-7 cells were transfected with 3 mg of plasm id DNA using the DEAE/dextran method. Seven days after transfection, supernatants were collected and stored at -20°C. The concentration of the receptor globulin chimeras was determined by enzyme-linked immunosorbent assay using a monoclonal rat anti-human IgG antibody conjugated to peroxidase. Round culture dishes (Falcon Primaria; BD Biosciences) were incubated with a spot of 50 µl of goat-anti-human IgG (Sigma) at a concentration of 10 mg/ml in 50 mM Tris, pH 9.5, for 90 minutes. Dishes were washed thrice with PBS (Seromed) and blocked with 1% BSA overnight at 4°C. The dishes were subsequently incubated with 1 ml of cell culture supernatant, containing 5 µg/ml E-selectin, ICAM-1, VCAM-1, or P-selectin IgG fusion protein for 30 minutes at 20°C. Dishes were then washed thrice, and tumor cells were resuspended at a density of 0.5 x 106 cells/ml in binding buffer for 30 minutes [9] and transferred to culture dishes. Thereafter, nonadherent cells were washed off, and the remaining cells were counted using a phase-contrast microscope. Five observation fields were chosen at random in each dish, and the mean value of the number of adherent cells per field was calculated.
Evaluation of CXCR4 Surface Expression
DU-145 or LNCaP cells were detached from culture flasks by accutase treatment, washed in blocking solution (PBS and 0.5% BSA), and then incubated for 60 minutes at 4°C with PE-labeled anti-CXCR4 monoclonal antibody. To separately analyze intracellular CXCR4 content, cells were fixed and permeabilized (Fix & Perm; Biozol-An der Grub Bioresearch, Eching, Germany) before adding the monoclonal antibody. CXCR4 expression on tumor cells was then measured using FACScan [FL-2H (log) channel histogram analysis; 1 x 104 cells/scan] and expressed as mean fluorescence units (MFU). Mouse IgG2a-PE (Cymbus Biotechnology, Hofheim, Germany) was used as isotype control.
To explore CXCR4 localization, tumor cells were transferred to round cover slips, which were placed in a 24-well multiplate. On reaching confluency, cell cultures were washed twice with PBS (with Ca2+ and Mg2+) and then fixed in cold (-20°C) methanol/acetone (60/40 vol/vol). Subsequently, cells were washed again with PBS (without Ca2+ and Mg2+) and afterward washed once with blocking buffer (0.5% BSA in PBS without Ca2+ and Mg2+). After removing the washing buffer, cells were incubated for 60 minutes with PE-conjugated anti-CXCR4 monoclonal antibody. To prevent photobleaching of the fluorescent dye, cover glasses with stained cells were taken out of the wells, and residual liquid was removed. The cells were then embedded in an antifade reagent/mounting medium mixture (ProLong Antifade Kit; MoBiTec, Göttingen, Germany) and mounted on slides. The slides were viewed using a confocal laser-scanning microscope (LSM 10; Zeiss, Jena, Germany) with a plan neofluar x 100/1.3 oil immersion objective.
Western Blot Analysis
CXCR4. Total CXCR4 content was evaluated by Western blot analysis. DU-145 or LNCaP cell lysates were applied to a 7 % polyacrylamide gel and electrophoresed for 90 minutes at 100 V. The protein was then transferred to nitrocellulose membranes. After blocking with nonfat dry milk for 1 hour, the membranes were incubated overnight with anti-CXCR4 antibody (dilution 1:100). HRP-conjugated goat-anti-mouse IgG (dilution 1:5000; Upstate Biotechnology, Lake Placid, NY) served as secondary antibody. The membranes were briefly incubated with ECL detection reagent (ECL; GE Healthcare, Freiburg, Germany) to visualize the proteins and were exposed to an X-ray film (Hyperfilm EC; Amersham).
Integrins and signaling proteins. Cell lysates were prepared from unstimulated cells or after stimulation with 500 ng/ml CXCL12. Western blot analysis was performed using the following monoclonal antibodies: anti-integrin β1 (1: 2500), anti-integrin β3 (1:2500), anti-integrin β4 (1:250), anti-integrin α2/VLA2α (1:250), anti-integrin α5 (1:5000), anti-integrin αL/LFA-1α (1:500), and anti-integrin αV (1:250) (all from BD Biosciences). Intracellular signaling cascade was evaluated using appropriate monoclonal antibodies that recognize the phosphorylated form of the proteins or total proteins (see above).
mRNA Expression of CXCR4
mRNA expression of CXCR and CXCL was evaluated by reverse transcriptase polymerase chain reaction (RT-PCR). Tumor cells were seeded in 50-ml culture flasks (growth area, 25 cm2; Falcon Primaria). Total RNA was extracted using RNeasy kit (Qiagen), and RNA samples were then treated with 80 U/ml RNAse-free DNAse I (Boehringer Mannheim) for 60 minutes at 37°C to eliminate amplifiable contaminating genomic DNA. Subsequently, samples were incubated for 10 minutes at 65°C to inactivate DNAse. Complementary DNA was synthesized from 1 µg of total RNA per sample with a 60-minute incubation at 42°C, using the Moloney murine leukemia virus reverse transcriptase (Invitrogen, Karlsruhe, Germany) and oligo-(dT) priming (Boehringer Mannheim). Amplification was carried out using gene-specific primers and Platinum-Taq polymerase (Invitrogen) in a Mastercycler Gradient thermocycler (Eppendorf, Hamburg, Germany). The primer sequences for CXCR4 were as follows: 5′ GGTGGTCTATGTTGGCGTCT 3′ (sense) and 5′ TGGAGTGTGACAGCTTGGAG 3′ (antisense). Internal controls for the RT-PCR reaction were performed by running parallel reaction mixtures with the housekeeping gene GAPDH: 5′ ATCTTCCAGGAGCGAGATCC 3′ (sense) and 5′ ACCACTGACACGTTGGCAGT 3′ (antisense). Reactions were performed in the presence of 0.5 µl of cDNA, with an initial incubation step at 94°C for 5 minutes. Cycling conditions consisted of denaturation at 94°C for 60 seconds, annealing at 60°C for 60 seconds, and extension at 72°C for 60 seconds over 35 cycles. The reaction was completed by another 10-minute incubation step at 72°C. PCR products were subjected to electrophoresis in 2% agarose gel and visualized by ethidium bromide.
Statistical Analysis
All experiments were performed three to six times. Statistical significance was investigated by the Wilcoxon-Mann-Whitney U test. Differences were considered statistically significant at P < .05.
Results
CXCR4 Expression Profile in DU-145 and LNCaP Cells
To follow the expression pattern of CXCR4 in prostate tumor cells, two different prostate tumor cell lines, DU-145 and LNCaP, were employed. In doing so, the CXCR4 “route”—analysis of the CXCR4-encoding mRNA, cytoplasmic accumulation of CXCR4 proteins, and membrane presentation of CXCR4 receptors—was traced. Strong CXCR4 mRNA activity was observed in DU-145 cells, whereas moderate CXCR4 mRNA activity in LNCaP cells was noted (Figure 1A). Western blot analysis revealed distinct amounts of CXCR4 proteins in DU-145 cells and a lower CXCR4 protein content in LNCaP cells (Figure 1B). The next step involved examining the CXCR4 surface expression level on both cell lines. Histogram plots revealed very limited fluorescence intensity in both cell lines, making data interpretation difficult (Figure 1C). To verify the specificity and integrity of the anti-CXCR4 antibody, experiments were repeated using HUVEC cultures as positive controls. According to earlier data [10,11], distinct amounts of CXCR4 have been detected on HUVEC (Figure 2A). In addition, DU-145 and LNCaP cells were permeabilized and then marked again with the anti-CXCR4 antibody. The procedure resulted in enhanced fluorescence signals, demonstrating high amounts of intracellular CXCR4 proteins (Figure 2, B and C), concordant with Western blot results. Thus, the integrity of the antibodies used has been proven, and we conclude that CXCR4 receptors are present on the tumor cell membrane, although at a very low level. Confocal microscopy of DU-145 cells showed intracellular localization of CXCR4, but also weak receptor accumulation along intercellular boundaries (Figure 3).
Figure 1.
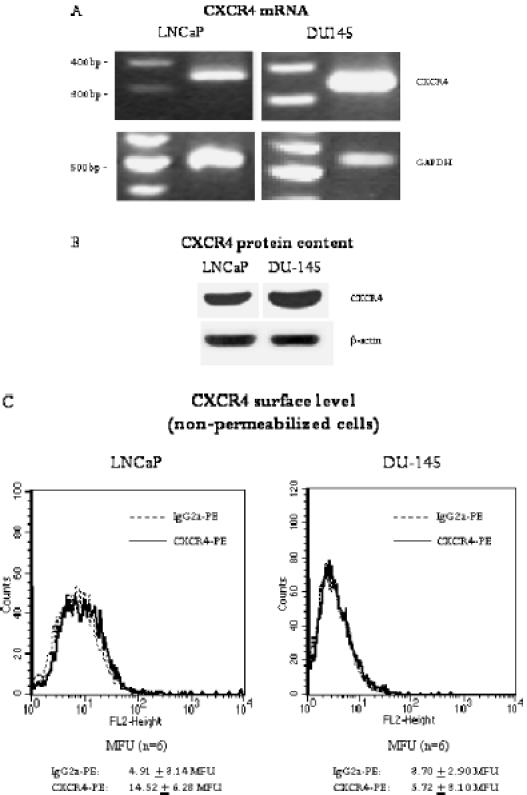
Expression of CXCR4 in LNCaP and DU-145 prostate tumor cells. (A) PCR analysis demonstrates strong CXCR4 mRNA expression in DU-145 cells and moderate CXCR4 mRNA expression in LNCaP cells (fragment length, 346 bp). Internal control for the RT-PCR reaction was performed by running parallel reaction mixtures with the housekeeping gene GAPDH (fragment length, 509 bp). The figure shows one of four representative experiments. (B) Western blot analysis of CXCR4 in LNCaP and DU-145 tumor cells. The monoclonal antibody clone 12G5 was used to recognize CXCR4. β-Actin served as internal control. One of three representative experiments is shown. (C) Fluorescence analysis of CXCR4 surface expression. A PE-conjugated monoclonal antibody anti-CXCR4, clone 12G5, was used to analyze CXCR level. A mouse IgG2a-PE served as isotype control. Fluorescence was analyzed using a FACScan flow cytometer, and a histogram plot (FL2, height) was generated to show PE fluorescence. Fluorescence was expressed as MFU. The mean values of MFU from six experiments are given below each representative histogram.
Figure 2.

Integrity of anti-CXCR4 monoclonal antibodies. HUVEC were used as positive controls, and CXCR4 surface expression of unfixed cells was evaluated by the PE-conjugated monoclonal antibody anti-CXCR4 clone 12G5 (A). Mouse IgG2a-PE served as isotype control. In the second part, DU-145 (B) or LNCaP (C) cells were permeabilized, and fluorescence analysis of intracellular CXCR4 was carried out thereafter. Each figure demonstrates a significant fluorescence shift after labeling the cells with CXCR4-PE. One of three representative experiments is shown. The mean values of MFU from six experiments are also given.
Figure 3.

Confocal analysis of CXCR4 distribution. DU-145 tumor cells were grown in standard medium. Unconjugated monoclonal antibody clone 12G5 was used to analyze CXCR4. Indocarbocyanine (Cy3)-conjugated goat-anti-mouse IgG was added as secondary antibody. The figure shows distinct CXCR4 expression at intercellular boundaries (arrows) and strong intracellular accumulation (scale, 10 µM; original magnification, x 100/1.3 oil immersion objective).
Functionality of CXCR4 Receptor
Migration experiments were carried out to test whether the few CXCR4 receptors detected on the prostate tumor cell membrane are functionally active. Dose-response analysis revealed a strong chemotactic activity of both DU-145 and LNCaP cells, which was maximal when 500 ng/ml CXCL12 was applied (data not shown). Therefore, we used this concentration in subsequent neutralization studies.
The number of LNCaP and DU-145 cells migrating in response to CXCL12 was significantly higher than that for cells not exposed to CXCL12 as a chemoattractant. CXCL12-dependent chemotaxis was neutralized by treatment with the anti-CXCR4 antibody, but not with anti-CXCR3 antibody (Figure 4). Tumor cells in which CXCR4 had been knocked down by siRNA did not respond to a CXCL12 stimulus, whereas cells treated with scrambled siRNA responded (Figure 4). Nonresponding cells remained viable, as confirmed by propidium iodide double-stranded DNA intercalation or quantitative fluorescence analysis of enzyme-catalyzed fluorescein-diacetate metabolism. These experiments demonstrated that CXCR4 is functionally active and that CXCL12 specifically acts on CXCR4.
Figure 4.

CXCR4 expressed on DU-145 and LNCaP cells is functionally active. Tumor cell migration toward CXCL12 was assessed in a Transwell chamber assay. DU-145 or LNCaP cells were seeded in the upper chamber, and 500 ng/ml CXCL12 was placed in the lower well. Cells that migrated to the lower surface of the membrane were stained by hematoxylin and counted. In control experiments, a medium without CXCL12 was used. Statistical significance was investigated by the Wilcoxon-Mann-Whitney U test. To demonstrate CXCR4 dependence, tumor cells whose CXCR4 was blocked by monoclonal antibodies or whose CXCR4 was knocked down by siRNA were also applied in parallel experiments. Scrambled siRNA or nonspecific IgG served as controls. Knockdown was controlled 48 hours after RT-PCR and Western blot analysis (right panel). One of six representative experiments is shown. *Significantly different from controls; #significantly different from nontreated cells moving toward CXCL12.
CXCR4-Driven Adhesion to Endothelial Cells and Extracellular Matrix
DU-145 or LNCaP cells strongly attached to HUVEC (DU-145 > LNCaP) after 60 minutes (Figure 5), whereas the adhesion rate of CXCR4 siRNA-transfected cells was significantly reduced. Tumor cells that were treated with scrambled siRNA attached to HUVEC to a similar extent as nontreated control cells.
Figure 5.

Adhesion of DU-145 or LNCaP cells to HUVEC depends on CXCR4. Control tumor cells, tumor cells treated with scrambled siRNA, or tumor cells whose CXCR4 was knocked down were activated with 500 ng/ml CXCL12 and then added at a density of 0.5 x 106 cells/well to HUVEC monolayers for 60 minutes. Nonadherent tumor cells were washed off in each sample; the remaining cells were fixed and counted in five different fields (5 x 0.25 mm2) using a phase-contrast microscope. Mean values were calculated from five counts. Mean adhesion capacity is depicted as counted cells per square millimeters. One of six representative experiments is shown. **Significantly different from controls (P < .01).
In a similar fashion, the binding of DU-145 or LNCaP cells to extracellular matrix components was also CXCR4-dependent. Figure 6 shows representative data obtained with DU-145 or LNCaP tumor cells. The percentage of adherent cells differed according to the matrix protein used. Maximum adhesion capacity was measured on fibronectin- and laminin-coated plates; a lower binding rate was seen when culture plates were precoated with collagen. Tumor cells that had lost their CXCR4 receptors by siRNA knockdown showed less binding activity than control tumor cells or cells pretreated with scrambled siRNA. The effect was independent of the matrix component used.
Figure 6.
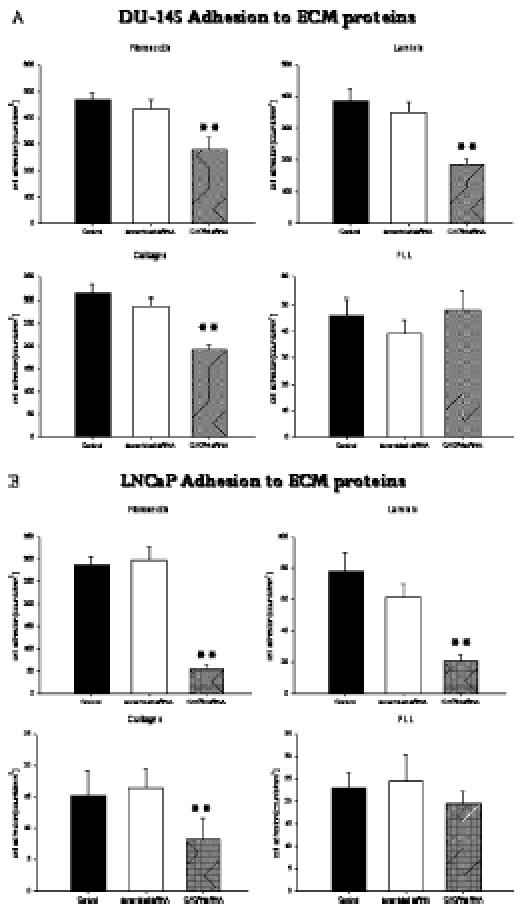
Adhesion of prostate tumor cells to extracellular matrix proteins depends on CXCR4. DU-145 (A) or LNCaP (B) cells treated with scrambled siRNA, or DU-145/LNCaP cells whose CXCR4 was knocked down were activated with 500 ng/ml CXCL12 and then added to immobilized fibronectin, laminin, collagen, or nonspecific poly-l-lysine (PLL) at a density of 0.5 x 106 cells/well for 60 minutes. Nontreated cells served as controls. Nonadherent tumor cells were washed off in each sample; the remaining cells were fixed and counted in five different fields (5 x 0.25 mm2) using a phase-contrast microscope. Mean values were calculated from five counts. Specific adhesion capacity (background adhesion on a plastic surface was subtracted from adhesion to matrix proteins) is depicted as counted cells per square millimeters. One of six representative experiments is shown. **Significantly different from controls (P < .01).
These data, therefore, indicate that the adhesion of prostate tumor cells to the endothelium or the matrix is mediated by CXCR4.
CXCR4 Receptors Serve as Signal Transmitters
The adhesion experiments demonstrated that CXCR4 participates in the interaction of prostate tumor cells with the endothelium or the extracellular matrix. However, they did not explain how CXCR4 contributed to the adhesion process. Two options seemed to be possible: 1) CXCR4 anchors the tumor cells to a specific ligand expressed on endothelial cells and matrix proteins, or 2) CXCR4 modifies further receptors expressed on tumor cells, which then regulate adhesion to endothelial cells and matrix proteins. To assess whether CXCR4 serves as an adhesion receptor itself or if CXCR4 activates further receptors relevant for the adhesion process, the following experimental strategy was employed: Tumor cells were stimulated with CXCL12 and subsequently neutralized with CXCR4 monoclonal antibodies, or they were initially blocked with anti-CXCR4 monoclonal antibodies and subsequently treated with CXCL12. Cells were then allowed to attach to endothelium or matrix proteins. We found that cells treated with CXCL12—before CXCR4—receptors were blocked, and their adhesion and binding rate were enhanced, similar to those cells that were treated with CXCL12 alone (Figure 7, representative of HUVEC and fibronectin). However, when CXCR4 receptors were blocked first, CXCL12 was unable to upregulate cell attachment. Obviously, CXCR4 is not required to attach the tumor cells to their counterparts. Rather, receptor engagement by CXCL12 is the event critical to managing cell adhesion. Therefore, CXCR4 does not directly modulate cell binding, but is necessary to transmit CXCL12-induced signals, which activate adhesion-specific receptors.
Figure 7.

CXCR4 does not anchor prostate tumor cells to HUVEC or matrix proteins, but transmits signals after receptor engagement by CXCL12, which then allows cell adhesion. The study design was created as follows: 1) DU-145 or LNCaP cells were activated with 500 ng/ml CXCL12 (control experiments were carried out without CXCL 12 activation); or 2) DU-145 or LNCaP cells were activated with 500 ng/ml CXCL 12, and CXCR4 was blocked thereafter by monoclonal antibodies (CXCL12-AB); or 3) DU-145 or LNCaP cells were first treated with anti-CXCR4 monoclonal antibodies and then activated with 500 ng/ml CXCL12 (AB-CXCL12). Tumor cells were then added to HUVEC or immobilized fibronectin. Mean adhesion from five different fields (5 x 0.25 mm2) was evaluated after 60 minutes. Adhesion capacity was strongly reduced in DU-145 or LNCaP cells when CXCR4 was blocked before CXCL12 was applied. Adhesion was not reduced when tumor cells were activated with CXCL12 followed by initiation of receptor blockade or when cells were treated with unspecific IgG. One of six representative experiments is shown. **Significantly different from controls (P < .01); ##significantly different from cells activated with CXCL12.
To identify these receptors, DU-145 or LNCaP cells were added to plates precoated with immobilized ICAM-1, VCAM-1, E-selectin, or P-selectin adhesion proteins. The proteins selected are expressed along the vessel wall and are possible candidates for tumor cell/endothelial cell interaction. Prostate cells mainly attached to E-selectin; only a few cells bound to ICAM-1, VCAM-1, or P-selectin (data not shown). The same binding behavior was observed when siRNA-treated tumor cells were used, indicating that CXCR4 does not regulate selectin or ICAM/VCAM-driven processes in our in vitro model.
Remarkably, β3 and α5 integrin subunits became strongly upregulated when prostate tumor cells were stimulated with CXCL12 (Figure 8A). The level of α2 (the expression of which was very low), β1, β4, and αV subunits did not change in CXCL12-treated cells compared to nontreated controls (data not shown). CXCL12 evoked ILK and FAK upregulation and enhanced FAK phosphorylation. Integrins mediated binding to the extracellular matrix and, according to our data, the CXCL12-induced adhesion of DU-145 cells to HUVEC, fibronectin, laminin, or collagen was inhibited by β3- and/or α5-blocking antibodies (Figure 8C). This suggests that CXCR4 alters β3 and α5 integrins and cell-endothelium and cell-matrix adhesion, which is dependent on β3 and α5. This process is specifically attributable to CXCR4 because blocking of CXCR3 receptors did not prevent CXCL12-evoked cell binding (data not shown).
Figure 8.

CXCL12 modulates integrin expression; ILK, FAK, and FAK phosphorylation (FAK phospho); and p38-dependent pathways. (A and B) DU-145 cells were incubated with 500 ng/ml CXCL12 for 4 hours. Cell lysates were analyzed by specific antibodies against integrin β3 or α5. FAK and ILK were examined by mouse IgG1 monoclonal antibodies (1:1000), as indicated in Materials and Methods section. Intracellular signaling cascade was evaluated using appropriate monoclonal antibodies recognizing the phosphorylated form of the p38 protein [p38 (pT180/pY182)] or p38 in total (p38α). β-Actin served as internal control. One of three representative experiments is shown. (C) The CXCL12-mediated adhesion of DU-145 cells to HUVEC or extracellular matrix proteins is β3- or α5-dependent. CXCL12-activated DU-145 cells were preincubated with β3- or α5-blocking antibodies or the corresponding IgG isotype control, and then added to HUVEC monolayers or immobilized collagen, laminin, or fibronectin. Adherent cells were counted after 60 minutes. Adhesion of cells not treated with monoclonal antibodies was set at 100%. Adhesion blockade diminished adhesion to HUVEC and to extracellular matrix proteins. One of three representative experiments is shown.
CXCR4 Downregulates p38 MAPK
In a further step, CXCL12-induced intracellular signaling in DU-145 tumor cells was analyzed. No difference was observed between the total amount of ERK1 and JNK proteins (CXCL12-activated versus untreated control cells), and only weak phosphorylation was measured. Phosphorylation did not change after CXCL12 incubation. However, p38 MAPK was reduced after CXCL12 treatment. p38 phosphorylation was strongly downregulated after CXCL12 stimulation in DU-145 cells (Figure 8B). CXCR4, therefore, strongly influences the p38 pathway.
Discussion
CXCL12 and its receptor CXCR4 may be involved in all stages of tumor development and progression. CXCL12 promotes the growth of gastrointestinal, pancreatic, breast, and ovarian cancer cells [12–15]. There is growing evidence that CXCL12 and CXCR4 regulate the migration and metastasis of small cell lung cancer cells [16] and are implicated in organ-specific metastases of head and neck squamous cell carcinoma [17]. CXCR4 expression is associated with recurrence, survival, and liver metastasis in colorectal cancer patients [18] and predicts poor prognosis in patients with malignant melanoma [19].
Although there is no doubt that CXCR4 also plays a role in prostate cancer, it is not clear how the CXCR4-CXCL12 axis functions. Analysis of clinical samples has demonstrated elevated CXCR4 protein expression in both localized and metastatic prostate cancers [7]. A study has revealed a higher CXCR4 expression rate in patients with bone metastasis than in those with no bone metastasis [20].
We speculate that CXCR4 directs tumor cell traffic to distant organs in a manner that does not correspond to CXCR4 expression level. Furthermore, chemotaxis toward a CXCL12 gradient may not exclusively explain the potential for neoplastic cells to migrate and invade other tissues. Tumor cell contact with the vessel wall and the underlying matrix must occur to allow the penetration and initiation of secondary tumors. Our results indicate that CXCR4/CXCL12 is an important mediator for the adherence of prostate tumor cells to the endothelium and for interactions with extracellular matrix proteins such as laminin, fibronectin, and collagen.
Evidence has shown that CXCR4 proteins preferentially accumulate in the cytoplasm of DU-145 and LNCaP tumor cells. However, the amount of CXCR4 appearing at the cell surface was limited, and confocal analysis was necessary to clearly demonstrate receptor localization along cell boundaries. CXCR4 surface receptors were functionally active, and chemoinvasion took place regardless of the receptor amount. Even more DU-145 cells responded to CXCL12 than LNCaP cells, although higher CXCR4-specific fluorescence was detected on LNCaP cells. In accordance with our observation, LNCaP and PC3 tumor cells have recently been shown to migrate in a comparable fashion and to invade through extracellular matrix components in response to CXCL12, at rates not corresponding to CXCR4 surface expression [2]. Obviously, receptor saturation occurs at a very low level and, consequently, quantitative receptor enhancement beyond a specific threshold will not accelerate migratory processes of prostate tumor cells. Therefore, it is not surprising that overexpression of CXCR4, induced by transfection, does not upregulate the chemotactic potential of tumor cells [5]. Presumably, CXCL12 release is critical for cell migration, and cancerous cells expressing CXCR4 are more likely to seed distant sites where high levels of CXCL12 are found. Indeed, our findings demonstrated CXCL12 concentration to be the limiting factor for chemotactic activity.
An argument against our hypothesis might be that enzymatic detachment of the cells might downregulate CXCR4 surface expression, leading to reduced fluorescence signals. However, pilot studies revealed that accutase treatment did not alter surface epitopes, in contrast to trypsin, which is most often used to detach cells. Therefore, although we cannot completely rule out slight CXCR4 modifications by accutase, this assumption seems not to be very likely.
The model of CXCR4-triggered chemotaxis might explain the improved motility and invasiveness acquired by the tumor cells after extravasation into the target tissue. We now document that CXCR4 plays a decisive role in controlling preinvasive binding events, as well as tumor cell interaction with the extracellular matrix. CXCL12-enhanced attachment of DU-145 and LNCaP cells to endothelial cells, laminin, fibronectin, and collagen is a process that can be antagonized by CXCR4-specific antibodies or CXCR4 knockdown. As we demonstrated in CXCL12 activation studies, CXCR4 did not regulate adhesion itself, but served as a signaling element to modulate integrin α5 and β3 expression. Blocking the integrin subunits led to a decrease of cell binding, indicating that these receptors are indeed involved in tumor cell/endothelial cell/extracellular matrix interaction.
Disseminated prostate tumors are characterized by altered integrin expression. In particular, αVβ3, which is not expressed in normal prostate tissue but is upregulated in prostatic adenocarcinoma, has been linked to invasive behavior [21]. Nemeth et al. [22] pointed to the importance of α5β3 integrins in controlling the growth and metastasis of prostate cancer cells in the bone, and overexpression of α5 subunits increased the adhesion of the cell line PC-3 to collagen type I, fibronectin, and laminin [23]. The regulation of α5- and β3-mediated tumor cell adhesion by CXCL12 could therefore play a key role during cell homing into, and trafficking inside, the bone.
This is the first report to demonstrate CXCR4-triggered integrin activation in prostate cancer. However, this observation might not be restricted to prostate cancer, and the concept of chemokine-integrin interplay may be valid for other tumor types. It has been postulated that adhesion of human melanoma cells to endothelial cells depends on crosstalk between CXCR4 and β1 integrin chains [24]. The binding of small cell lung cancer cells to the extracellular matrix seems to be mediated by α2, α4, α5, and β1 integrins, along with CXCR4 activation [16]. Based on available data, we hypothesize that CXCR4 represents a ubiquitous receptor molecule expressed on normal and neoplastic tissues. However, integrin equipment might be different among normal and metastatic cells and between specific tumor cell types, allowing tumor transmigration after the occupation of CXCR4 by CXCL12 had taken place.
Surprisingly, our experiments did not reveal any effects of CXCL12 on tumor cell binding to immobilized VCAM, ICAM, E-selectin, and P-selectin, although CXCR4 knockdown or CXCR4 receptor blockade significantly reduced the adhesion of DU-145 or LNCaP cells to HUVEC. Obviously, the interaction of endothelial CAM or selectins with their respective ligands will not be influenced by the CXCR4-CXCL12 axis. CXCL12 was found to be a rapid and potent stimulator of CD34+ stem/progenitor cells, leading to the formation of actin-containing protrusions with CD44 adhesion receptors located at their tips [25]. It cannot be ruled out that a similar crosstalk exists between CD44 and CXCR4 signaling in prostate tumor cells. Nevertheless, we should be aware that α5- and β3-blocking antibodies partially prevented tumor binding to HUVEC; therefore, these integrins also seem involved in tumor cell/endothelial cell interaction. However, as integrins predominantly connect tumor cells to the extracellular matrix, we speculate that downregulation of adhesive capacity might be caused by preventing tumor cell anchorage to matrix proteins expressed on endothelial cells rather than to the endothelial cells themselves. Indeed, HUVEC cultures produce and build a complex matrix network consisting of fibronectin, laminin, and collagen [26].
Looking at the signaling components participating in CXCR4-mediated cell adhesion, we found that enhanced α5 and β3 integrin expression was paralleled by reduced protein expression and phosphorylation of p38 MAPK. Taichman et al. [6] observed a rapid phosphorylation of ERK proteins in PC-3 cells within 5 minutes of CXCL12 stimulation, and CXCR4-mediated activation of both p38 MAPK and ERK has been ascribed to human embryonic kidney 293 cells [27]. This might conflict with our results. However, both citations are based on a short-course stimulus. Our experimental strategy was designed to evaluate intracellular signaling at the same time point that integrin upregulation became obvious (i.e., 4 hours after adding CXCL12). Presumably, CXCR4-mediated activation and protein increase of MAPK molecules reveal an early intracellular event, whereas specific downregulation of p38 and p38 phosphorylation might occur later. In fact, ERK phosphorylation returned to baseline after 30 minutes in the Taichman et al. study. In a murine pre-B cell line, p38 was slightly enhanced 30 minutes following CXCL12 binding to CXCR4, but was reduced below controls thereafter [28].
p38 has long been attributed to be a proapoptotic factor, the downregulation of which triggers enhanced cell survival and growth [29,30]. However, novel reports also point out the role of p38 in cell invasion processes. In this context, the adhesion of prostate carcinoma cell lines becomes reduced in the presence of a p38 inhibitor [31,32]. Very recently, Huang et al. [33] observed that genistein blocks the activation of p38, thereby inhibiting processes closely linked to metastasis.
This might speak for a sensitive balance between tumor cell invasion and tumor cell proliferation, channeled by p38. The engagement of CXCR4 evoked distinct modifications of p38 content and activity in our experiments, although our results do not explain if reduction of p38 contributes to enhanced α5 and β3 integrin synthesis or if integrin elevation creates a negative feedback loop that downregulates p38. Interestingly, high α5β1 integrin expression in several tumor cell lines has been found to be accompanied by low p38 activity [34]. These authors concluded from their study that α5β1 integrins are responsible for blocking p38 activity, which drives tumor cells toward persistent growth. According to this hypothesis, new data highlight the critical contributions of p38 in the negative regulation of cell cycle progression, the attenuation of oncogenic signals, and the positive regulation of several tumor-suppressor pathways [35–37].
Based on this, it seems plausible that α5 and β3 integrins participate in the interaction of prostate tumor cells with extracellular matrix proteins that allow solitary cells or small groups of cells to establish metastases and, after a period of time, shift from invasive to proliferative behavior.
Our results show that CXCR4 receptors are expressed on prostate tumor cells, enabling the cells to migrate toward a CXCL12 gradient and to contact endothelial cells and extracellular matrix proteins. The effects observed did not depend on CXCR4 surface expression level. CXCR4-mediated adhesion was established by α5 and β3 integrin subunits and took place in the presence of reduced p38 and p38 phosphorylation. Presumably, reduced p38 prevents apoptosis and allows rapid tumor growth necessary for survival in a distant organ.
Acknowledgement
The authors are grateful to Karen Nelson for critically reading the manuscript.
Footnotes
This work was supported by the Horst Müggenburg-Stiftung, the Matthias Lackas-Stiftung, and the Heinrich und Erna Schaufler-Stiftung.
References
- 1.Arya M, Patel HR, McGurk C, Tatoud R, Klocker H, Masters J, Williamson M. The importance of the CXCL12-CXCR4 chemokine ligand-receptor interaction in prostate cancer metastasis. J Exp Ther Oncol. 2004;4:291–303. [PubMed] [Google Scholar]
- 2.Singh S, Singh UP, Grizzle WE, Lillard JW., Jr CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84:1666–1676. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 3.Hart CA, Brown M, Bagley S, Sharrard M, Clarke NW. Invasive characteristics of human prostatic epithelial cells: understanding the metastatic process. Br J Cancer. 2005;92:503–512. doi: 10.1038/sj.bjc.6602325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaday GG, Hua SB, Peehl DM, Pauling MH, Lin YH, Zhu L, Lawrence DM, Foda HD, Zucker S. CXCR4 and CXCL12 (SDF-1) in prostate cancer: inhibitory effects of human single chain Fv antibodies. Clin Cancer Res. 2004;10:5630–5639. doi: 10.1158/1078-0432.CCR-03-0633. [DOI] [PubMed] [Google Scholar]
- 5.Darash-Yahana M, Pikarsky E, Abramovitch R, Zeira E, Pal B, Karplus R, Beider K, Avniel S, Kasem S, Galun E, et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004;18:1240–1242. doi: 10.1096/fj.03-0935fje. [DOI] [PubMed] [Google Scholar]
- 6.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–1837. [PubMed] [Google Scholar]
- 7.Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, Pienta KJ, Taichman RS. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462–473. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 8.Wittig BM, Thees R, Kaulen H, Gott K, Bartnik E, Schmitt C, Meyer zum Buschenfelde KH, Dippold W. Alpha(1,3)fucosyltransferase expression in E-selectin-mediated binding of gastrointestinal tumor cells. Int J Cancer. 1996;67:80–85. doi: 10.1002/(SICI)1097-0215(19960703)67:1<80::AID-IJC14>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Blaheta RA, Hailer NP, Brude N, Wittig B, Leckel K, Oppermann E, Bachmann M, Harder S, Cinatl J, Scholz M, et al. In vitro analysis of verapamil-induced immunosuppression: potent inhibition of T cell motility and lymphocytic transmigration through allogeneic endothelial cells. Transplantation. 2000;69:588–597. doi: 10.1097/00007890-200002270-00021. [DOI] [PubMed] [Google Scholar]
- 10.Melchionna R, Porcelli D, Mangoni A, Carlini D, Liuzzo G, Spinetti G, Antonini A, Capogrossi MC, Napolitano M. Laminar shear stress inhibits CXCR4 expression on endothelial cells: functional consequences for atherogenesis. FASEB J. 2005;19:629–631. doi: 10.1096/fj.04-2219fje. [DOI] [PubMed] [Google Scholar]
- 11.Salmaggi A, Gelati M, Pollo B, Frigerio S, Eoli M, Silvani A, Broggi G, Ciusani E, Croci D, Boiardi A, et al. CXCL12 in malignant glial tumors: a possible role in angiogenesis and cross-talk between endothelial and tumoral cells. J Neuro-Oncol. 2004;67:305–317. doi: 10.1023/b:neon.0000024241.05346.24. [DOI] [PubMed] [Google Scholar]
- 12.Guleng B, Tateishi K, Ohta M, Kanai F, Jazag A, Ijichi H, Tanaka Y, Washida M, Morikane K, Fukushima Y, et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res. 2005;65:5864–5871. doi: 10.1158/0008-5472.CAN-04-3833. [DOI] [PubMed] [Google Scholar]
- 13.Marchesi F, Monti P, Leone BE, Zerbi A, Vecchi A, Piemonti L, Mantovani A, Allavena P. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004;64:8420–8427. doi: 10.1158/0008-5472.CAN-04-1343. [DOI] [PubMed] [Google Scholar]
- 14.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 15.Porcile C, Bajetto A, Barbieri F, Barbero S, Bonavia R, Biglieri M, Pirani P, Florio T, Schettini G. Stromal cell-derived factor-1 alpha (SDF-1alpha/CXCL12) stimulates ovarian cancer cell growth through the EGF receptor transactivation. Exp Cell Res. 2005;308:241–253. doi: 10.1016/j.yexcr.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann TN, Burger JA, Glodek A, Fujii N, Burger M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene. 2005;24:4462–4471. doi: 10.1038/sj.onc.1208621. [DOI] [PubMed] [Google Scholar]
- 17.Samara GJ, Lawrence DM, Chiarelli CJ, Valentino MD, Lyubsky S, Zucker S, Vaday GG. CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous cell carcinoma. Cancer Lett. 2004;214:231–241. doi: 10.1016/j.canlet.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 18.Kim J, Takeuchi H, Lam ST, Turner RR, Wang HJ, Kuo C, Foshag L, Bilchik AJ, Hoon DS. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J Clin Oncol. 2005;23:2744–2753. doi: 10.1200/JCO.2005.07.078. [DOI] [PubMed] [Google Scholar]
- 19.Scala S, Ottaiano A, Ascierto PA, Cavalli M, Simeone E, Giuliano P, Napolitano M, Franco R, Botti G, Castello G. Expression of CXCR4 predicts poor prognosis in patients with malignant melanoma. Clin Cancer Res. 2005;11:1835–1841. doi: 10.1158/1078-0432.CCR-04-1887. [DOI] [PubMed] [Google Scholar]
- 20.Mochizuki H, Matsubara A, Teishima J, Mutaguchi K, Yasumoto H, Dahiya R, Usui T, Kamiya K. Interaction of ligand-receptor system between stromal-cell-derived factor-1 and CXC chemokine receptor 4 in human prostate cancer: a possible predictor of metastasis. Biochem Biophys Res Commun. 2004;320:656–663. doi: 10.1016/j.bbrc.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Zheng D-Q, Woodward AS, Fornaro M, Tallini G, Languino LR. Prostatic carcinoma cell migration via αVβ3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 1999;59:1655–1664. [PubMed] [Google Scholar]
- 22.Nemeth JA, Cher ML, Zhou Z, Mullins C, Bhagat S, Trikha M. Inhibition of alpha(v)beta3 integrin reduces angiogenesis, bone turnover, and tumor cell proliferation in experimental prostate cancer bone metastases. Clin Exp Metastasis. 2003;20:413–420. doi: 10.1023/a:1025461507027. [DOI] [PubMed] [Google Scholar]
- 23.Shen X, Falzon M. Parathyroid hormone-related protein upregulates integrin expression via an intracrine pathway in PC-3 prostate cancer cells. Regul Pept. 2003;113:17–29. doi: 10.1016/s0167-0115(02)00293-8. [DOI] [PubMed] [Google Scholar]
- 24.Cardones AR, Murakami T, Hwang ST. CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer Res. 2003;63:6751–6757. [PubMed] [Google Scholar]
- 25.Avigdor A, Goichberg P, Shivtiel S, Dar A, Peled A, Samira S, Kollet O, Hershkoviz R, Alon R, Hardan I, et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood. 2004;103:2981–2989. doi: 10.1182/blood-2003-10-3611. [DOI] [PubMed] [Google Scholar]
- 26.Leroy-Dudal J, Demeilliers C, Gallet O, Pauthe E, Dutoit S, Agniel R, Gauduchon P, Carreiras F. Transmigration of human ovarian adenocarcinoma cells through endothelial extracellular matrix involves alphav integrins and the participation of MMP2. Int J Cancer. 2005;114:531–543. doi: 10.1002/ijc.20778. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 28.Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE. The alpha-chemokine, stromal cell-derived factor-1 alpha, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003;278:33753–33762. doi: 10.1074/jbc.M303313200. [DOI] [PubMed] [Google Scholar]
- 30.Edlund S, Bu S, Schuster N, Aspenstrom P, Heuchel R, Heldin NE, ten Dijke P, Heldin CH, Landstrom M. Transforming growth factor-beta 1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 byTGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol Biol Cell. 2003;14:529–544. doi: 10.1091/mbc.02-03-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, He HY, Li HM, Zheng J, Heng WJ, You JF, Fang WG. ERK1/2 and p38 pathways are required for P2Y receptor-mediated prostate cancer invasion. Cancer Lett. 2004;215:239–247. doi: 10.1016/j.canlet.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 32.Hayes SA, Huang X, Kambhampati S, Platanias LC, Bergan RC. p38 MAP kinase modulates Smad-dependent changes in human prostate cell adhesion. Oncogene. 2003;22:4841–4850. doi: 10.1038/sj.onc.1206730. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Chen S, Xu L, Liu Y, Deb DK, Platanias LC, Bergan RC. Genistein inhibits p38 map kinase activation, matrix metalloproteinase type 2, and cell invasion in human prostate epithelial cells. Cancer Res. 2005;65:3470–3478. doi: 10.1158/0008-5472.CAN-04-2807. [DOI] [PubMed] [Google Scholar]
- 34.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK (MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- 35.Bulavin DV, Fornace AJ., Jr p38 MAP kinase's emerging role as a tumor suppressor. Adv Cancer Res. 2004;92:95–118. doi: 10.1016/S0065-230X(04)92005-2. [DOI] [PubMed] [Google Scholar]
- 36.Dasgupta P, Betts V, Rastogi S, Joshi B, Morris M, Brennan B, Ordonez-Ercan D, Chellappan S. Direct binding of apoptosis signal-regulating kinase 1 to retinoblastoma protein: novel links between apoptotic signaling and cell cycle machinery. J Biol Chem. 2004;279:38762–38769. doi: 10.1074/jbc.M312273200. [DOI] [PubMed] [Google Scholar]
- 37.Khaled AR, Bulavin DV, Kittipatarin C, Li WQ, Alvarez M, Kim K, Young HA, Fornace AJ, Durum SK. Cytokine-driven cell cycling is mediated through Cdc25A. J Cell Biol. 2005;169:755–763. doi: 10.1083/jcb.200409099. [DOI] [PMC free article] [PubMed] [Google Scholar]