

Abstract
Malignant pleural mesotheliomas (MPMs) are usually wild type for the p53 gene but contain homozygous deletions in the INK4A locus that encodes p14ARF, an inhibitor of p53-MDM2 interaction. Previous findings suggest that lack of p14ARF expression and the presence of SV40 large T antigen (L-Tag) result in p53 inactivation in MPM. We did not detect SV40 L-Tag mRNA in either MPM cell lines or primary cultures, and treatment of p14ARF-deficient cells with cisplatin (CDDP) increased both total and phosphorylated p53 and enhanced p53 DNA-binding activity. On incubation with CDDP, levels of positively regulated p53 transcriptional targets p21WAF, PIG3, MDM2, Bax, and PUMA increased in p14ARF-deficient cells, whereas negatively regulated survivin decreased. Significantly, p53-induced apoptosis was activated by CDDP in p14ARF-deficient cells, and treatment with p53-specific siRNA rendered them more CDDP-resistant. p53 was also activated by: 1) inhibition of MDM2 (using nutlin-3); 2) transient overexpression of p14ARF; and 3) targeting of survivin using antisense oligonucleotides. However, it is noteworthy that only survivin downregulation sensitized cells to CDDP-induced apoptosis. These results suggest that p53 is functional in the absence of p14ARF in MPM and that targeting of the downstream apoptosis inhibitor survivin can sensitize to CDDP-induced apoptosis.
Keywords: Mesothelioma, cisplatin, p53, p14ARF, survivin
Introduction
Malignant pleural mesothelioma (MPM) is a cancer of mesothelial cells that line lung pleural membranes. The incidence of MPM is highly associated with exposure to asbestos [1]. Due to the long latency period of the disease, projections suggest that the number of MPM-related deaths in western Europe will double each year until 2018 [2]. MPM is a treatment-resistant tumor with a very poor prognosis [3]. Although there is no standard treatment for MPM, current multimodality interventions include combined chemotherapy with cisplatin (CDDP) and pemetrexed or gemcitabine [4].
p53 has been termed as the “guardian of the genomes” because it mediates the effects of DNA damage and, depending on cellular context, induces cell cycle arrest and DNA repair or death. p53-induced apoptosis involves the transcription of proapoptotic genes (such as Bax and PUMA) and the repression of antiapoptotic genes (such as survivin and Bcl-2) [5]. p53 also displays transcription-independent proapoptotic functions by interacting directly with Bcl-xL and Bax and by inducing the mitochondrial pathway of apoptosis [6]. In resting cells, p53 regulates its own expression by inducing the synthesis of the inhibitor MDM2, which binds p53 and blocks DNA trans-activation. MDM2 also has E3 ligase activity that promotes the ubiquitination and degradation of p53 through the 26S proteosome [7]. Recognition of DNA damage leads to the stabilization of p53 through posttranslational modifications such as phosphorylation, acetylation, and sumoylation, which interfere with p53-MDM2 interaction [5]. During oncogenic stress, inhibition of p53 by MDM2 is also abrogated by the tumor-suppressor protein p14ARF, which binds to MDM2, sequesters it in the nucleolus, and blocks its E3 ligase activity [8]. p14ARF expression has also been described to play a role in the response of p53 to DNA damage [9,10], although these results are controversial [11,12].
Most cancers have evolved mechanisms to deregulate p53, and inactivating mutations in the p53 gene arise in approximately 50% of human tumors [13]. In those tumors with wild-type p53, there are often alterations in genes that regulate p53, such as amplifications of the MDM2 gene or, as is the case for MPM, homozygous deletions in the INK4A locus encoding p14ARF [14]. Yang et al. [15] proposed that p53 is inactive in MPM cells as a result of p14ARF deficiency because MPM cells were sensitive to infection by the oncolytic virus ONYX-015. In addition, the SV40 large T antigen (L-Tag) has been proposed to block p53 function in MPM [16]; however, recent studies question the frequency of SV40 infection in MPM tumors [17].
In the present study, we investigated the status of p53 in MPM cells lacking p14ARF. Our findings suggest that p53 is functional in MPM in the absence of p14ARF and that it is activated by DNA damage.
Materials and Methods
Cell Culture
The MPM cell lines SPC212, ZL5, ZL55, and ZL34 were generated in our laboratory and have been described previously [18]. The Met5A cell line (Dr. J. F. Lechner; NCI, Bethesda, MD) corresponds to normal human mesothelial cells immortalized by transfection with an SV40 early region plasmid [19], and the NCI-H28 cell line was obtained from ATCC (Manassas, VA). Primary MPM cultures were derived from MPM pleural effusions, as described previously [18], and were characterized by the positive staining of cells with antibodies specific for calretinin. All cells were grown in RPMI 1640 (Sigma, St. Louis, MI) supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 10% fetal bovine serum, and 1% (wt/vol) penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Where indicated, CDDP (Bristol-Myers Squibb AG, Baar, Switzerland) and nutlin-3 (Alexis Corporation, Lausen, Switzerland) were added. Nutlin-3 was dissolved in dimethyl sulfoxide (DMSO) and compared to cells treated with similar volumes of DMSO alone.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RNA was isolated from cell lines using the RNeasy kit (Qiagen, Basel, Switzerland), according to the manufacturer's instructions. One microgram of RNA was used in RT-PCR using the One-Step RT-PCR kit (Qiagen). The following primers were used: SV40 T antigen: 5′ primer (5′-AGTCCTCGAGTCTTTGCAGCTAATGGACCT) and 3′ primer (5′-AGTCTCTAGATCCTTTGTGGTGTAAATAGC); p14ARF: 5′ primer (5′-TGCTCACCTCTGGTGCCAAAG) and 3′ primer (5′-TGGTCTTCTAGGAAGCGGCTG); GAPDH: 5′ primer (5′-GAGTCAACGGATTTGGTCGT) and 3′ primer (5′-TGACAAAGTGGTCGTTGAGG).
PCR cycle conditions were as follows: 35 cycles of 94°C for 40 seconds, 58°C for 40 seconds, and 72°C for 40 seconds for SV40 L-Tag; and 28 cycles of 94°C for 30 seconds, 64°C for 30 seconds, and 72°C for 50 seconds for p14ARF and GAPDH. Ten microliters of PCR products was analyzed by electrophoresis on 2.5% agarose gels, and GAPDH band was used as standardized control.
p53 DNA-Binding Activity
Nuclear extracts were prepared using the Nuclear Extract kit (Active Motif, Carlsbad, CA), and p53 DNA-binding activity was assayed using the TransAM p53 kit (Active Motif), according to the manufacturer's instructions. Binding to p53 target DNA sequence was measured by colorimetric assay, and absorbance was read at 450 nm. Samples were performed in quadruplicate, and the mean and standard deviations were calculated. Experiments were performed in triplicate, and representative data are shown.
p53 Sequencing
DNA sequencing was performed on DNA isolated from SPC212 cells using Affymetrix p53 Gene Chip Array and Sequencing Analysis (BRT Laboratories, Inc., Baltimore, MD).
Western Blot Analysis
Cells were lysed with RIPA buffer (Upstate Cell Signaling Solutions, Dundee, UK) in the presence of phosphatase inhibitors (Sigma) for 30 minutes on ice. Lysates were clarified by centrifugation (10,000g for 30 minutes at 4°C). Western blot analysis was performed as described previously [20]. Antibodies with specificity for survivin (R&D Systems, Inc., Minneapolis, MN), β-actin (C4; ICN Biomedicals, Inc., Aurora, OH), phospho-p53 (ser15) (Cell Signaling Technology, Inc., Beverly, MA), p14ARF (C18), p21WAF (H-164), p53 (DO-1), MDM2 (N-20), PIG-3 (C20), Bax (N-20), and PUMA (N-19) (Santa Cruz Biotechnology, Santa Cruz) were used.
Flow Cytometry
Both adherent and floating cells were collected from samples and washed in phosphate-buffered saline (PBS). Cells were then fixed in 50% ethanol and incubated with propidium iodide (PI)/RNase (BD Pharmingen, San Diego CA) for 15 minutes at room temperature. Alternatively, live cells were resuspended in binding buffer and stained with Annexin V-FITC (Calbiochem, Darmstadt, Germany) for 15 minutes at room temperature. Ten thousand events were analyzed by flow cytometry using a FACScalibur flow cytometer (Becton Dickinson, Mountain View, CA). For PI staining, doublets and aggregates were excluded by gating, and Modfit software was used to identify cell populations in different phases of the cell cycle. The percentage of apoptotic cells was determined after PI or Annexin V labeling by gating on untreated samples.
Caspase-3-Like Protease Activity
Both adherent and floating cells were collected from samples and washed in PBS. Caspase-3-like protease activity in cell lysates was analyzed by colorimetric assay, according to the manufacturer's recommendations (Alexis Corporation). Cleavage of the labeled substrate DEVD-pNA was monitored at 405 nm using a SPECTRAmax 340 microplate reader (Paul Bucher Analytik und Biotechnologie, Basel, Switzerland). The caspase-3-like protease activity in lysates was calculated as fold increase of the absorbance signal obtained with lysates of untreated (viable) cells kept under identical conditions.
Transfections
The p14ARF-pcDNA3.1 plasmid was a kind gift from Prof. Gordon Peters (ICRF, London, UK). p53-specific siRNA and control siRNA were purchased from Cell Signaling Technology, Inc., and Dharmacon, Inc. (Chicago, IL), respectively. Transfections with plasmids or siRNA were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer's recommendations. Cells were plated at a concentration of 150,000 cells/ml in medium containing 10% fetal calf serum (FCS) but containing no antibiotics 1 day before transfection and were transfected for 24 hours using OptiMEM (Invitrogen). Thereafter, the medium was changed to a complete medium containing 10% FCS with indicated concentrations of CDDP for a further 48 hours.
Measurement of Cell Growth
Cell growth was determined using a colorimetric cell viability assay based on the reduction of the tetrazolium salt MTT, as described [21]. Cells were plated in quadruplicate in 96-well plates (7500 cells/well), and absorbance was measured at 570 nm using a SPECTRAmax 340 microplate reader. Cell growth was calculated as a percentage of the absorbance signal obtained with wells of untreated (viable) cells kept under identical conditions.
Antisense Oligonucleotides
Survivin phosphothioate antisense oligonucleotides with the sequence 5′-CCCAGCCTTCCAGCTCCTTG-3′ targeting nucleotides 233 to 253 of the survivin mRNA and the control sequence 5′-CCTAGCCTTCCAGGTCCTAG-3′ (mismatches underlined) were purchased from Microsynth AG (Balgoch, Switzerland). Cells were transfected with Oligofectamine (Invitrogen), according to the manufacturer's protocol. Cells were plated at a concentration of 150,000 cells/ml 1 day before transfection and were transfected for 6 hours; the medium was changed, and cells were incubated for a further 48 hours with the indicated concentration of CDDP.
Results
MPM Cells Are Negative for p14ARF Protein Expression and SV40 L-Tag mRNA
Homozygous deletions in the INK4A locus of MPM occur at a frequency of at least 70%, resulting in the absence of p14ARF expression [14]. We investigated p14ARF expression in five established MPM cell lines and three primary cultures. Three MPM cell lines (NCI-H28, ZL5, and ZL34) and three MPM primary cultures (SDM4, SDM5, and SDM6) were negative for p14ARF mRNA, whereas it could be detected in SPC212 and ZL55 cells and in an SV40-transformed mesothelial cell line, Met5A (Figure 1). However, in contrast to Met5A cells, no p14ARF protein was detected in any of the MPM primary cultures and cell lines tested. The presence of SV40 viral DNA sequences in MPM tumors has given rise to the suggestion that p53 is inactivated in MPM by the SV40 L-Tag [16]. MPM cell lines and primary cultures were therefore tested for SV40 L-Tag mRNA by RT-PCR, but it could only be detected in the SV40-transformed Met5A cell line.
Figure 1.

Expression levels of p14ARF in MPM cell lines. RNA was isolated from MPM primary cultures and cell lines and SV40-transformed Met5A cells, and RT-PCR was performed to detect p14ARF, SV40 L-Tag and GAPDH mRNA. Lysates of the same cell lines were analyzed by immunoblotting using anti-p14ARF C-terminal antibodies. Staining of actin was used as equal loading control.
p53 Can Be Activated in MPM in the Absence of p14ARF
To determine whether p53 is functional in the absence of p14ARF expression, the status of p53 in MPM cell lines and primary cultures was investigated in response to CDDP treatment. The ability of active p53 protein to bind to its target DNA sequence was measured using an enzyme-linked immunosorbent assay (ELISA)-based assay, which demonstrated that seven of eight MPM cell lines and primary cultures had increased levels of p53-specific DNA binding in response CDDP treatment (Figure 2A). In contrast, SPC212 cells had no detectable p53 activity, and sequencing of the p53 gene in these cells revealed a missense mutation in the hotspot codon 175. ZL55 cells had low levels of p53 DNA-binding activity in response to CDDP, regardless of p14ARF mRNA expression.
CDDP induced an increase in basal levels of p53 protein in ZL5, ZL34, and NCI-H28 cells, whereas in SPC212 cells, the levels of p53 remained consistently high (Figure 2B). Residue ser15 in the transactivation domain of p53 is phosphorylated by protein kinases in response to CDDP, resulting in the disruption of p53-MDM2 interaction and increased p53 activity [22,23]. Levels of phospho-ser15 were found to increase in response to CDDP treatment in all MPM cell lines, including p53 mutant SPC212 cells. These results confirm those of others, which demonstrate that both wild-type and mutant p53 can be phosphorylated on ser15 [24].
Figure 2.
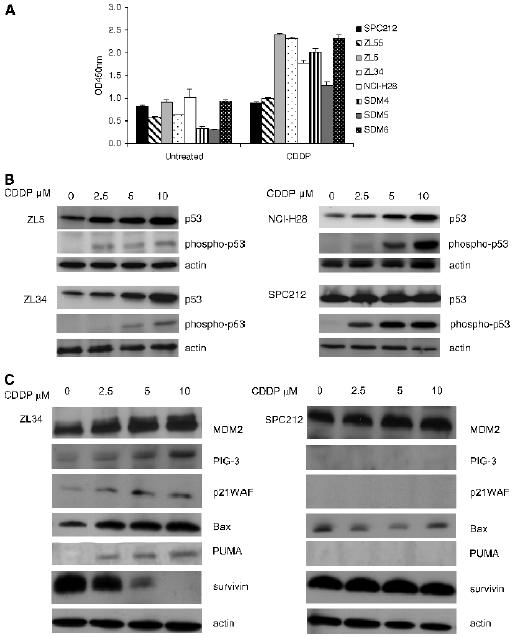
p53 is activated in p14ARF-deficient MPM cell lines. (A) MPM primary cultures and cell lines were cultured with 5 µM CDDP for 48 hours or were left untreated. Nuclear extracts were prepared, and binding to the p53 recognition sequence was measured by ELISA, as described in Materials and Methods section. Data are presented as mean ± SD OD450 nm absorbance values. (B) MPM cell lines were cultured with the indicated concentrations of CDDP for 48 hours. Lysates were analyzed by immunoblotting using antibodies specific for p53 and phospho-p53 (ser15). Staining of actin was used as loading control. (C) MPM cell lines were cultured with the indicated concentrations of CDDP for 48 hours. Lysates were analyzed by immunoblotting using antibodies specific for MDM2, PIG3, p21WAF, Bax, PUMA, and survivin. Staining of actin was used as loading control.
In ZL34 cells, the expression of p53 transcriptional targets MDM2, PIG3, p21WAF, Bax, and PUMA was increased by CDDP treatment, and similar profiles were observed for the ZL5 and NCI-H28 cell lines (data not shown), whereas no induction was seen for SPC212 cells (Figure 2C). Survivin expression is negatively regulated by p53 [25], and immunoblotting demonstrated that, in ZL34 cells, survivin levels decreased with increasing concentrations of CDDP, whereas they remained unchanged in the p53 mutant cell line, SPC212 (Figure 2C). No increase in p14ARF expression was detected after CDDP treatment (data not shown). Together, these results suggest that p53-mediated transcription regulation is fully functional in MPM cells in the absence of p14ARF.
p53-Induced Apoptosis Is Activated in p14ARF-Deficient MPM Cells
MPM cell lines lacking p14ARF were investigated for their ability to undergo apoptosis and/or cell cycle arrest in response to CDDP. Treatment of MPM cells with CDDP leads to an increase in the percentage of cells in the sub-G1 phase of the cell cycle for both ZL5 and ZL34 cell lines, but not for SPC212 cells (Figure 3, A and B). ZL34 and SPC212 cells, but not ZL5, cells accumulated on the S phase on CDDP treatment (Figure 3A), which has been observed previously and occurs independently of p53 status [26]. Apoptosis was also detected by the measurement of Annexin V staining and caspase-3-like activity in ZL5 and ZL34 cells in response to CDDP treatment, but not in the p53 mutant SPC212 cell line (Figure 3, C and D).
Figure 3.

CDDP induces apoptosis in MPM cell lines. (A) Flow diagrams of untreated cells or of cells treated with 10 µM CDDP, fixed, and stained with PI. Panels represent the proportion of cells in different phases of the cell cycle. Representative data of three independent experiments are shown. (B, C, and D) Cells were incubated with the indicated concentrations of CDDP for 48 hours. Standard error bars represent the standard deviation of triplicate samples. (B) Percentage of apoptotic cells in sub-G1 as measured by PI staining and flow cytometry. (C) Percentage of apoptotic cells as measured by Annexin V-FITC staining and flow cytometry. (D) The caspase-3-like protease activity in cell lysates was calculated as fold increase of the absorbance signal obtained with lysates of untreated cells.
To demonstrate the role of p53 in CDDP-induced death in MPM, cells were treated with p53-specific siRNA. Incubation of ZL34 cells with p53 siRNA resulted in a dose-dependent decrease in p53, p21WAF, and Bax levels and in increased survivin relative to control siRNA (Figure 4A). Treatment of cells with p53 siRNA and 5 µM CDDP also resulted in decreased p53, p21WAF, and Bax and in increased survivin relative to control siRNA (Figure 4A). Transfection of ZL34 cells with p53-specific siRNA resulted in less CDDP-induced growth inhibition and decreased apoptosis compared to control siRNA (Figure 4B). A comparable increase in resistance to CDDP was observed when ZL5 cells were treated with the p53 inhibitor, pifithrin-α (data not shown). These results demonstrate that p53 contributes to CDDP-induced apoptosis in MPM.
Figure 4.

p53-specific death is induced by CDDP in MPM cells. (A) ZL34 cells were transfected with the indicated concentrations of p53 siRNA or control siRNA for 24 hours, or were treated with 50 nM p53 or control siRNA for 24 hours and incubated with 5 µM CDDP for a further 48 hours. Lysates were analyzed by immunoblotting using antibodies specific for p53, p21WAF, Bax, and survivin. Staining of actin was used as loading control. (B) ZL34 cells were transfected with 50 nM p53 siRNA or control siRNA for 24 hours and were subsequently incubated for 48 hours with 5 µM CDDP. Cell growth inhibition was calculated as the percentage relative to untreated controls. Representative data of three independent experiments are shown. Caspase-3-like activity and the percentage of apoptotic cells were determined by Annexin V staining and flow cytometry, as described in Figure 3. Student's t test was used to determine the significance between the p53 siRNA + CDDP group and the control siRNA + CDDP group.
Inhibition of MDM2 Induces G1 Arrest, But Not Apoptosis
Nutlin-3, a small-molecule inhibitor of MDM2 that binds the p53-binding pocket and prevents its interaction with p53 [27], was used to investigate the role of MDM2 in the p53 response observed in p14ARF-deficient MPM cells. Incubation with nutlin-3 resulted in increased levels of p53 and its transcriptional targets MDM2 and p21WAF in ZL34 cells, suggesting that p53 was activated. However, as reported by Thompson et al. [28], no increase in phospho-p53 (ser15) was induced (Figure 5A). The effect of nutlin-3 was p53-dependent, as expression of p53 target genes was not altered in SPC212 cells. Nutlin-3 treatment induced a G1 cell cycle arrest in ZL34 cells (Figure 5B), but no caspase-3-like activity was detected and CDDP-induced apoptosis was abrogated in the presence of nutlin-3 (Figure 5C). Nutlin-3 had no effect on the sensitivity of SPC212 cells to CDDP, whereas ZL5 and NCI-H28 behaved similarly to ZL34 (data not shown). These results suggest that, although inhibition of MDM2 by nutlin-3 can activate p53-induced cell cycle arrest in MPM cells, apoptosis is not triggered and CDDP-induced apoptosis is inhibited.
Figure 5.

Nutlin-3 induces p53 activity and cell cycle arrest, but not apoptosis. (A) ZL34 and SPC212 cells were cultured for 24 hours with the indicated concentrations of nutlin-3. Lysates were analyzed by immunoblotting using antibodies specific for p53, phospho-p53 (ser15), MDM2, and p21WAF. Staining of actin was used as loading control. (B) ZL34 cells were cultured with 10 µM nutlin-3 for 48 hours. Cells were fixed and stained with PI before FACS staining of nuclei. Representative data of three independent experiments are shown. (C) ZL34 cells were cultured with 10 µM nutlin-3 or the same volume of DMSO in the presence of 1.2 µM CDDP for 48 hours. Cells were lysed, and caspase-3-like protease activity was measured as in Figure 3D.
p53 Activation by Survivin Targeting, But Not p14ARF Expression, Sensitizes Cells to CDDP-Induced Apoptosis
Others have shown that overexpression of p14ARF using adenoviruses in MPM cells activates p53 and induces cell cycle arrest and apoptosis [29]. In contrast, expression of p14ARF at more physiological levels in several different cancer types induced cell cycle arrest but not apoptosis [30]. We introduced p14ARF into p14ARF-deficient cells by transient transfection and determined their effect on p53 activation and apoptosis. ZL34 cells expressing p14ARF had increased levels of p53 compared to the empty vector control; however, in response to CDDP, p53 levels were only slightly enhanced by the presence of p14ARF (Figure 6A). In addition, the difference in caspase-3-like activity between controls and cells expressing p14ARF treated with CDDP were not significantly different (Figure 6B). This suggests that expression of p14ARF does not sensitize cells to CDDP-induced apoptosis.
Figure 6.

Survivin antisense oligonucleotides, but not p14ARF expression, activates p53-induced apoptosis. ZL34 cells were transfected with the plasmid pcDNA3 (vector) or pcDNA3-p14ARF (p14ARF) for 6 hours, then the indicated concentrations of CDDP were added for a further 48 hours. Cells were then (A) harvested for immunoblotting using p14ARF, p53, or actin antibodies, or (B) lysed, and caspase-3-like protease activity was measured using colorimetric assay, as in Figure 3D. Representative data of three independent experiments are shown. Absorbance values obtained with untreated cells maintained under identical experimental conditions were taken as 100%. (C) ZL34 cells were transfected with survivin antisense oligonucleotide 4003 or mismatch control 4003mis for 6 hours and were harvested 20 hours after the start of transfection. Lysates were analyzed by immunoblotting using antibodies specific for survivin, p53, phospho-p53 (ser15), p21WAF, and MDM2. Staining of actin was used as loading control. (D) ZL34 cells were transfected with transfection reagent alone (OF), survivin antisense oligonucleotide 4003, or mismatch control 4003mis for 6 hours and then for a further 48 hours with 2.5 µM CDDP. Lysates were analyzed by immunoblotting using antibodies specific for survivin and p53. Staining of actin was used as loading control. (E) ZL34 cells were transfected with 200 nM survivin antisense oligonucleotide 4003 or the 4003mis mismatch control for 6 hours, and then 1.2 µM CDDP was added for a further 48 hours. Cells were lysed and caspase-3-like protease activity was measured as in Figure 3D. Student's t test was used to determine the significance between the 4003 + CDDP group and the 4003mis + CDDP group.
Survivin is highly expressed in many cancers, including MPM [31], and is a negative prognostic marker in non-small cell lung cancer (NSCLC) [32]. Although survivin expression is negatively regulated by p53 [25], it has also been demonstrated that, in turn, survivin may negatively control p53 function [33,34]. ZL34 cells were incubated with an antisense oligonucleotide 4003 targeting survivin, and a dose-dependent decrease in survivin protein levels—in addition to increased levels of p53, phospho-p53 (ser15), p21WAF, MDM2, and its 60-kDa cleavage product relative to the mismatch control oligonucleotide 4003mis—was observed (Figure 6C). A combination of 4003 with CDDP further increased levels of p53 compared to the mismatch control (Figure 6D). Treatment with survivin antisense oligonucleotides has been shown by Olie et al. [21] and Xia et al. [35] to induce apoptosis in NSCLC and MPM cells, respectively. CDDP-induced caspase-3-like activity was significantly enhanced by survivin downregulation in ZL34 cells (Figure 6E).
Discussion
Previous studies have proposed that p53 is inactive in MPM, as a result of either p14ARF deletion [15,29,36] or SV40 infection [37]. The aim of the present study was to determine whether p53 functions in the absence of p14ARF and whether it contributes to the apoptotic response to CDDP. Studies using the oncolytic virus ONYX-015, which is thought to selectively infect those cells expressing nonfunctional p53 [38], showed that MPM cell lines lacking p14ARF were sensitive to viral infection and that expression of exogenous p14ARF rendered cells more resistant to infection [15]. These results lead to the conclusion that the p53 pathway is disrupted in MPM cells lacking p14ARF; however, more recent reports suggest that the ability of ONYX-015 to infect cancer cells is determined by late viral RNA transport rather than by p53 status [39,40]. Our study demonstrates that p53 is functional in MPM in the absence of p14ARF, as it can activate the transcription of target genes and can contribute to apoptotic response. It is also noteworthy that a recent large-scale transcriptional analysis of MPM human tumors revealed that p53 and several of its target genes are upregulated in MPM tumors relative to normal tissues in the absence of p14ARF [41].
Although studies with mice containing targeted deletions in p19ARF (the murine equivalent of p14ARF) give evidence for its role as a tumor suppressor [8,11], it is less clear whether p19ARF is an essential component of the p53 response to DNA damage. Khan et al. [10] showed that p53 response to DNA damage is defective in p19ARF knockout mice, whereas Schmitt et al. [12] showed that, although loss of p19ARF completely disabled p53 during lymphomagenesis, p53 was activated in response to DNA damage in transplanted murine Eµ-Myc lymphomas. In the present study, using human MPM cells deficient in p14ARF expression, we found that, although transient overexpression of p14ARF activated p53, it did not sensitize cells to CDDP-induced apoptosis, suggesting that it is not required for the DNA damage response of p53 in MPM.
The disruption of wild-type p53 has been shown in some systems to increase resistance to DNA damage-induced apoptosis [45,46], whereas in other systems, increased sensitivity has been demonstrated [42–44]. It is probable that the type of response to DNA damage induced in cells where p53 has been inactivated depends on cell type, genetic status, and the type of drug used to damage DNA. It is, however, generally accepted that p53 status contributes to sensitivity to CDDP [47], and our results are in agreement with this, as treatment of MPM cells with p53-specific siRNA increased CDDP resistance to apoptosis.
The small-molecule inhibitor nutlin-3 perturbs p53-MDM2 interaction, resulting in the activation of wild-type p53 and, in some cases, apoptosis [27]. In MPM cells, nutlin-3 did not induce apoptosis, and the level of CDDP-induced apoptosis decreased on combination with nutlin-3. Similarly, a recent study showed that treatment of cancer cell lines with nutlin-3 induced a strong G1 cell cycle arrest, but only low levels of apoptosis [48]. One explanation for these findings is that the cell cycle arrest induced by nutlin-3 has an inhibitory effect on apoptosis [49,50]. In addition, expression of downstream inhibitors could attenuate the apoptotic response induced by nutlin-3; indeed, recent data have shown that p53 is directly inhibited by Bcl-xL [6]. Because MPM cells express high levels of Bcl-xL, these may limit transactivation-independent apoptosis induction by p53, as well as limit Bax/Bak oligomerization. We have shown previously that antisense oligonucleotides targeting Bcl-xL and Bcl-2 sensitize MPM cells to CDDP-induced apoptosis [51], and we are currently investigating the interaction between Bcl-xL and p53.
Similar to Bcl-xL, survivin is highly expressed in MPM [31] and in other cancers, where its expression is associated with aggressive tumor behavior and poor prognosis [52,53]. Survivin inhibits caspase activation [54] and plays an important role in cell division, allowing cells to progress through mitosis [55]. Survivin expression is negatively regulated by p53; indeed, p53-specific siRNA treatment leads to increased survivin expression in MPM cells, which may contribute to the observed increased resistance to CDDP. Conversely, survivin has also been shown to negatively regulate p53, either by inhibiting caspase-mediated cleavage of MDM2 [34] or by safeguarding against mitotic catastrophe and the resulting DNA damage [56]. Targeting of survivin with antisense oligonucleotides in MPM resulted in the appearance of a cleavage product of MDM2 after antisense treatment, suggesting that survivin may indeed stabilize MDM2 [34]. In addition, survivin downregulation also induced p53 phosphorylation, which suggests that DNA damage response is also activated.
In conclusion, this study demonstrates that p53 is functional in MPM cells and contributes to the apoptotic response induced by CDDP. It also demonstrates that removal of downstream inhibitors such as survivin can enhance this response more successfully than the disruption of p53-MDM2 interaction by p14ARF or nutlin-3.
Acknowledgement
We would like to thank Professor Gordon Peters (ICRF) for the pcDNA-p14ARF plasmid.
Abbreviations
- MPM
malignant pleural mesothelioma
- CDDP
cisplatin
Footnotes
This study was funded by the Swiss League of Cancer (OCS-01491-02-2004).
References
- 1.Carbone M, Kratzke RA, Testa JR. The pathogenesis of mesothelioma. Semin Oncol. 2002;29:2–17. doi: 10.1053/sonc.2002.30227. [DOI] [PubMed] [Google Scholar]
- 2.Peto J, Decarli A, La Vecchia C, Levi F, Negri E. The European mesothelioma epidemic. Br J Cancer. 1999;79:666–672. doi: 10.1038/sj.bjc.6690105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steele JPC. Prognostic factors in mesothelioma. Semin Oncol. 2002;29:36–40. doi: 10.1053/sonc.2002.30299. [DOI] [PubMed] [Google Scholar]
- 4.Steele JP, Klabatsa A. Chemotherapy options and new advances in malignant pleural mesothelioma. Ann Oncol. 2005;16:345–351. doi: 10.1093/annonc/mdi094. [DOI] [PubMed] [Google Scholar]
- 5.Michalak E, Villunger A, Erlacher M, Strasser A. Death squads enlisted by the tumour suppressor p53. Biochem Biophys Res Commun. 2005;331:786–798. doi: 10.1016/j.bbrc.2005.03.183. [DOI] [PubMed] [Google Scholar]
- 6.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 7.Kohn KW, Pommier Y. Molecular interaction map of the p53 and Mdm2 logic elements, which control the off-on switch of p53 in response to DNA damage. Biochem Biophys Res Commun. 2005;331:816–827. doi: 10.1016/j.bbrc.2005.03.186. [DOI] [PubMed] [Google Scholar]
- 8.Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutat Res. 2005;576:22–38. doi: 10.1016/j.mrfmmm.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Khan S, Guevara C, Fujii G, Parry D. p14ARF is a component of the p53 response following ionizing irradiation of normal human fibroblasts. Oncogene. 2004;23:6040–6046. doi: 10.1038/sj.onc.1207824. [DOI] [PubMed] [Google Scholar]
- 10.Khan SH, Moritsugu J, Wahl GM. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci USA. 2000;97:3266–3271. doi: 10.1073/pnas.050560997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 12.Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- 13.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 14.Prins JB, Williamson KA, Kamp MM, Van Hezik EJ, Van der Kwast TH, Hagemeijer A, Versnel MA. The gene forthe cyclin-dependent kinase-4 inhibitor, CDKN2A, is preferentially deleted in malignant mesothelioma. Int J Cancer. 1998;75:649–653. doi: 10.1002/(sici)1097-0215(19980209)75:4<649::aid-ijc25>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang CT, You L, Uematsu K, Yeh CC, McCormick F, Jablons DM. p14(ARF) modulates the cytolytic effect of ONYX-015 in mesothelioma cells with wild-type p53. Cancer Res. 2001;61:5959–5963. [PubMed] [Google Scholar]
- 16.Testa JR, Giordano A. SV40 and cell cycle perturbations in malignant mesothelioma. Semin Cancer Biol. 2001;11:31–38. doi: 10.1006/scbi.2000.0344. [DOI] [PubMed] [Google Scholar]
- 17.Lopez-Rios F, Illei PB, Rusch V, Ladanyi M. Evidence against a role for SV40 infection in human mesotheliomas and high risk of false-positive PCR results owing to presence of SV40 sequences in common laboratory plasmids. Lancet. 2004;364:1157–1166. doi: 10.1016/S0140-6736(04)17102-X. [DOI] [PubMed] [Google Scholar]
- 18.Schmitter D, Lauber B, Fagg B, Stahel RA. Hematopoietic growth factors secreted by seven human pleural mesothelioma cell lines: interleukin-6 production as a common feature. Int J Cancer. 1992;51:296–301. doi: 10.1002/ijc.2910510220. [DOI] [PubMed] [Google Scholar]
- 19.Ke Y, Reddel RR, Gerwin BI, Reddel HK, Somers AN, McMenamin MG, LaVeck MA, Stahel RA, Lechner JF, Harris CC. Establishment of a human in vitro mesothelial cell model system for investigating mechanisms of asbestos-induced mesothelioma. Am J Pathol. 1989;134:979–991. [PMC free article] [PubMed] [Google Scholar]
- 20.Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003;10:356–364. doi: 10.1038/sj.cdd.4401157. [DOI] [PubMed] [Google Scholar]
- 21.Olie RA, Simoes-Wust AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60:2805–2809. [PubMed] [Google Scholar]
- 22.Damia G, Filiberti L, Vikhanskaya F, Carrassa L, Taya Y, D'Incalci M, Broggini M. Cisplatinum and Taxol induce different patterns of p53 phosphorylation. Neoplasia. 2001;3:10–16. doi: 10.1038/sj.neo.7900122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 24.Minamoto T, Buschmann T, Habelhah H, Matusevich E, Tahara H, Boerresen-Dale AL, Harris C, Sidransky D, Ronai Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene. 2001;20:3341–3347. doi: 10.1038/sj.onc.1204458. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 26.Attardi LD, de Vries A, Jacks T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene. 2004;23:973–980. doi: 10.1038/sj.onc.1207026. [DOI] [PubMed] [Google Scholar]
- 27.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 28.Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- 29.Yang CT, You L, Yeh CC, Chang JW, Zhang F, McCormick F, Jablons DM. Adenovirus-mediated p14(ARF) gene transfer in human mesothelioma cells. J Natl Cancer Inst. 2000;92:636–641. doi: 10.1093/jnci/92.8.636. [DOI] [PubMed] [Google Scholar]
- 30.Gallagher S, Kefford RF, Rizos H. Enforced expression of p14ARF induces p53-dependent cell cycle arrest but not apoptosis. Cell Cycle. 2005;4:465–472. doi: 10.4161/cc.4.3.1526. [DOI] [PubMed] [Google Scholar]
- 31.Falleni M, Pellegrini C, Marchetti A, Roncalli M, Nosotti M, Palleschi A, Santambrogio L, Coggi G, Bosari S. Quantitative evaluation of the apoptosis regulating genes Survivin, Bcl-2 and Bax in inflammatory and malignant pleural lesions. Lung Cancer. 2005;48:211–216. doi: 10.1016/j.lungcan.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Kren L, Brazdil J, Hermanova M, Goncharuk VN, Kallakury BV, Kaur P, Ross JS. Prognostic significance of anti-apoptosis proteins survivin and bcl-2 in non-small cell lung carcinomas: a clinicopathologic study of 102 cases. Appl Immunohistochem Mol Morphol. 2004;12:44–49. doi: 10.1097/00129039-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 33.Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, Ciofani M, Rottapel R, Zuniga-Pflucker JC, et al. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med. 2004;199:399–410. doi: 10.1084/jem.20032092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23:8146–8153. doi: 10.1038/sj.onc.1207992. [DOI] [PubMed] [Google Scholar]
- 35.Xia C, Xu Z, Yuan X, Uematsu K, You L, Li K, Li L, McCormick F, Jablons DM. Induction of apoptosis in mesothelioma cells by antisurvivin oligonucleotides. Mol Cancer Ther. 2002;1:687–694. [PubMed] [Google Scholar]
- 36.Altomare DA, Vaslet CA, Skele KL, De Rienzo A, Devarajan K, Jhanwar SC, McClatchey AI, Kane AB, Testa JR. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res. 2005;65:8090–8095. doi: 10.1158/0008-5472.CAN-05-2312. [DOI] [PubMed] [Google Scholar]
- 37.Carbone M, Rizzo P, Grimley PM, Procopio A, Mew DJ, Shridhar V, de Bartolomeis A, Esposito V, Giuliano MT, Steinberg SM, et al. Simian virus-40 large-T antigen binds p53 in human mesotheliomas. Nat Med. 1997;3:908–912. doi: 10.1038/nm0897-908. [DOI] [PubMed] [Google Scholar]
- 38.McCormick F. Cancer-specific viruses and the development of ONYX-015. Cancer Biol Ther. 2003;2:S157–S160. [PubMed] [Google Scholar]
- 39.Edwards SJ, Dix BR, Myers CJ, Dobson-Le D, Huschtscha L, Hibma M, Royds J, Braithwaite AW. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14(ARF) tumor suppressor genes. J Virol. 2002;76:12483–12490. doi: 10.1128/JVI.76.24.12483-12490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 41.Gordon GJ, Rockwell GN, Jensen RV, Rheinwald JG, Glickman JN, Aronson JP, Pottorf BJ, Nitz MD, Richards WG, Sugarbaker DJ, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol. 2005;166:1827–1840. doi: 10.1016/S0002-9440(10)62492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerley-Hamilton JS, Pike AM, Li N, DiRenzo J, Spinella MJ. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embryonal carcinoma. Oncogene. 2005;24:6090–6100. doi: 10.1038/sj.onc.1208755. [DOI] [PubMed] [Google Scholar]
- 43.Xu GW, Mymryk JS, Cairncross JG. Inactivation of p53 sensitizes astrocytic glioma cells to BCNU and temozolomide, but not cisplatin. J Neuro-Oncol. 2005;74:141–149. doi: 10.1007/s11060-004-6601-3. [DOI] [PubMed] [Google Scholar]
- 44.Hawkins DS, Demers GW, Galloway DA. Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res. 1996;56:892–898. [PubMed] [Google Scholar]
- 45.Fan S, Smith ML, Rivet DJ, II, Duba D, Zhan Q, Kohn KW, Fornace AJ, Jr, O'Connor PM. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995;55:1649–1654. [PubMed] [Google Scholar]
- 46.Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 48.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. From the cover: Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. Apoptosis inhibitory activity of cytoplasmic p21 (Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le HV, Minn AJ, Massague J. Cyclin-dependent kinase inhibitors uncouple cell cycle progression from mitochondrial apoptotic functions in DNA-damaged cancer cells. J Biol Chem. 2005;280:32018–32025. doi: 10.1074/jbc.M504689200. [DOI] [PubMed] [Google Scholar]
- 51.Hopkins-Donaldson S, Cathomas R, Simoes-Wust AP, Kurtz S, Belyanskya L, Stahel RA, Zangemeister-Wittke U. Induction of apoptosis and chemosensitization of mesothelioma cells by Bcl-2 and Bcl-xL antisense treatment. Int J Cancer. 2003;106:160–166. doi: 10.1002/ijc.11209. [DOI] [PubMed] [Google Scholar]
- 52.Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 53.Salz W, Eisenberg D, Plescia J, Garlick DS, Weiss RM, Wu XR, Sun TT, Altieri DC. A survivin gene signature predicts aggressive tumor behavior. Cancer Res. 2005;65:3531–3534. doi: 10.1158/0008-5472.CAN-04-4284. [DOI] [PubMed] [Google Scholar]
- 54.Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during Taxol-induced apoptosis. J Biol Chem. 2003;278:23130–23140. doi: 10.1074/jbc.M300957200. [DOI] [PubMed] [Google Scholar]
- 55.Altieri DC. Cytokinesis, apoptosis and survivin: three for tango? Cell Death Differ. 2001;8:4–5. doi: 10.1038/sj.cdd.4400795. [DOI] [PubMed] [Google Scholar]
- 56.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]