

Abstract
Early experience has a particularly great effect on most organisms. Normal development may be disrupted by early environmental influences; individuals that survive have to cope with the damaging consequences. Additionally, the responses required to cope with environmental challenges in early life may have long-term effects on the adult organism. A further set of processes, those of developmental plasticity, may induce a phenotype that is adapted to the adult environment predicted by the conditions of early life. A mismatch between prediction and subsequent reality can cause severe health problems in those human societies where economic circumstances and nutrition are rapidly improving. Understanding the underlying mechanisms of plasticity is, therefore, clinically important. However, to conduct research in this area, developmental plasticity must be disentangled from disruption and the adverse long-term effects of coping. The paper reviews these concepts and explores ways in which such distinctions may be made in practice.
Keywords: developmental plasticity, developmental disruption, adaptive responses, foetus, human disease
1. Introduction
The early phase of life in many species is characterized by the presence of responses to environmental stimuli. This developmental plasticity is important in generating a range of phenotypes suitable for different environments and, thus, promoting the perpetuation of the genotype. Developmental plasticity may, however, also play a role in the generation of human disease (Bateson 2001; Bateson et al. 2004).
The human foetus and infant can respond to unbalanced nutrition and other adverse influences by changing their developmental and growth trajectories (Gluckman & Hanson 2004b). The processes involved may include induction of attributes that adapt the individual for the type of environment in which he or she is likely to live later in life. People who are born particularly small have health disadvantages both in infancy and in later life (Barker 1998). But even within the normal range of birth sizes, birth weight is inversely related to the risk of developing coronary heart disease, Type 2 diabetes and hypertension (Barker 2001). Developmental pathways may affect the incidence of human disease in other ways. Those born as heavier babies and similarly brought up in affluent environments generally enjoy a reduced risk of such chronic disease as adults (excluding those whose increased size is owing to maternal pathological conditions, such as diabetes mellitus; Barker 2001; Barker et al. 2001; Eriksson et al. 2001). Postnatal ill-effects arising from these distinct developmental pathways observed in humans appear to result from the mismatch between the postnatal environmental state and the phenotype developmentally selected in utero or in early infancy. The relationship between constrained or impaired foetal or infant development and fast growth in childhood may be responsible for the increased risks of the diseases of affluence (Eriksson et al. 2001; Gluckman & Hanson 2004a). The epidemiological observations that led to this view (Barker & Osmond 1986; Rich-Edwards et al. 1997), and which remain somewhat controversial (reviewed in Gluckman & Hanson 2004d), suggest that such disease is not simply the result of adult lifestyle, but that it has important developmental determinants that are not genetic in origin. Such observations have important implications, given the increasing concern about the epidemic of cardiovascular and metabolic disease worldwide (Popkin 2001; Fall 2003). The one estimate of the size of the effects of the early environment on later disease risk (Barker et al. 2002) suggests that it is very substantial.
Several explanations have been offered for the long-term effects on the offspring of an altered maternal environment (Hattersley & Tooke 1999; Hales & Barker 2001; Singhal et al. 2003; Gluckman & Hanson 2004a). In this paper we identify these alternative hypotheses using examples of direct and indirect changes in nutrient availability. Since these explanations are not necessarily mutually exclusive, we explore how the different influences on adult characteristics are confounded and how they may be disentangled from each other. Clarity in understanding these issues may assist in resolving the continuing debate about the relative importance of developmental factors in the genesis of adult disease. For convenience, and because of the plethora of good examples, we shall usually limit our discussion to mammalian—especially human—development, but many of our general points are applicable to a wide range of animal and plant species.
2. Developmental responses to environmental influences
Development of an individual obviously depends on both its genes and its environment, but the precise character of the interplay between the developing individual and its environment is critical. At each phase of development the organism may be sensitive to particular environmental cues with the effects of its responses impacting on subsequent stages of development. In sheep, the nutritional status of the mother at around conception can influence how the late gestation foetus responds to subsequent maternal nutritional stress (Harding 1997). The setting of the hypothalamic pituitary adrenal axis may also respond in parallel and, hence, lead to changes in the postnatal responses to external stressors (Hawkins et al. 2001; Bloomfield et al. 2004). In the rat, prenatal under-nutrition changes how the offspring responds to different levels of nutrition (Vickers et al. 2000; Ozanne et al. 2004). Such processes affect many physiological systems, such as appetite, nutrient partitioning, cardiovascular control and responses to stress, each of which may confer some survival advantage in the appropriate environment. In table 1, we classify the types of responses the developing organism can make to an environmental influence.
Table 1.
Types of developmental response to environmental influences
| nature of the response | time of conferred advantage | outcome | examples |
|---|---|---|---|
| developmental disruption | nil | survivors may be severely dysfunctional | neural tube defect owing to maternal folate deficiency (Kalter 2003) |
| immediately adaptive | immediate | survival but with possible long-term costs (‘coping’) to Darwinian fitness | premature delivery following maternal infection—benefit is leaving infected intrauterine environment early while cost is permanent stunting (Hofman et al. 2004; Keelan et al. 2003; Irving et al. 2000) |
| predictively adaptive | not necessarily immediate, anticipated to be advantageous at a later period | advantageous if predicted future and actual future environments match | coat thickness in meadow vole pup (Lee & Zucker 1988); low sweat gland innervation in a hot environment (Kawahata & Sakamoto 1951; Diamond 1991) |
| disadvantageous if predicted and future actual environments mismatch | impaired metabolic responses to a nutritionally rich adult environment of prenatally constrained individuals within the normal birth weight range (Gluckman & Hanson 2004a) |
(a) Developmental disruption
Developmentally disruptive events in response to environmental stimuli irreversibly interfere with embryonic development and, depending on their nature, may have deleterious effects either in utero and/or after birth. Generally, such cues act by interfering with a developmental process during periods of vulnerability, such that structural deficits emerge. The stimulus may be a drug, ionizing radiation, a major environmental shift such as hyperthermia or hypoxia, disease, or a gross nutritional disruption (e.g. folate deficiency leads to neural tube defects) in humans and other mammals.
(b) Immediately adaptive responses
Effects on homeostatic responses can also be developmentally-specific, representing examples of specific adaptations to the conditions of foetal life. For example, the availability of oxygen to the foetus is limited by the capacity of the placenta to transfer oxygen, and the foetus has no mechanism by which to increase oxygen availability. Therefore, under low oxygen conditions, the foetus must conserve oxygen, by redistributing blood flow to vital organs such as the heart and brain at the expense of others such as muscle, gut and kidney, and by reducing oxidative metabolism. After birth, the initial response of all terrestrial vertebrates to shortage of oxygen is to increase pulmonary ventilation. The foetus does not use its lungs for ventilation, but it does make rhythmic breathing-like movements that are essential to prenatal lung growth in preparation for birth. However, these movements are energy consuming and need not be continuous. When oxygen supplies are poor, the foetus uses a specific descending neural pathway to inhibit breathing movements to conserve oxygen for high priority functions (i.e. cardiac and brain utilisation). Postnatally, this inhibitory pathway must be inactivated so that ventilation can be enhanced during conditions of low oxygen (Johnston et al. 1990). A further example is that the neural mechanisms underlying infant suckling and associated behaviour are different from those underlying adult feeding (Williams et al. 1979). These examples illustrate adaptive responses made by the developing organism that have a clear, immediate value in utero or in the infant period.
(c) Predictive adaptive responses
Some responses made by the developing organism to environmental conditions may not have immediate adaptive value, but manifest their benefits later in life. It needs to be emphasized that these are not simply the effects of constraint in utero, but rather mechanisms by which the foetus uses an early environmental cue to ‘predict its future’ and adopts a developmental pathway that might best suit it to its expected postnatal or adult environment. The efficacy of this sort of plastic strategy will depend on whether the prediction made is correct (i.e. well-matched to actual environmental conditions in later life) or not (mismatched; Bateson et al. 2004; Gluckman & Hanson 2004a,c). The evolution of the ability to mount a predictive and adaptive plastic response will probably depend on a number of features, such as the accuracy of the cue and the frequencies of the various environmental states, as well as the consequences of a mismatch (Gluckman & Hanson 2004c) and the intrinsic costs of plasticity itself (DeWitt et al. 1998).
An example of such a predictive adaptive response is coat thickness in the meadow vole (Microtus pennsylvanicus; Lee & Zucker 1988). The vole pup is born with a thicker coat in autumn than in spring. Given the constancy of both the intrauterine and the nesting thermal environments, development of coat thickness, which is induced by trans-placental transfer of melatonin signalling day-length (Lee et al. 1989), can only be made in expectation of the predicted postnatal thermal environment. Such predictive responses may be an important component of mammalian development. Through them, the foetus predicts its postnatal environment based on maternal cues transduced via the placenta and sets its physiological homeostatic mechanisms to match that postnatal environment. Clearly, a considerable amount depends on the accuracy of the prediction. When a match between the predicted and actual environment occurs, the offspring will almost certainly thrive. Where the prediction is incorrect, however, the organism is left with a postnatal physiology that is mismatched and inappropriate, putting it at increased risk from predation or disease (Gluckman & Hanson 2004a). Such errors in prediction might arise either because the postnatal environment has shifted or because the foetus has received faulty information on which to base its prediction. The latter is most likely to happen in the presence of maternal disease or placental dysfunction, but also as a result of exaggerated maternal constraint (see below; Gluckman & Hanson 2004b).
Predictive responses can be also induced in neonates. The reported effects of the infant thermal environment on the number of active sweat glands may represent a human example (Kawahata & Sakamoto 1951; Diamond 1991). The mechanisms underlying predictive adaptive responses are likely to involve epigenetic change such as DNA methylation and other functional and structural changes, upon which subsequent development is contingent, occurring during sensitive periods in development (Waterland & Jirtle 2004).
While we have described each of the three possible types of response as distinct processes, they have characteristics that may overlap and grade into each other.
3. Coping and tradeoffs
Between the pathological and adaptive extremes, the developing organism might be forced by the environmental exposure to ‘cope’ in ways that might involve homeostatic mechanisms or more substantial adjustments in physiology and morphology to ensure survival. Sometimes this coping will have long-term effects on Darwinian fitness by imposing costs that impact on the individual at a later stage in its life. Coping responses allow the foetus to survive a potentially deleterious environment until birth. For example, a general response of the foetus to maternal nutritional deprivation is to reduce foetal growth (reviewed in Harding & Gluckman 2001). The foetus cannot outgrow the supply of nutrients delivered across the placenta, and matching growth to supply is therefore an important and inevitable aspect of the regulation of foetal growth. This supply is limiting even under physiological conditions and this limitation is termed maternal constraint (Gluckman & Hanson 2004b). An impaired growth trajectory may become irreversible if foetal under-nutrition is prolonged (Mellor & Murray 1982). This change is clearly of benefit to the foetus, enabling it to optimize the use of limited nutrients but, as we have discussed, it may have postnatal costs. Pancreatic development, and thereby insulin release, is altered (Fowden & Hill 2001), the pathways by which insulin exerts its effects on substrate uptake into tissues are changed (Ozanne et al. 2003), and so is the growth of blood vessels supplying nutrients to highly metabolically active tissues (Bennis-Taleb et al. 1999). All these latter processes will lead to a level of ‘insulin resistance’, which economizes on energy consumption. If the offspring is reared in an energy-poor environment, then such phenotypic attributes confer advantage and the individual is more likely to survive to reproduce. Conversely, if the individual is reared in a nutritionally enriched postnatal environment, then it may be adversely affected by having insulin resistance, with pathological consequences. This example shows how a prenatal survival strategy may have long-term costs in some environments.
If the developing organism increases its chances of survival by, for example, economizing on the building of structures that would have been helpful to it in later life, the short-term gain may be at the expense of its subsequent reproductive success. In polygynous species, such as red deer, a small male may be unable to compete with larger males for mates and, as a consequence, have a low chance of fathering any offspring (Albon et al. 1987; Kruuk et al. 1999). In such cases the risks of failing to survive through devoting too many resources to growth have to be balanced against the risks of failing to reproduce. The balance, of course, is achieved during the course of evolution, with those individuals that find the optimal trade-off being most likely to have descendants.
The developing organism is not like a machine that is still being constructed and it does not have to be useful while being built. Rather, the living organism has to function at each stage of development and may play an active role in its own survival and further development. Consequently, it is likely to be equipped with specialized anatomical and physiological adaptations enabling it to deal with its foetal (if it is a mammal), neonatal and juvenile environments. The particularities of the foetal circulation provide an anatomical example. Foetal oxygenation is provided by the placenta rather than by the lungs. Two unique vessels, the ductus venosus and ductus arteriosus, together with the foramen ovale (a prenatal perforation in the inter-atrial septum of the heart), create a pattern of circulation in the foetus so that a substantial fraction of the most oxygenated blood bypasses the foetal liver and lungs to supply preferentially the foetal brain and heart. Shortly after birth, these ducts and the foramen ovale close and perfusion of the lungs increases enormously to allow oxygenation via the lungs.
4. Timing of developmental influences
In mammals, environmental influences that have long-term consequences can operate even on the pre-ovulatory oocyte and pre-implantation embryo. For example, the allocation of embryonic stem cells to inner cell mass versus trophectoderm is altered in blastocysts taken from rats fed a low protein diet for only the first 4.5 days of pregnancy; the offspring develop a higher blood pressure postnatally (Kwong et al. 2000). Comparable effects have been reported in larger animals, where the in vitro culture conditions of sheep embryos can induce dramatic subsequent effects such as very large offspring (Young et al. 2001; Walker et al. 1996). Increasing attention is focused on the possible long-term effects of human in vitro fertilization (Jackson et al. 2004).
Postnatal manipulation in infancy also has long-term consequences. Handling of the neonatal rat pup has long-term consequences for the neural regulation of the glucocorticoid axis and, thus, to corticosterone release, an important mediator of the stress response (Weaver et al. 2004). Different feeding regimes in human neonates, particularly in those born prematurely (Singhal et al. 2003), have long-term metabolic consequences. Different infant growth patterns may lead to different disease risks in adult life (Eriksson et al. 2001).
Clearly, the effects of an environmental stressor may be developmentally stage-dependent. For example, studies of survivors of the Dutch winter famine of 1944–1945 show specific long-term consequences of nutrient deprivation at different stages in human pregnancy (Ravelli et al. 1998; Ravelli et al. 1999).
Some developmental responses may have multi-generational effects. For instance, manipulation of maternal dietary methyl group provision in agouti mice around conception alters the degree of DNA methylation at the agouti locus in the pre-implantation embryo and the colour of the subsequently developing coat (Wolff et al. 1998; Waterland & Jirtle 2003). Such intergenerational effects in mammals are further confounders in interpreting experimental and clinical observations. For example, the growth of descendants of rats undernourished in utero did not return to normal for several generations (Stewart et al. 1980). The female offspring of women exposed to famine in Holland in 1944/45 in turn gave birth to growth-impaired infants (Lumey 1992). The abnormal vascular responses of rodent foetuses exposed to abnormal maternal nutrition in utero persist into the second generation (Brawley et al. 2003). At least three possible mechanisms could be involved in such intergenerational effects. The first is epigenetic change, by which DNA is modified by environmental effects, either via methylation at CpG islands or by changes in histone acetylation (Jablonka et al. 1992). While previously considered to be restricted to a single generation owing to reversal by demethylation after meiosis, recent evidence suggests that this clearing of a ‘memory’ via methylation in some alleles is incomplete and that the effect may persist beyond one generation (Waterland & Jirtle 2003). Secondly, the female reproductive tract develops in the first half of human foetal life. Girls that were growth retarded in utero have reduced uterine size and this reduction may lead to impaired uterine–placental function when, in time, these women become pregnant (Ibanez et al. 2000). Thirdly, changes in metabolic homeostasis induced in the foetus/neonate may affect metabolic and cardiovascular function when these offspring in turn become pregnant; these outcomes may then induce particular phenotypes in the next generation of offspring (Drake & Walker 2004; Gluckman & Hanson 2004c).
At the molecular level, adaptive responses will be based on physiologically regulated mechanisms with appropriate changes in gene expression. Suites of genes are obviously involved in any major phenotypic outcome induced by developmental plasticity (Evans & Wheeler 2001). However, the induction of such changes is likely to be triggered by epigenetic effects on the genome. Thus epigenetic approaches may be particularly informative. Manipulating the behavioural environment of the neonatal rat produces highly specific changes in DNA methylation that affect gene expression permanently, with consequent endocrine and behavioural changes persisting into adulthood (Weaver et al. 2004). This type of epigenetic change (involving biochemical modification of DNA) is probably a fundamentally important mechanism in developmental plasticity. In contrast, developmental disruption owing to very poor levels of nutrition or other extreme environmental changes is not regulated.
5. Implications for empirical work
These considerations raise some important questions in the interpretation of empirical data. For example, if an early environmental cue has long-term effects, one must consider whether they have arisen because of developmental disruption or whether the response is a consequence of developmental plasticity and involves an adaptive and regulated physiological response. In the former case, the effects need not be related to the triggering event. In the latter case, the responses should relate functionally to the environmental events that triggered them, having immediate benefit or long-term predictive and adaptive significance (see table 1).
Studies of the reduction in nephron number in the kidneys are illustrative of these issues. Nephron number is reduced in the offspring of rodents in which the maternal diet was protein-restricted during pregnancy (Langley-Evans et al. 1999, 2003). Reduced nephron number may play a role in the aetiology of human hypertension, especially in those born with reduced body weight (Ingelfinger 2003; Keller et al. 2003). Is this reduced nephron number simply developmental disruption induced by a severe nutritional stimulus? Is it an appropriate adaptive response in utero to conserve energy under conditions of foetal nutrient limitation? Or is it a predictive response without adaptive value in utero, but induced in the expectation of a limited postnatal environment because the kidney has a large postnatal energy demand and nephron capacity is generally in excess? If the postnatal environment is richer than expected and the animal shows marked postnatal growth, potentially exceeding the limited renal reserve, is this outcome an example of an inappropriate prediction, simply a trade-off, or the consequences of a developmental disruption? The currently available evidence does not easily separate these possibilities.
What studies could help to tease out the different possible effects of environment on early mammalian development? By way of an example, consider a range of foetal nutritional conditions ranging from very low to extremely high (figure 1). Developmentally disruptive effects are most likely to be seen under very poor nutritional conditions; coping responses that have long-term adverse effects are most likely to be seen in low but not extreme conditions; predictive adaptive responses are likely to be expressed from moderately low to moderately high condition; and coping and disruptive effects may again be expressed in excessively high conditions. These effects are likely to grade into each other. Empirical programmes of research need to establish the precise shape of the function depicted in figure 1. Research design must consider the potentially confusing effects of trans-generational or within-generation changes in nutrition.
Figure 1.
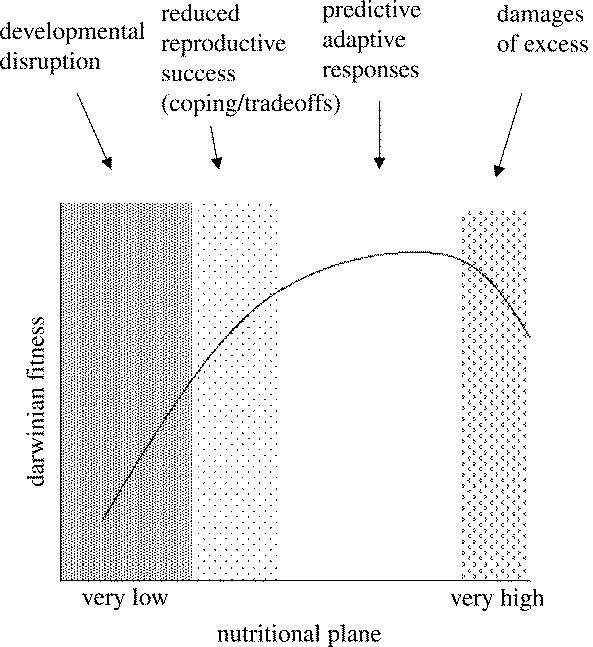
Proposed relationship between the environmental (nutritional) plane to which the developing organism is exposed to during the phase of developmental plasticity and the consequences for its Darwinian fitness in adult life. The relationship is determined by different mechanisms according to the degree of shift in the environmental state from the optimal, as suggested by the shaded columns.
6. The human perspective
The longer-term effects of environmental influences in early life may, however, become more important as longevity becomes a greater feature of the human species. A functional and evolutionary approach suggests that the pregnant woman may signal to her unborn baby about its future world. If the foetal forecast is wrong, however, the offspring is at greater health risk because its physiological adaptations are mismatched with the environment in which he or she will live. Gluckman & Hanson (2004b) suggested that the foetal environment was likely to match the postnatal environment during most of hunter–gatherer evolution. Further, the combination of maternal constraint on foetal growth and adaptive response mechanisms promoted fitness by creating an asymmetry that allowed a greater probability of survival through periods of poor nutrition. The rapid advances in longevity in recent times, however, together with the extreme changes in the postnatal nutritional/energy environment, have created a greater probability of mismatch (Gluckman & Hanson 2004c). Similarly, recent suggestions that rapid infant growth is the key risk factor (Barker et al. 2002; Eriksson et al. 2001) can be interpreted as permissive release from the constraint of foetal life in the presence of high nutrient availability (Gluckman & Hanson 2004c; Eriksson et al. 1999).
The diversity in past and present ecological conditions is likely to introduce complexity into the relationship between developmental prediction and later health. For example, some populations will have adapted to conditions of nutritional stress, especially seasonal food shortages, over a long time span (Truswell & Hansen 1976), while others will have been buffered from such effects leading to genetic differences between populations (Hattersley & Tooke 1999; Neel 1999; Diamond 2003). Recent studies on polymorphisms in a regulatory pathway affecting insulin sensitivity (Eriksson et al. 2002, 2003) demonstrate that the capacity of early environmental influences to affect later disease risk can be influenced by genotype. The possibility of a ‘thrifty genotype’ (Neel 1962) well adapted to harsh conditions is not incompatible with the plastic induction of deprivation-adapted phenotypes from a pool of uniform genotypes. Indeed, we would suggest that environmental factors acting on development must be considered alongside genetic factors in understanding the origins of many human diseases.
In summary, developmental cues may impact on long-term outcomes for the organism in one of several ways. These possibilities have implications for understanding the ecology of human disease. Distinctions should be made between (i) developmental disruption in response to environmental cues acting during the phase of developmental plasticity, (ii) immediately adaptive responses with long-term consequences and (iii) those adaptive responses induced early in development, but conferring long-term advantage. The empirical data, whether clinical or experimental, need to be interpreted in light of these considerations. Doing so will greatly improve our resolution of the important issue of the relative weight of developmental factors in the origin of health and disease.
Acknowledgments
We thank Dr C. Pinal for her assistance with the manuscript. Many of these concepts were discussed at a workshop held in February 2004 at the Liggins Institute, University of Auckland sponsored by the International Society for the Developmental Origins of Health and Disease and we thank the other participants for their contributions to the discussion.
Footnotes
As this paper exceeds the maximum length normally permitted, the authors have agreed to contribute to production costs.
References
- Albon S.D, Clutton-Brock T.H, Guinness F.E. Early development and population dynamics in red deer. II. Density-independent effects and cohort variation. J. Anim. Ecol. 1987;56:69–81. [Google Scholar]
- Barker D.J.P. 2nd edn. Churchill Livingstone; Edinburgh: 1998. Mothers, babies and health in later life. [Google Scholar]
- Barker D.J.P. 1st edn. Marcel Dekker; New York: 2001. Fetal origins of cardiovascular and lung disease. [Google Scholar]
- Barker D.J, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker D.J.P, Forsen T, Uutela A, Osmond C, Eriksson J.G. Size at birth and resilience to effects of poor living conditions in adult life: longitudinal study. Br. Med. J. 2001;323:1273–1276. doi: 10.1136/bmj.323.7324.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D.J.P, Eriksson J.G, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int. J. Epidemiol. 2002;31:1235–1239. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- Bateson P. Fetal experience and good adult design. Int. J. Epidemiol. 2001;30:928–934. doi: 10.1093/ije/30.5.928. [DOI] [PubMed] [Google Scholar]
- Bateson P, et al. Developmental plasticity and human health. Nature. 2004;430:419–421. doi: 10.1038/nature02725. [DOI] [PubMed] [Google Scholar]
- Bennis-Taleb N, Remacle C, Hoet J.J, Reusens B. A low-protein isocaloric diet during gestation affects brain development and alters permanently cerebral cortex blood vessels in rat offspring. J. Nutr. 1999;129:1613–1619. doi: 10.1093/jn/129.8.1613. [DOI] [PubMed] [Google Scholar]
- Bloomfield F.H, Oliver M.H, Hawkins P, Holloway A.C, Campbell M, Gluckman P.D, Harding J.E, Challis J.R. Periconceptional undernutrition in sheep accelerates maturation of the fetal hypothalamic–pituitary–adrenal axis in late gestation. Endocrinology. 2004;145:4278–4285. doi: 10.1210/en.2004-0424. [DOI] [PubMed] [Google Scholar]
- Brawley L, Poston L, Hanson M. Mechanisms underlying the programming of small artery dysfunction: review of the model using low protein diet in pregnancy in the rat. Arch. Physiol. Biochem. 2003;111:25–35. doi: 10.1076/apab.111.1.23.15138. [DOI] [PubMed] [Google Scholar]
- DeWitt T.J, Sih A, Wilson D.S. Costs and limits of phenotypic plasticity. Trends Ecol. Evol. 1998;13:77–81. doi: 10.1016/s0169-5347(97)01274-3. [DOI] [PubMed] [Google Scholar]
- Diamond J. Pearl Harbor and the Emperor's physiologists. Nat. Hist. 1991;100:2–7. [PubMed] [Google Scholar]
- Diamond J. The double puzzle of diabetes. Nature. 2003;423:599–602. doi: 10.1038/423599a. [DOI] [PubMed] [Google Scholar]
- Drake A.J, Walker B.R. The intergenerational effects of fetal programming: non-genomic mechanisms for the inheritance of low birth weight and cardiovascular risk. J. Endocrinol. 2004;180:1–16. doi: 10.1677/joe.0.1800001. [DOI] [PubMed] [Google Scholar]
- Eriksson J.G, Forsen T, Tuomilehto J, Winter P.D, Osmond C, Barker D.J. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. Br. Med. J. 1999;318:427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J.G, Forsen T, Tuomilehto J, Osmond C, Barker D.J. Early growth and coronary heart disease in later life: longitudinal study. Br. Med. J. 2001;322:949–953. doi: 10.1136/bmj.322.7292.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J.G, Lindi V, Uusitupa M, Forsen T.J, Laakso M, Osmond C, Barker D.J. The effects of the Pro12Ala polymorphism of the peroxisome proliferator-activated receptor-gamma2 gene on insulin sensitivity and insulin metabolism interact with size at birth. Diabetes. 2002;51:2321–2324. doi: 10.2337/diabetes.51.7.2321. [DOI] [PubMed] [Google Scholar]
- Eriksson J.G, Osmond C, Lindi V, Uusitupa M, Forsen T, Laakso M, Barker D. Interactions between peroxisome proliferator-activated receptor gene polymorphism and birth length influence risk for type 2 diabetes. Diabetes Care. 2003;26:2476–2477. doi: 10.2337/diacare.26.8.2476-a. [DOI] [PubMed] [Google Scholar]
- Evans J.D, Wheeler D.E. Gene expression and the evolution of insect polymorphisms. Bioessays. 2001;23:62–68. doi: 10.1002/1521-1878(200101)23:1<62::AID-BIES1008>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Fall C.H.D. The fetal and early life origins of adult disease. Indian Pediatr. 2003;40:480–502. [PubMed] [Google Scholar]
- Fowden, A. L. & Hill, D. J. 2001 Intra-uterine programming of the endocrine pancreas. Br. Med. Bull.60, 123–142. [DOI] [PubMed]
- Gluckman P.D, Hanson M.A. The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 2004a;15:183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D, Hanson M.A. Maternal constraint of fetal growth and its consequences. Semin. Fetal Neonat. Med. 2004b;9:419–425. doi: 10.1016/j.siny.2004.03.001. in press. [DOI] [PubMed] [Google Scholar]
- Gluckman P.D, Hanson M.A. Cambridge University Press; Cambridge, UK: 2004c. The fetal matrix: evolution, development, and disease. [Google Scholar]
- Gluckman P.D, Hanson M.A. Living with the past: evolution, development and patterns of disease. Science. 2004d;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Hales C.N, Barker D.J.P. The thrifty genotype hypothesis. Br. Med. Bull. 2001;60:5–20. doi: 10.1093/bmb/60.1.5. [DOI] [PubMed] [Google Scholar]
- Harding J.E. Periconceptual nutrition determines the fetal growth response to acute maternal undernutrition in fetal sheep of late gestation. Prenat. Neonat. Med. 1997;2:310–319. [Google Scholar]
- Harding J.E, Gluckman P.D. Growth, metabolic and endocrine adaptations to fetal undernutrition. In: Barker J.D.P, editor. Fetal origins of cardiovascular and lung disease. Marcel Dekker; New York: 2001. pp. 181–197. [Google Scholar]
- Hattersley A.T, Tooke J.E. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet. 1999;353:1789–1792. doi: 10.1016/S0140-6736(98)07546-1. [DOI] [PubMed] [Google Scholar]
- Hawkins P, Hanson M.A, Matthews S.G. Maternal undernutrition in early gestation alters molecular regulation of the hypothalamic–pituitary–adrenal axis in the ovine fetus. J. Neuroendocrinol. 2001;13:855–861. doi: 10.1046/j.1365-2826.2001.00709.x. [DOI] [PubMed] [Google Scholar]
- Hofman P.L, Regan F, Harris M, Robinson E, Jackson W, Cutfield W.S. The metabolic consequences of prematurity. Growth Horm. IGF Res. 2004;14:S136–S139. doi: 10.1016/j.ghir.2004.03.029. [DOI] [PubMed] [Google Scholar]
- Ibanez L, Potau N, Enriquez G, de Zegher F. Reduced uterine and ovarian size in adolescent girls born small for gestational age. Pediatr. Res. 2000;47:575–577. doi: 10.1203/00006450-200005000-00003. [DOI] [PubMed] [Google Scholar]
- Ingelfinger J.R. Is microanatomy destiny? N. Engl. J. Med. 2003;348:99–100. doi: 10.1056/NEJMp020168. [DOI] [PubMed] [Google Scholar]
- Irving R.J, Belton N.R, Elton R.A, Walker B.R. Adult cardiovascular risk factors in premature babies. Lancet. 2000;355:2135–2136. doi: 10.1016/S0140-6736(00)02384-9. [DOI] [PubMed] [Google Scholar]
- Jablonka E, Lachmann M, Lamb M.J. Evidence, mechanisms and models for the inheritance of acquired characters. J. Theor. Biol. 1992;158:245–268. [Google Scholar]
- Jackson R.A, Gibson K.A, Wu Y.W, Croughan M.S. Perinatal outcomes in singletons following in vitro fertilization: a meta analysis. Obstet. Gynecol. 2004;103:551–563. doi: 10.1097/01.AOG.0000114989.84822.51. [DOI] [PubMed] [Google Scholar]
- Johnston B.M, Moore P.J, Bennet L, Hanson M.A, Gluckman P.D. Almitrine mimics hypoxia in fetal sheep with lateral pontine lesions. J. Appl. Physiol. 1990;69:1330–1335. doi: 10.1152/jappl.1990.69.4.1330. [DOI] [PubMed] [Google Scholar]
- Kalter H. Teratology in the 20th century. Environmental causes of congenital malformations in humans and how they were established. Neurotoxicol. Teratol. 2003;25:131–282. [PubMed] [Google Scholar]
- Kawahata A, Sakamoto H. Some observations on sweating of the Aino. Jpn. J. Physiol. 1951;2:166–169. doi: 10.2170/jjphysiol.2.166. [DOI] [PubMed] [Google Scholar]
- Keelan J.A, Blumentstein M, Helliwell R.J, Sato T.A, Marvin K.W, Mitchell M.D. Cytokines, prostaglandins and parturition—a review. Placenta. 2003;24:S33–S36. doi: 10.1053/plac.2002.0948. [DOI] [PubMed] [Google Scholar]
- Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003;348:101–108. doi: 10.1056/NEJMoa020549. [DOI] [PubMed] [Google Scholar]
- Kruuk L.E.B, Clutton-Brock T.H, Rose K.E, Guinness F.E. Early determinants of lifetime reproductive success differ between the sexes in red deer. Proc. R. Soc. B. 1999;266:1655–1661. doi: 10.1098/rspb.1999.0828. (doi:10.1098/rspb.1999.0828) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong W.Y, Wild A.E, Roberts P, Willis A.C, Fleming T.P. Maternal undernutrition during the preimplantation period of rat development causes blastocyst abnormalities and programming of postnatal hypertension. Development. 2000;127:4195–4202. doi: 10.1242/dev.127.19.4195. [DOI] [PubMed] [Google Scholar]
- Langley-Evans S.C, Welham S.J, Jackson A.A. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci. 1999;64:965–974. doi: 10.1016/s0024-3205(99)00022-3. [DOI] [PubMed] [Google Scholar]
- Langley-Evans S.C, Langley-Evans A.J, Marchand M.C. Nutritional programming of blood pressure and renal morphology. Arch. Physiol. Biochem. 2003;111:8–16. doi: 10.1076/apab.111.1.8.15136. [DOI] [PubMed] [Google Scholar]
- Lee T.M, Zucker I. Vole infant development is influenced perinatally by maternal photoperiodic history. Am. J. Physiol. 1988;255:R831–R838. doi: 10.1152/ajpregu.1988.255.5.R831. [DOI] [PubMed] [Google Scholar]
- Lee T.M, Spears N, Tuthill C.R, Zucker I. Maternal melatonin treatment influences rates of neonatal development of meadow vole pups. Biol. Reprod. 1989;40:495–502. doi: 10.1095/biolreprod40.3.495. [DOI] [PubMed] [Google Scholar]
- Lumey L.H. Decreased birthweights in infants after maternal in utero exposure to the Dutch famine of 1944–1945. Paediatr. Perinat. Epidemiol. 1992;6:240–253. doi: 10.1111/j.1365-3016.1992.tb00764.x. [DOI] [PubMed] [Google Scholar]
- Mellor D.J, Murray L. Effects of long term undernutrition of the ewe on the growth rates of individual fetuses during late pregnancy. Res. Vet. Sci. 1982;32:177–180. [PubMed] [Google Scholar]
- Neel J.V. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 1962;14:353–362. [PMC free article] [PubMed] [Google Scholar]
- Neel J.V. The “thrifty genotype” in 1998. Nutr. Rev. 1999;57:s2–s9. doi: 10.1111/j.1753-4887.1999.tb01782.x. [DOI] [PubMed] [Google Scholar]
- Ozanne S.E, et al. Early growth restriction leads to down regulation of protein kinase C zeta and insulin resistance in skeletal muscle. J. Endocrinol. 2003;177:235–241. doi: 10.1677/joe.0.1770235. [DOI] [PubMed] [Google Scholar]
- Ozanne S.E, Lewis R, Jennings B.J, Hales C.N. Early programming of weight gain in mice prevents the induction of obesity by a highly palatable diet. Clin. Sci. 2004;106:141–145. doi: 10.1042/CS20030278. [DOI] [PubMed] [Google Scholar]
- Popkin B.M. Nutrition in transition: the changing global nutrition challenge. Asia Pac. J. Clin. Nutr. 2001;10:S13–S18. [PubMed] [Google Scholar]
- Ravelli A.C, van der Meulen J.H, Michels R.P, Osmond C, Barker D.J, Hales C.N, Bleker O.P. Glucose tolerance in adults after prenatal exposure to famine. Lancet. 1998;351:173–177. doi: 10.1016/s0140-6736(97)07244-9. [DOI] [PubMed] [Google Scholar]
- Ravelli A.C, van der Meulen J.H, Osmond C, Barker D.J, Bleker O.P. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am. J. Clin. Nutr. 1999;70:811–816. doi: 10.1093/ajcn/70.5.811. [DOI] [PubMed] [Google Scholar]
- Rich-Edwards J.W, Stampfer M.J, Manson J.E, Rosner B, Hankinson S.E, Colditz G.A, Willett W.C, Hennekens C.H. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. Br. Med. J. 1997;315:396–400. doi: 10.1136/bmj.315.7105.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A, Fewtrel M, Cole T.J, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet. 2003;361:1089–1097. doi: 10.1016/S0140-6736(03)12895-4. [DOI] [PubMed] [Google Scholar]
- Stewart R.J.C, Sheppard H, Preece R, Waterlow J.C. The effect of rehabilitation at different stages of development of rats marginally malnourished for ten to twelve generations. Br. J. Nutr. 1980;43:403–412. doi: 10.1079/bjn19800108. [DOI] [PubMed] [Google Scholar]
- Truswell, A. S. & Hansen, J. D. 1976 Medical research among the !Kung. In Kalahari hunter-gatherers. Studies of the !Kung San and their neighbors (ed. R. B. Lee & I. DeVore), pp. 168–195. Cambridge, MA: Harvard University Press.
- Vickers M.H, Breier B.H, Cutfield W.S, Hofman P.L, Gluckman P.D. Fetal origins of hyperphagia, obesity and hypertension and its postnatal amplification by hypercaloric nutrition. Am. J. Physiol. 2000;279:E83–E87. doi: 10.1152/ajpendo.2000.279.1.E83. [DOI] [PubMed] [Google Scholar]
- Walker S.K, Hartwich K.M, Seamark R.F. The production of unusually large offspring following embryo manipulation: concepts and challenges. Theriogenology. 1996;45:111–120. [Google Scholar]
- Waterland R.A, Jirtle R.L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland R.A, Jirtle R.L. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Weaver I.C.G, Cervoni N, Champagne F.A, D'Alessio A.C, Sharma S, Seckl J.R, Dymov S, Szyf M, Meaney M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Williams C.L, Rosenblatt J.S, Hall W.G. Inhibition of suckling in weaning-age rats: a possible serotonergic mechanism. J. Comp. Physiol. Psychol. 1979;93:414–429. doi: 10.1037/h0077592. [DOI] [PubMed] [Google Scholar]
- Wolff G.L, Kodell R.L, Moore S.R, Cooney C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12:949–957. [PubMed] [Google Scholar]
- Young L.E, et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 2001;27:153–154. doi: 10.1038/84769. [DOI] [PubMed] [Google Scholar]