

Abstract
We introduce a new Bayesian clustering algorithm for studying population structure using individually geo-referenced multilocus data sets. The algorithm is based on the concept of hidden Markov random field, which models the spatial dependencies at the cluster membership level. We argue that (i) a Markov chain Monte Carlo procedure can implement the algorithm efficiently, (ii) it can detect significant geographical discontinuities in allele frequencies and regulate the number of clusters, (iii) it can check whether the clusters obtained without the use of spatial priors are robust to the hypothesis of discontinuous geographical variation in allele frequencies, and (iv) it can reduce the number of loci required to obtain accurate assignments. We illustrate and discuss the implementation issues with the Scandinavian brown bear and the human CEPH diversity panel data set.
IT has been a recent matter of debate to decide whether clusters identified by Bayesian algorithms were artificially detected structures emerging from uneven sampling along clines or were actually well-differentiated groups (Serre and Pääbo 2004; Rosenberg et al. 2005). It has indeed been suggested that uneven sampling during the experimental design might influence clustering patterns and that the degree of clustering might be diminished by use of samples with greater spatial homogeneity. This dilemma has even introduced doubt about whether Bayesian clustering algorithms are appropriate tools for studying genetic structure in populations with continuous variation of allele frequencies.
Such issues have been reported after a study of genetic structure of human populations by Rosenberg et al. (2002). Without the use of predefined populations, this study inferred the geographical ancestries of individuals from 52 worldwide samples with individuals genotyped at 377 microsatellite loci. Using the Bayesian clustering program STRUCTURE (Pritchard et al. 2000) and increasing the number of loci from 377 to 993, Rosenberg et al. (2005) have shown that the six clusters found in their previous study are robust and, at the notable exception of the genetic isolate Kalash, that they match with the major geographic regions in the world. These clusters were interpreted as arising from small discontinuities in allele frequencies when geographical barriers are crossed.
In the latter and other applications of clustering algorithms, the spatial data are actually treated off line and are not part of the modeling. Bayesian models such as those developed by Pritchard et al. (2000), Dawson and Belkhir (2001), or Corander et al. (2003) nevertheless offer a natural and appropriate framework for including spatial prior information when assigning an individual to a fixed number of clusters. For example, a recent study by Guillot et al. (2005) used spatial explicit priors in a full-Bayes perspective and successfully identified genetic barriers in a wolverine population. An assignment method was also used by Wasser et al. (2004) to infer the spatial origin of African elephants. Here we argue that modified Bayesian algorithms can provide additional evidence to solve cline/cluster dilemmas such as those discussed in Rosenberg et al. (2005). A natural way to proceed is to include priors on continuous variation of genetic diversity in the Bayesian model used by STRUCTURE and check whether or not the previously discussed clusters are robust.
In this study, we present a new hierarchical Bayes algorithm that incorporates models for geographical continuity of allele frequencies. This is achieved by using hidden Markov random fields (HMRFs) as prior distributions on cluster membership. An informal definition of HMRFs states that allele frequencies at a specific geographical site are more likely to be close to the allele frequencies at neighboring sites than at distant sites. The problem of local differentiation may also be studied in terms of change in correlation with distance as considered by Malécot (1948), where “individuals living nearby tend to be more alike than those living far apart” (Kimura and Weiss 1964, p. 561). The HMRF is basically another formulation of the same idea with statistical correlation hidden at the cluster membership level.
We illustrate some applications of HMRFs in a Bayesian context. First, in populations with presumed continuous variation in allele frequencies, we argue that HMRFs are powerful when detecting geographical discontinuities in allele frequencies and regulating the number of clusters. Then, we address the cline/cluster dilemma with HMRFs using a subsample of the CEPH human polymorphism data set and check that the main clusters obtained with STRUCTURE are robust to the inclusion of continuous variation in allele frequencies through space. In addition, we show that an accuracy similar to the one obtained with nonspatial methods can be achieved while using a smaller number of genetic markers.
THE POTTS–DIRICHLET MODEL
In this study we borrow from the toolbox of statistical physics the concept of Markov random field (MRF), also called the Potts model (Potts 1952; Preston 1974; Wu 1982. The model has been coined to handle stochastic networks where particles in identical states evolve in patches larger than expected under an absence of interactions. Guttorp (1995) gives a recent review of the Potts model at a fairly introductory level. Since the 1970s, MRFs have a long tradition in image analysis, where the color of pixels is correlated to the color of neighboring pixels (see, e.g., Geman and Geman 1984; Besag 1986; Ripley 1988). In this context MRFs account for the property that adjacent pixels are more likely to be of the same color than nonadjacent pixels. HMRFs are relatively recent, but they have been successfully applied in several domains (Zhang et al. 2001; Green and Richardson 2002; Destrempes et al. 2005). Ideas from Bayesian spatial genetics were also used in association studies (Thomas et al. 2003). In analogy with image analysis, MRF can model the fact that individuals from spatially continuous populations are more likely to share cluster membership with their close neighbors than with distant representatives. They seem therefore relevant to study populations for which continuous variation of allele frequencies may be used as a postulate.
Devising MRF models raises a difficulty when the study design is irregular. While the definition of neighborhood is immediate in the case of lattice observations, it is less obvious in the case of irregular sampling, because many choices are available. In this study, we use the natural neighborhood structure obtained from the so-called Dirichlet tiling. Denoting by (si), 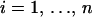 , the set of observation sites for n individuals, each si is surrounded by points that are closer to si than to any other sampling site. This set of points is known as the Dirichlet cell (or tile). Two sampling sites are neighbors if their cells share a common edge. The use of the sampling locations to define cells is natural unless the sampling locations are unrepresentative of the individual spatial distribution. However, the method works in principle for any fixed tiling, as soon as the user can define a neighborhood structure to incorporate in the Potts model. In the sequel, we refer to the Potts model build on the Dirichlet tiling generated by sampling sites as the Potts–Dirichlet model.
, the set of observation sites for n individuals, each si is surrounded by points that are closer to si than to any other sampling site. This set of points is known as the Dirichlet cell (or tile). Two sampling sites are neighbors if their cells share a common edge. The use of the sampling locations to define cells is natural unless the sampling locations are unrepresentative of the individual spatial distribution. However, the method works in principle for any fixed tiling, as soon as the user can define a neighborhood structure to incorporate in the Potts model. In the sequel, we refer to the Potts model build on the Dirichlet tiling generated by sampling sites as the Potts–Dirichlet model.
We denote by ci the cluster from which the individual i originates, and we assume the existence of at most Kmax clusters. As we shall see later, the constant Kmax should indeed be considered to be larger than the true (or presumed true) number of clusters, K. We let c = (ci) denote the cluster configuration, i.e., a map that takes all cells and specifies the clusters to which they belong. In addition we let U(c) denote the number of neighboring pairs with the same labels in c. Formally, we have
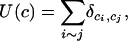 |
(1) |
where i ∼ j indicates that i and j are neighbors, and the Kronecker symbol 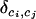 takes the value 1 if ci = cj and otherwise 0. Large values of U(c) correspond to spatial patterns with large patches of individuals belonging to the same cluster. Small values of U(c) (maybe equal to 0) correspond to patterns that do not display any sort of spatial organization.
takes the value 1 if ci = cj and otherwise 0. Large values of U(c) correspond to spatial patterns with large patches of individuals belonging to the same cluster. Small values of U(c) (maybe equal to 0) correspond to patterns that do not display any sort of spatial organization.
The Potts model is a probability distribution on the set of cluster configurations. Given n observation sites, the probability of configuration c is written as
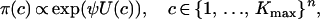 |
(2) |
where ψ is a nonnegative parameter called the interaction parameter. The value ψ = 0 corresponds to the uniform distribution on the configuration space. Large values of ψ make more likely the observation of largely clustered configurations corresponding to large U(c). Two simulations of the Potts–Dirichlet model are displayed in Figure 1 for Kmax = 3, ψ = 0.1, ψ = 0.9, where the sites were generated from the uniform distribution on a square domain. For Kmax = 3–6, simulations (not reported) showed that the value ψ = 1.0 can be considered a high level of spatial interaction, for which the probability that pairs of neighbors are in the same cluster is close to one. In contrast, values of ψ ≤ 0.4 correspond to weak interactions. In this case the probability that pairs of neighbors are in the same cluster is <0.3. Values of ψ around ψ ≈ 0.6–0.7 are suitable for observing the coexistence of several clusters, while for larger values the model has a tendency to form a single cluster. We also note that the Potts model does not assume connected clusters, and the number K of observed clusters may be lower than Kmax.
Figure 1.—

Two cluster configurations from the three-states Potts–Dirichlet model. For ψ = 0.1, no spatial structure can be observed (the situation is close to the noninformative prior used by STRUCTURE). For ψ = 0.9, a number of nonnecessarily connected random clusters can be observed.
To work with a well-defined probability distribution, the requirement that probabilities sum to one must be fulfilled. This is achieved by taking
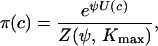 |
(3) |
where Z(ψ, Kmax) is a normalizing constant called the partition function:
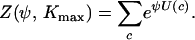 |
(4) |
Computing the partition function of the Potts model and performing perfect sampling for an arbitrary graph is feasible if there are only a few sampling sites; otherwise it is a highly difficult problem. Historically the Metropolis algorithm got around the issue by using an ingenious cancellation of this constant term (Metropolis et al. 1953).
In addition to providing a flexible way to model a spatially organized population, the Potts model satisfies a spatial Markov property that states that the conditional probability for membership in ci given the configuration at all other sites c−i = (cj)j≠i is equal to the conditional probability given the state of its neighbors c∂i = (cj)j∼i. Mathematically, this property can be written as
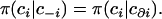 |
(5) |
More specifically, we have
 |
(6) |
The above conditional probabilities involve local computations only, and the sum 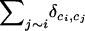 can be interpreted as the sum of influences of all neighbors of i. The Markov property is a basis for implementing fast simulation and inference algorithms.
can be interpreted as the sum of influences of all neighbors of i. The Markov property is a basis for implementing fast simulation and inference algorithms.
HIERARCHICAL BAYES
Model:
In this section, we present the hierarchical Bayes model based on an HMRF. With ψ = 0, the HMRF model assumes a noninformative spatial prior and then encompasses the classical Bayesian clustering models of Pritchard et al. (2000), Dawson and Belkhir (2001), and Corander et al. (2003), which can be seen as particular cases. In addition to a spatial prior, a second modification of the standard Bayesian clustering model includes departures from the HW equilibrium caused by inbreeding. Inbreeding coefficients represent the probability that two homologous genes are identical by descent. To implement the modification, inbreeding coefficients can be considered as additional statistical parameters φk. We use notations similar to those used in the previous works: L is the number of loci, Jℓ is the number of alleles at locus ℓ, and z is the collection of all genotypes (the data). Given that the individual i originates from the cluster ci = k and given the allele frequencies fk.. in this cluster, the conditional probability of observing the genotype ziℓ = (aiℓ, biℓ) at locus ℓ is
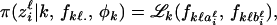 |
(7) |
where 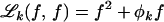 and
and 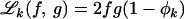 for f ≠ g (see, e.g., Hartl and Clark 1997). Diploidy is also assumed.
for f ≠ g (see, e.g., Hartl and Clark 1997). Diploidy is also assumed.
We write the set of all parameters as θ = (ψ, c, f, φ) with ψ the interaction parameter; c the cluster configuration; f = (fkℓj), k = 1, … , Kmax, ℓ = 1, … , L, j = 1, … , Jℓ, the allele frequencies; and 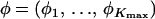 the inbreeding coefficients in each subpopulation. As in STRUCTURE, the priors on allele frequencies are Dirichlet distributions
the inbreeding coefficients in each subpopulation. As in STRUCTURE, the priors on allele frequencies are Dirichlet distributions 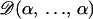 . The prior distributions on the φk's are beta
. The prior distributions on the φk's are beta 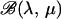 distributions. Although we have included ψ in the parameter list to implement a full-Bayes approach, the estimation of ψ nevertheless generates specific computational difficulties due to the exponential number of terms involved in the partition function Z (Gelman and Meng 1998). For this reason, we often consider fixed values for this parameter with typical values within the range (0.1, 1.0). This can be formulated with prior distributions on the rescaled interaction parameters ψ/ψmax being either beta distributions or constant (Dirac) distributions. The prior distribution on θ reflects the hierarchy of the model and takes the following form:
distributions. Although we have included ψ in the parameter list to implement a full-Bayes approach, the estimation of ψ nevertheless generates specific computational difficulties due to the exponential number of terms involved in the partition function Z (Gelman and Meng 1998). For this reason, we often consider fixed values for this parameter with typical values within the range (0.1, 1.0). This can be formulated with prior distributions on the rescaled interaction parameters ψ/ψmax being either beta distributions or constant (Dirac) distributions. The prior distribution on θ reflects the hierarchy of the model and takes the following form:
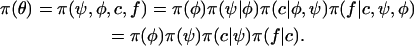 |
(8) |
Assuming linkage equilibrium between loci, the likelihood is defined as
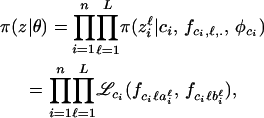 |
(9) |
where 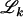 is defined in Equation 7.
is defined in Equation 7.
Inference using Markov chain Monte Carlo:
Inferences on θ are carried out by simulating the posterior distribution π(θ | z) through a Markov chain Monte Carlo (MCMC) sampling algorithm. In this algorithm, we combine sequential updates of blocks of parameters, each block of parameters being either fully or partially updated. The description of the MCMC steps is detailed in the appendix. A complete update of all blocks of parameters is referred to as a cycle.
Estimating the number of clusters:
As other Bayesian clustering methods do, the HMRF model refers implicitly to an unknown number of clusters K. In practice this number K has to be estimated. Previous approaches typically fall into two categories: (1) maximizing the likelihood modified with a penalty that decreases with model complexity (e.g., Bayes information criteria and deviance information criteria) and (2) choosing a prior distribution on K and maximizing the posterior distribution using transdimensional MCMC computations (which are usually time-consuming to develop and to run). Although these methods have proved effective in many cases, we use an alternative approach known as regularization in statistics. For this terminology, we refer to the book by Ripley (1996, Chap. 4.3, p. 136). The rationale for regularization and the relationship with the algorithm implemented in STRUCTURE can be explained as follows. Let Ls(z, f, c) denote the log-probability for the complete data (observed plus unobserved) in the original approach of Pritchard et al. (2000). When we refer to this approach, we mean the no-admixture model with uncorrelated allele frequencies. Assuming absence of inbreeding, the log-probability of the HMRF model can be expressed as
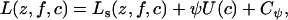 |
(10) |
where the term U(c) represents the contribution from the spatial prior, and Cψ is a constant that depends on ψ. For the value ψ = 0, the model implemented in STRUCTURE is then recovered. In fact, Equation 10 corresponds to the Lagrangian formulation of an optimization problem where ψ can be viewed as the Lagrange multiplier. With the data in hand, the optimization problem seeks the most likely cluster assignments under the constraint that a maximal number of neighboring pairs should fall in the same clusters. For small ψ's (ψ < 0.3), the constraint is weak, and the results are expected to be close to those produced by STRUCTURE. For larger values the results are generally expected to differ.
In the regularization approach, Kmax is a value presumed larger than the true number of clusters K. When the algorithm is started, the cluster configuration c spans arbitrary values between 1 and Kmax. As the chain runs, the program attempts to reduce the number of nonempty clusters that is finally considered as an estimate of K. In practice, one starts with runs with small values of Kmax and increases Kmax unless the estimated K is strictly lower than Kmax. Then, one checks that the result remains identical when higher values of Kmax are used. Practice also shows that repeating shorter runs and performing estimation from the runs with the highest likelihood is a reasonable strategy.
The connections between model selection and regularization have been emphasized several times in the statistical literature. Indeed, regularization is a key argument in statistical procedures such as ridge regression (Hoerl and Kennard 1970), lasso estimators (Tibshirani 1996), and feedforward neural networks weight decay (Bishop 1995). Such methods were successful in various areas such as text mining or gene selection from large transcriptomic data sets. Nevertheless, we are not aware of any published statistical methods that have used regularization in a hidden context as is done here. The relevance of the regularization principle is carefully assessed in simulation study.
SIMULATION STUDY
In this section we report results from an intensive simulation study. The goals of our experiments are (i) to give evidence that the MCMC implementation is correct, (ii) to assess the value of predictions obtained from the HMRF model with particular attention paid to estimation of the unknown number of populations K and the cluster configuration c, and (iii) to compare the HMRF model with a nonspatial approach and to a lesser extent with the Bayesian clustering algorithm GENELAND developed by Guillot et al. (2005).
Estimating the number of clusters:
To check the validity of the HMRF model, we performed inferences for 300 simulated data sets obtained as replicates from the model prior distributions. Individual geographical coordinates were generated from a two-dimensional uniform distribution on a square domain. Genotypes with 10 loci and 10 alleles per locus were simulated using multinomial sampling from the Dirichlet 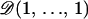 distribution. The interaction parameter ψ was simulated according to a uniform distribution on
distribution. The interaction parameter ψ was simulated according to a uniform distribution on 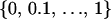 . The inbreeding coefficients were simulated according to a beta
. The inbreeding coefficients were simulated according to a beta  distribution. The hidden cluster configurations c were generated from the Potts–Dirichlet model with K = Kmax = 1, 2, 3 classes. Replicates with K = 1, 2, 3 classes were simulated for n = 50, 100, 150 individuals, respectively.
distribution. The hidden cluster configurations c were generated from the Potts–Dirichlet model with K = Kmax = 1, 2, 3 classes. Replicates with K = 1, 2, 3 classes were simulated for n = 50, 100, 150 individuals, respectively.
In the full-Bayes inference method (inference of ψ), the computation of the partition function Z(ψ, Kmax) involved preliminary off-line runs. They were carried out with 20,000 cycles of a Gibbs sampler with a thinning period of 10 cycles. The maximal number of clusters was fixed to Kmax = 5, and 30,000 cycles, a burn-in period of 20,000, and a thinning period of 10 cycles were used. The parameter ψ was kept equal to 0 during the first 5000 cycles (see Updating the interaction parameter ψ in the appendix for more details).
The estimation errors are summarized in Figure 2. This figure displays histograms for the three types of data sets K = 1, 2, 3. For data sets made of a single population, the HMRF model estimated 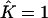 in almost all replicates. Data sets made of K = 2 clusters were also identified as being so for >80 replicates (of 100), and, in the data sets for which we had
in almost all replicates. Data sets made of K = 2 clusters were also identified as being so for >80 replicates (of 100), and, in the data sets for which we had 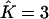 instead of
instead of 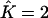 , the third cluster consisted of less than two individuals. For data sets made of K = 3 populations, perfect estimation dropped to 55%, but a closer look at the results for which we had
, the third cluster consisted of less than two individuals. For data sets made of K = 3 populations, perfect estimation dropped to 55%, but a closer look at the results for which we had 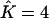 instead of
instead of 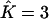 revealed that the third cluster consisted of less than four individuals. In these cases, a longer run might empty the spurious cluster (but we did not evaluate how long this might take). In all simulations, each extra cluster consisted of at most six individuals. Furthermore, K was never underestimated. These results are summarized in Table 1.
revealed that the third cluster consisted of less than four individuals. In these cases, a longer run might empty the spurious cluster (but we did not evaluate how long this might take). In all simulations, each extra cluster consisted of at most six individuals. Furthermore, K was never underestimated. These results are summarized in Table 1.
Figure 2.—

Distributions of the number of clusters estimated by the HRMF model. Data sets were simulated from the prior distributions of the HMRF model. The vertical lines indicate the true number of populations.
TABLE 1.
Proportions of individuals assigned to extra clusters given the number of estimated clusters 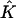 and their true number K
and their true number K
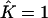 |
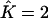 |
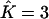 |
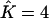 |
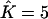 |
|
|---|---|---|---|---|---|
| True K = 1 | 0 | 0.02 | 0 | 0 | 0 |
| True K = 2 | — | 0 | 0.0136 | — | 0.03 |
| True K = 3 | — | — | 0 | 0.0096 | 0.0267 |
— indicates cases that never occurred during the simulation study.
Estimating cluster membership probabilities:
We now turn to the accuracy of inference in terms of correct assignments. We denote by (xij) the n × n matrix whose entries are xij = 1 if ci = cj and 0 otherwise. Similarly we denote by 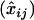 the corresponding matrix obtained from the estimated cluster configuration
the corresponding matrix obtained from the estimated cluster configuration 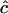 . We assessed the accuracy of cluster assignment through the error rate in coassignment (ERCA) defined as
. We assessed the accuracy of cluster assignment through the error rate in coassignment (ERCA) defined as
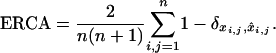 |
This pair-based measure has the advantage over individual-based indexes of being insensitive to the issue of (cluster) label switching.
To assess the benefit of our approach as compared to models accounting neither for inbreeding nor for spatial structure, we carried out additional experiments from the HMRF model at ψ = 0 and φ = 0 (Hardy–Weinberg equilibrium assumed). The assumptions of this simpler model (referred to as the nonspatial model) were similar to those made in the programs STRUCTURE (Pritchard et al. 2000), PARTITION (Dawson and Belkhir 2001), and BAPS (Corander et al. 2003). The HMRF model with fixed parameters ψ = 0 and φ = 0 was used instead of these programs to avoid potential biases due to specific computer implementations. Typical cluster configurations at low and high ψ's are portrayed in Figure 1 for K = 3. They correspond to low and high levels of spatial organization (ψ = 0.1 and 0.9). In this section similar situations were reproduced with K = 2.
We simulated 200 data sets from the HMRF model prior distributions with Kmax = 2, using simulations from the MCMC program without data (1000 cycles). Running the program for a fixed number of cycles did not warrant the convergence of the MCMC sampler. As the aim of the simulation study was the retrieval of previously stored allele frequencies and cluster memberships, this shortcoming did not affect the performance study. In the sampled data, individuals were occasionally grouped in a single cluster (for values of ψ > 0.8). The clusters had no predefined size and might consist of very few (<10) individuals. The ERCA rates are reported in Table 2. In this table, the rates were averaged either over all data sets or over subsets of data that corresponded to different levels of pairwise FST, interaction parameter ψ, and inbreeding coefficients (φ1, φ2).
TABLE 2.
Error rate in coassignments (ERCA) for 200 simulated data sets (n = 100, L = 10, Jℓ = 10) with Kmax = 2
| Genetic structure: | Spatial structure: | Inbreeding | |||
|---|---|---|---|---|---|
| FST | ψ | (φ1, φ2) | Nonspatial model | HMRF model | GENELAND |
| All | All | All | 16.1 | 0.7 | 3.2 |
| FST ≤ 0.08 | All | All | 26.3 | 1.6 | 6.6 |
| 0.08 < FST ≤ 0.09 | All | All | 7.6 | 0.6 | 1.4 |
| 0.09 < FST ≤ 0.1 | All | All | 8 | 0.6 | 1.4 |
| FST > 0.1 | All | All | 8.3 | 0.2 | 1.1 |
| All | ψ ≤ 0.2 | All | 1.1 | 1 | 1.1 |
| All | 0.2 < ψ ≤ 0.4 | All | 1 | 0.8 | 1.6 |
| All | 0.4 < ψ ≤ 0.6 | All | 2.7 | 0.7 | 0.9 |
| All | 0.6 < ψ ≤ 0.8 | All | 28.2 | 0.4 | 4.7 |
| All | ψ > 0.8 | All | 42.4 | 0.5 | 6.9 |
| All | All | (<0.06, <0.06) | 17.2 | 0.3 | 0.7 |
| All | All | (<0.06, >0.1) or (>0.1, <0.06) | 10 | 0.5 | 1.9 |
| All | All | (>0.1, >0.1) | 12.3 | 1 | 1.5 |
| FST ≤ 0.08 | ψ ≤ 0.4 | All | 2.7 | 2.1 | 2.8 |
| FST ≤ 0.08 | 0.6 < ψ ≤ 1 | All | 41.8 | 0.9 | 9.4 |
| FST > 0.1 | ψ ≤ 0.4 | All | 0.2 | 0.1 | 0.4 |
| FST > 0.1 | 0.6 < ψ ≤ 1 | All | 23.7 | 0.3 | 2.4 |
The three models were initialized at Kmax = 2.
The results provided evidence that the HMRF model increased the number of correct assignments compared to the nonspatial model. A more detailed look at subsets of simulated data revealed that the HMRF model always performed better than the other models whatever the levels of spatial interaction or inbreeding. The highest improvements were obtained at low levels of differentiation (FST ≤ 0.08) and high levels of spatial structure (ψ > 0.6). The HMRF model achieved the smallest improvements over the other models for high levels of inbreeding, although it still gave very accurate results. In these cases, the inbreeding coefficients were correctly estimated (results not shown).
The error rates of the nonspatial model were in some cases very high. This was indeed the case for large values of ψ. These results may be explained as data sets generated from large ψ sometimes contained a single cluster. Due to the regularization procedure, this cluster was successfully detected by the HMRF model (and also by GENELAND) but not by the nonspatial model, which split the unique population into two arbitrary parts.
These results carried information about the performance of the HMRF model when the initial number of clusters was close to the true number (Kmax = 2, K = 1 or 2). We repeated the inference study on the same 200 data sets with Kmax = 5. The global ERCA was ∼10%, which was still a low misclassification rate.
REAL DATA ANALYSIS
Scandinavian brown bears:
The Scandinavian brown bear (Ursus arctos) is an example of a wild population with strong female phylopatry and male-mediated gene flows. We analyzed the same data set as in two previous studies (Waits et al. 2000; Manel et al. 2004) from 366 geo-referenced individuals genotyped at 19 microsatellite loci. We first used the full-Bayes HMRF model implemented with the same prior distributions as in the simulation study and ran the algorithm with Kmax = 4–7. After 30,000 cycles, the HMRF model with Kmax = 4 converged to the same clusters as described in the previous study. We referred to these clusters as the south (S), middle (M), north–west–north (NWN), and north–north (NN) areas. With Kmax = 5–7, the HMRF model yielded five clusters, three of which coincided with the Kmax = 4 run while the fourth (S) was split into two subsets with random shapes. The spatial interaction parameter ψ had posterior mode within the range (0.6, 0.8) (95% credible interval). However, the random shapes of the two S subclusters were an indicator that the MCMC runs might have not converged, perhaps due to the large amount of computational resource spent in the estimation of ψ. Therefore we performed 10 additional runs of the algorithm for two values of the interaction parameter ψ = 0.7–0.8. The runs that reached the highest likelihood resulted in the same four clusters as previously observed (see Figure 3). Inferences carried out under a fixed large value of ψ usually favor cluster configurations made of few large clusters. The fact that the HRMF model obtained the same clusters as STRUCTURE gave evidence that these original clusters were robust to the inclusion of a spatial prior. A by-product of the HMRF model is its ability to infer inbreeding coefficients. The inbreeding coefficients posterior estimates were computed as φNN = 0.022, φNWN = 0.006, φM = 0.013, and φS = 0.007. These small values were consistent with the observation that STRUCTURE worked well for this data set. The HMRF model with fixed parameter setting converged faster than the full-Bayes version. (We used 1000 cycles for Kmax = 4 and 20,000 cycles for Kmax = 7.) GENELAND runs at fixed K = 4–6 produced the same assignment results as the HMRF model (5000 cycles). Using reversible jumps, the posterior distribution of K exhibited a mode at K = 5 and a 95% credible interval K ∈ (4, 8) (50,000 cycles).
Figure 3.—

Estimated cluster configuration for the Scandinavian brown bear data set in North Sweden using the HMRF model (four clusters).
Human data:
We used the Human Genome Diversity Panel–Centre d'Etude du Polymorphisme Humain (HGDP–CEPH) (Cann et al. 2002) to further assess the influence and the benefit of including spatial continuity prior hypotheses in the analysis of multilocus genotypes. The HGDP–CEPH diversity panel data set contains 1056 individuals genotyped at 377 autosomal microsatellite loci. It was first studied with the software STRUCTURE by Rosenberg et al. (2002). Without using predefined populations, six main genetic clusters were identified, five of which corresponded to major geographic regions. Here we restricted the study to the Eurasian and East Asian populations, including samples with distinct origins, 8 from Pakistan, 16 from China, and 1 from Siberia, Japan, and Cambodia (451 individuals). Two reasons could be given for limiting the study to Eurasian and East Asian populations. First, these populations contained two of the five main clusters as well as the sixth cluster found by Rosenberg et al. (2002). Second, the 27 populations live on a same mainland, which justified using the Dirichlet tiling without modifying the neighborhood structure (although our computer program makes this possible). Coordinates of individuals in each sample were not known explicitly. Instead they were available as sample intervals from Cann et al. (2002). For instance, the Kalash from Pakistan have longitudes in the range 35°–37° E and latitudes in the range 71°–72° N. Individual coordinates were generated randomly within the specified intervals. We checked that the results presented here were rather independent of the individual coordinates within each sample (not reported).
To evaluate the inclusion of geographic continuity prior, subsets of data containing 20, 10, and 5 random loci were extracted from the original data set (20 subsamples for each number of loci). The HMRF model was initialized with Kmax = 3 clusters and then run for 50,000 cycles, with a burn-in period of 500 cycles and a thinning interval of 5 cycles. The interaction parameter ψ was either estimated from the same prior distributions as in the simulation study (full Bayes) or fixed to ψ = 0.6. With 20 loci, all outputs contained two clusters (Pakistan including Kalash, 8 samples, against the other Asian populations) regardless of the estimation strategy of the interaction parameter ψ. With 10 loci the HMRF model identified the two main clusters in 18 of the 20 runs. With 5 loci no successful run was observed. The nonspatial version (ψ = 0) led to the same outputs when the number of clusters was set to Kmax = 2.
To further highlight the potential of the HMRF model, we focused on the Pakistan data set and the retrieval of the Kalash cluster. The Kalash sample contains 25 of the 200 individuals from the eight Pakistan samples. Ranges for sample spatial coordinates are reported in Table 3 (Cann et al. 2002), and a representation of the resampled individual locations is displayed in Figure 4. In this study, data sets with 40, 30, and 20 randomly chosen loci were extracted from the Pakistan data set. The idea here is to use the results from a large number of loci as the “correct” answer and then see which methods are able to get this correct answer with fewer loci. Because all the extracted data sets did not contain the same amount of information about genetic structure, we distinguished three distinct levels of potential difficulty (strong clustering, SC; weak clustering, WC; and no cluster, NC) according to the following classification. For each subset, we preliminarily computed a neighbor-joining (NJ) tree using the shared allele distance (see Nei and Kumar 2000), which separated the Pakistan samples in two sister clades. Data sets for which one clade contained >20 Kalash grouped against the remaining Pakistan representatives were classified as SC. Such data sets were expected to be easy for Bayesian clustering algorithms, because a more basic analysis gives a correct answer. As well there were data sets for which no obvious clusters could be directly inferred from the NJ tree. These data sets were classified as NC, and they were expected to be difficult for Bayesian clustering algorithms. We added an intermediate class, WC, for which the Kalash sample generally formed a cluster in the NJ tree, but this was done in association with other samples such as Pathan or Balochi/Brahui. With 40 randomly chosen loci, ∼38% of all data sets were in the SC category, 24% were classified as WC, and the remaining 38% were NC. One NJ tree clustered the Balochi/Brahui against the rest of Pakistan. With 30 loci, these numbers changed to SC + WC = 42% and NC = 58%, and NC increased to 76% in the 20-loci data sets. These ratios were obtained from 300 distinct data sets.
TABLE 3.
Latitudes and longitudes for the eight Pakistan samples (from Cann et al. 2002)
| Sample name | Latitude | Longitude | Sample size |
|---|---|---|---|
| Brahui | 30°–31° N | 66°–67° E | 25 |
| Balochi | 30°–31° N | 66°–67° E | 25 |
| Hazara | 33°–34° N | 70° E | 25 |
| Makrani | 26° N | 62°–66° E | 25 |
| Shindi | 24°–27° N | 68°–70° E | 25 |
| Pathan | 32°–35° N | 69°–72° E | 25 |
| Kalash | 35°–37° N | 71°–72° E | 25 |
| Burusho | 36°–37° N | 73°–75° E | 25 |
Figure 4.—

Sampled geographical coordinates of 70 individuals from the Pakistan data set and the associated Dirichlet tiling. (The full sample was not shown but a similar spatial distribution was assumed for the 200 individuals.)
We performed 10 runs of the HMRF model for 42 subsets (21 subsets with 40 loci and 21 subsets with 30 loci). The HMRF model was first run for 200 cycles at ψ = 0.4, and these cycles were followed by a further 500–1000 cycles at ψ = 0.6. For the CEPH diversity panel data set, this strategy appeared more efficient than the full-Bayes approach, which was statistically unable to identify the Kalash (we attributed this failure to the algorithmic complications and the approximations made in estimating ψ). The run with the highest likelihood was saved as the final result. The same strategy was also used at ψ = 0 with a larger total number of cycles (up to 2000). Small burn-in (10 cycles) and thinning (1 cycle) periods were implemented. We first used Kmax = 2 in both the HMRF and nonspatial versions. To compare with published results, we also assumed absence of inbreeding.
For SC data sets with 40 loci, the HMRF model and the nonspatial versions performed similarly and retrieved the Kalash sample. Similar results were reported for STRUCTURE in the literature (Bamshad et al. 2003; Ramachandran et al. 2004). The HMRF model failed to identify the Kalash in a single WC subset whereas the nonspatial version failed twice in this category. The HMRF model identified the Kalash successfully in 75% of NC samples whereas the nonspatial version failed in the same ratio (75%). The divergence between the spatial and nonspatial version increased as we reproduced the study with 30 loci. The HMRF algorithm failed to identify the Kalash in 37% of the NC cases. The global success rate of the HMRF model was, however, >85% (including SC, WC, and NC cases) whereas this global rate dropped to 47% in the nonspatial algorithm. With 20 loci, both algorithms failed in a majority of the NC cases. For all loci, the Kmax = 3 results were in strict concordance with the Kmax = 2 results for the spatial version although >10 runs were sometimes necessary in the NC cases.
DISCUSSION
Detecting population subdivision is a subject of great interest to population geneticists, and a large body of approaches have been developed for this. In this study, we have presented a Bayesian clustering algorithm that incorporates hidden Markov random fields as prior distributions on cluster configurations. Markov random fields are mathematical models that account for the “continuity” of discrete random variables on a graph or a network (for a rigorous definition of continuity in this context, refer to Preston 1974). The term hidden means that the cluster configuration is unobserved and is instead reconstructed from an MCMC algorithm. In spatial population genetics the term continuous population usually refers to S. Wright's famous concept of isolation by distance (Wright 1943), which can in turn be understood in terms of the stepping-stone model (Kimura and Weiss 1964; Rousset 2004). Because it considers interacting demes on a lattice, the stepping-stone model exhibits the same type of spatial Markov property as does the Potts model. Inserting the stepping-stone model into a Bayesian framework generates conceptual difficulties because its stationary distribution has no known formulation. However, the HMRF model may capture its essential properties.
While STRUCTURE has recently become prominent among clustering algorithms, another recent approach includes spatially explicit priors in a highly structured statistical framework (Guillot et al. 2005). The approach developed by Guillot et al. (2005) nevertheless differs from the HMRF model significantly. In Guillot et al. (2005), population territories are viewed as unions of polygons. A full-Bayes algorithm estimates the number of populations using the reversible-jump MCMC machinery. The simulation study carried out by Guillot et al. (2005) suggests that their model performs well when genetic discontinuities occur as very simple polygonal lines are crossed (e.g., straight lines). A field study and a subsequent analysis by Coulon et al. (2006) also support these observations. Although simple shaped territories are likely to be quite common, there are also important cases where these assumptions do not hold (for example, limited gene flows in areas with complex geography, mountain ranges, worldwide studies). In the HMRF model, spatial dependencies are prescribed at the individual level directly. The advantage of the HRMF approach is that it can assign individuals when the hidden cluster configurations are too complex to be summarized by simple polygonal regions.
The HMRF model involves an interaction parameter ψ that corresponds to the intensity with which two neighbors belong to the same cluster. Estimates of ψ may be interpreted as local measures of spatial clusteredness for the studied sample. The higher ψ is the more likely that the population may consist of a unique cluster with a high level of genetic continuity (e.g., slow clinal variation). Estimates of ψ found in the studied (real) data sets were generally >0.5, which indicated the presence of continuous organization. Nevertheless, interpretations of such parameters would lead us far beyond the scope of this study, because the connection to statistical physics is not so direct in this context. In addition, we have also claimed that ψ may play a more important role as a Lagrange multiplier in a constrained optimization problem where the nonspatial likelihood is optimized while the algorithm attempts to assign a maximal number of neighbor pairs to a same cluster. We have indeed argued that the HMRF algorithm then contains an implicit way for deciding the number of clusters, a major issue in such statistical mixtures algorithms. From this perspective, maintaining fixed values of the interaction parameter ψ may be preferable to estimating this parameter and has the additional advantage of avoiding difficult computational issues (Gelman and Meng 1998). The simulation study evaluated the use of the full-Bayes HRMF algorithm (estimation of ψ) only. This was done because simulations and inferences with fixed ψ would have biased the results toward very low ERCAs and very optimistic conclusions. During the analysis of real data, versions of the HMRF model at fixed values of ψ (∼0.5–0.7) nevertheless achieved better performances and were considerably faster than the full-Bayes version.
The use of the HMRF model has been illustrated in two previously published data sets. The Scandinavian brown bear is an example of a population with a strong female phylopatry. Scandinavian bears were almost exterminated at the beginning of the 20th century. After efforts to protect the species in Sweden, the bear population has recovered from four female concentration areas. Until recently these areas were believed to represent the surviving relict subpopulations after the 1930s bottleneck (see, e.g., Waits et al. 2000). Using two independent methods (neighbor-joining trees and the Bayesian clustering algorithm STRUCTURE), Manel et al. (2004) found four genetic clusters that matched with geographical clusters, but two of them were distinct from the original female concentration areas. Using a coalescent approach, Blum et al. (2004) computed the female dispersal rate and found an estimate of 9 km per generation. Because of the low dispersal rate in this population, local genetic similarities can be considered as a reasonable assumption to be included in a Bayesian model for brown bear genetic diversity. The HMRF model has been used for detecting geographical discontinuities in allele frequencies. The results confirmed previously published results and provided reasonable estimates for the number of clusters.
Using the human CEPH diversity panel data set, we checked whether the clusters obtained without spatial priors were robust to the hypothesis of continuous geographical variation in allele frequencies. The results presented here reconciled the two apparently divergent perspectives of Rosenberg et al. (2002, 2005) and Serre and Pääbo (2004), which brought into conflict clines and clusters regarding variation of human diversity. Restricting to Eurasian and Asian populations and working with a prior on continuous variation (ψ ≈ 0.6), we recovered the three main clusters found by the algorithm STRUCTURE. Some important facts must be mentioned at this stage:
The two main clusters (Pakistan/non-Pakistan) were identified with <20 randomly chosen loci. The Kalash cluster was identified using <50 loci.
More importantly, the algorithm was unable to confirm the presence of other clusters in the Pakistan and East Asia areas, perhaps due to the simultaneous effects of reducing the number of loci (<120 loci) and imposing the continuity prior. The combination of these effects may have led to the neglect of some very small discontinuities that were previously detected when STRUCTURE was used with large values of K and a larger number of loci. We performed 10 additional runs of the HMRF model using the full set of loci. Regarding the Pakistan data, we were also not able to retrieve other clusters. Regarding the East Asia data set, we identified one additional cluster in the northeastern area that matched with the Yakut–Japanese samples. This cluster was also apparent in the NJ tree.
The weight given to the prior distribution was a moderate value that also corresponded to the posterior mean estimated from the full-Bayes algorithm when it converged [ψ ≈ 0.6, 95% credible interval (0.5, 0.9)].
A stronger level of prior interaction (e.g., ψ ≈ 1) led to a unique cluster and gave strong support to Serre and Pääbo's hypothesis of clinal variation within a unique cluster.
Weaker levels of prior interaction (e.g., ψ ≈ 0.2) led to the same results as STRUCTURE and supported Rosenberg's small discontinuities hypothesis.
Here we supported the intermediate view of clinal variation of allele frequencies with a number of discontinuities smaller than those estimated by Rosenberg et al. (2002). See Figure 5 for a picture of the reconciliation.
Figure 5.—

The reconciliation illustrated. At the left of the ψ-axis, a clustering analysis does not account for the spatial continuity of allele frequencies and may detect more clusters than actually exist. At the right, the pure continuity hypothesis assumes no cluster. Here the vision is intermediate, with the main discontinuities confirmed, but some small clusters may be considered nonsignificant.
In conclusion we have shown that the HMRF model can achieve accuracy similar to that obtained with nonspatial methods while using a smaller number of genetic markers. Consequently the use of HMRF algorithms could be advocated in cases where the number of polymorphic loci available to the study is limited, and a prior knowledge about continuous spatial structure could be incorporated with certainty.
The source codes used in this study are available as an R package that also provides additional visual displays and the data sets used during this study. The R package was mainly developed by S. Ancelet, and a version supporting Linux OS and R 3.1.1. can be downloaded from S. Ancelet's or O. François's website. A multiple-platform software will be made available within a few months.
Acknowledgments
We are grateful to Noah Rosenberg for his suggestions on an early version of this manuscript. We thank Stephanie Manel, Oscar Gaggiotti, and Chibiao Chen for fruitful discussions and Mathieu Emily for his help with simulations of the Potts model on a Dirichlet tiling. We are also grateful to two anonymous reviewers for their constructive comments. O.F. was supported by grants from the Algorithmes et populations biologiques-Institut Informatique et Mathématiques Appliquées de Grenoble project and the French ministry of research Action Concertée Incitative-Interface Mathématique physique Biologie project.
APPENDIX: DETAILS OF MARKOV CHAIN MONTE CARLO COMPUTATIONS
We iterated updates of blocks of parameters where the basic update was as follows.
Updating allele frequencies fkℓj:
We used a componentwise Metropolis–Hastings Markov chain simulation algorithm. For the cluster labeled k and locus labeled ℓ, an update of 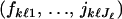 selected two alleles at random with indexes j and j′ and proposed to change their frequencies fkℓj and fkℓj′ as follows. Denoting
selected two alleles at random with indexes j and j′ and proposed to change their frequencies fkℓj and fkℓj′ as follows. Denoting 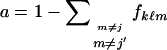 , new frequencies fkℓj* and fkℓj′* are proposed as fkℓj* = aBf and fkℓj′*= a − fkℓj*, where Bf is sampled from a beta
, new frequencies fkℓj* and fkℓj′* are proposed as fkℓj* = aBf and fkℓj′*= a − fkℓj*, where Bf is sampled from a beta 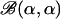 distribution (often α = 1). This move was accepted with probability
distribution (often α = 1). This move was accepted with probability
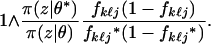 |
(A1) |
The update was based on the conditional distribution of the Dirichlet distribution (Gibbs sampler). The complete update of allele frequencies replicated this basic step for each locus and in all clusters. Typical values of α were α = 1 or 2.
Updating inbreeding coefficients φk:
We implemented a componentwise independent Metropolis–Hastings sampler. For each population we iterated the following basic update. A new inbreeding coefficient φk* was sampled from a  distribution. We assumed a beta
distribution. We assumed a beta 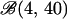 prior distribution on each φk; hence φk* was accepted with probability
prior distribution on each φk; hence φk* was accepted with probability
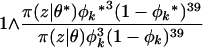 |
(A2) |
as we assumed a uniform prior on φk and made a symmetric proposal.
Updating the cluster configuration c:
We used sequential updates for all i ∈ {1, … , n}, where all sites were visited in order. At the ith step, a new value ci* was drawn from a uniform distribution over all possible cluster labels {1, … , Kmax}. This new state was accepted with probability
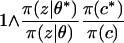 |
(A3) |
and then it replaced the current cluster label ci. The ratio π(c*)/π(c) can be calculated from a local variation of the function U(c) very easily as
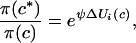 |
where
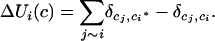 |
Although this has not received much space in this article, we also conducted numerical checks on the correctness of the MCMC sampler. In particular we checked that the results were consistent with those obtained with STRUCTURE at ψ = 0, and we checked that prior distributions were well recovered when the algorithm was implemented without data.
Updating the interaction parameter ψ (full-Bayes only):
Metropolis–Hastings updates of ψ required evaluating ratios of distributions of the form π(c | ψ*)/π(c | ψ) for ψ* the new value. From Equation 3, this computation involved the ratio 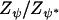 , which was computationally intractable. To avoid this difficulty, we implemented a statistical physics approach known as thermodynamic integration (Gelman and Meng 1998) previously used by Green and Richardson (2002) in the context of spatial epidemiology studies and also described in detail in Hurn et al. (2003). The method consisted of approximating the continuous interval (0, ψmax) by a discrete set of values {δ, 2δ, … , ψmax} and evaluating Z(ψ, Kmax) for each ψ using importance sampling. Here, we used δ = 0.1 and the maximal value of the interaction parameter was ψmax = 1. The importance sampling method used MCMC computations based on the simulation of the Potts model with 50,000 cycles (thinning period of 100 cycles).
, which was computationally intractable. To avoid this difficulty, we implemented a statistical physics approach known as thermodynamic integration (Gelman and Meng 1998) previously used by Green and Richardson (2002) in the context of spatial epidemiology studies and also described in detail in Hurn et al. (2003). The method consisted of approximating the continuous interval (0, ψmax) by a discrete set of values {δ, 2δ, … , ψmax} and evaluating Z(ψ, Kmax) for each ψ using importance sampling. Here, we used δ = 0.1 and the maximal value of the interaction parameter was ψmax = 1. The importance sampling method used MCMC computations based on the simulation of the Potts model with 50,000 cycles (thinning period of 100 cycles).
The values Z(ψ, Kmax) were stored in a look-up table and were used in all additional computations with the same graph topology. Updates of ψ were then carried out by a standard Metropolis–Hastings Markov chain.
References
- Bamshad, M., S. Wooding, W. Watkins, C. Ostler and M. e. a. Batzer, 2003. Human population genetic structure and inference of group membership. Am. J. Hum. Genet. 72: 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besag, J., 1986. On the statistical analysis of dirty pictures. J. R. Stat. Soc. Ser. B 48(3): 259–302. [Google Scholar]
- Bishop, C., 1995. Neural Networks for Pattern Recognition. Clarendon Press, Oxford.
- Blum, M. G. B., C. Damerval, S. Manel and O. François, 2004. Brownian models and coalescent structures. Theor. Popul. Biol. 65: 249–261. [DOI] [PubMed] [Google Scholar]
- Cann, H., C. Toma, L. Cazes, M. Legrand, V. Morel et al., 2002. A human genome diversity cell line panel. Science 296: 261–262. [DOI] [PubMed] [Google Scholar]
- Corander, J., P. Waldmann and M. Sillanpää, 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon, A., G. Guillot, J. Cosson, J. Angibault, S. Aulagnier et al., 2006. Genetics structure is influenced by lansdcape features. Empirical evidence from a roe deer population. Mol. Ecol. 15: 1669–1679. [DOI] [PubMed] [Google Scholar]
- Dawson, K., and K. Belkhir, 2001. A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet. Res. 78: 59–77. [DOI] [PubMed] [Google Scholar]
- Destrempes, F., M. Mignotte and J.-F. Angers, 2005. A stochastic method for Bayesian estimation of hidden Markov random field models with application to a color model. IEEE Trans. Image Proc. 14: 1097–1108. [DOI] [PubMed] [Google Scholar]
- Gelman, A., and X. Meng, 1998. Simulating normalizing constants: from importance sampling to bridge sampling to path sampling. Stat. Sci. 13: 163–185. [Google Scholar]
- Geman, S., and D. Geman, 1984. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans. Patt. Anal. Machine Intell. 6: 721–741. [DOI] [PubMed] [Google Scholar]
- Green, P., and S. Richardson, 2002. Hidden Markov models and disease mapping. J. Am. Stat. Assoc. 97(460): 1055–1070. [Google Scholar]
- Guillot, G., A. Estoup, F. Mortier and J. Cosson, 2005. A spatial statistical model for landscape genetics. Genetics 170: 1261–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot, G., F. Mortier and A. Estoup, 2005. Geneland: a computer package for landscape genetics. Mol. Ecol. Notes 5(3): 708–711. [Google Scholar]
- Guttorp, P., 1995. Stochastic Modelling of Scientific Data. Chapman & Hall, London/New York.
- Hartl, D., and G. Clark, 1997. Principles of Population Genetics. Sinauer Associates, Sunderland MA.
- Hoerl, A., and R. Kennard, 1970. Ridge regression: biased estimation for nonorthogonal problems. Technometrics 12: 55–67. [Google Scholar]
- Hurn, M., O. Husby and H. Rue, 2003. A tutorial in image analysis, pp. 87–141 in Spatial Statistics and Computational Methods (Lecture Notes in Statistics), edited by J. Møller. Springer, Berlin/Heidelberg, Germany/New York.
- Kimura, N., and G. Weiss, 1964. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malécot, G., 1948. Les Mathématiques de l'Hérédité. Masson, Paris.
- Manel, S., E. Bellemain, J. Swenson and O. François, 2004. Assumed and inferred spatial structure of populations: the Scandinavian brown bears revisited. Mol. Ecol. 13: 1327–1331. [DOI] [PubMed] [Google Scholar]
- Metropolis, N., A. Rosenbluth, M. Rosenbluth, A. Teller and E. Teller, 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21: 1087–1092. [Google Scholar]
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, London/New York/Oxford.
- Potts, R., 1952. Some generalized order-disorder transformations. Proc. Camb. Philos. Soc. 48: 106–118. [Google Scholar]
- Preston, C., 1974. Gibbs States on Countable State Space. Cambridge University Press, Cambridge, UK.
- Pritchard, J., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, S., N. Rosenberg, L. Zhivotovsky and M. Feldman, 2004. Robustness of the inference of human population structure: a comparison of X-chromosomal and autosomal microsatellites. Hum. Genomics 1: 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley, B., 1988. Statistical Inference for Spatial Processes. Cambridge University Press, Cambridge, UK.
- Ripley, B., 1996. Pattern Recognition and Neural Networks. Oxford University Press, Oxford.
- Rosenberg, N., J. Pritchard, J. Weber, H. Cann, K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2981–2985. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N., S. Saurabh, S. Ramachandran, C. Zhao, J. Pritchard et al., 2005. Clines, clusters, and the effect of study design on the influence of human population structure. PLoS Genet. 1(6): 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset, F., 2004. Genetic Structure and Selection in Subdivided Populations. Princeton University Press, Princeton, NJ.
- Serre, D., and S. Pääbo, 2004. Evidence for gradients of human genetic diversity within and among continents. Genome Res. 14: 1679–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D., D. Stram, D. Conti, J. Molitor and P. Marjoram, 2003. Bayesian spatial modeling of haplotype associations. Hum. Hered. 56: 32–40. [DOI] [PubMed] [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 58: 267–288. [Google Scholar]
- Waits, L., P. Taberlet, J. Swenson, F. Sandegren and R. Franzen, 2000. Nuclear DNA microsatellite analysis of genetic diversity and gene flow in the Scandinavian brown bear Ursus arctos. Mol. Ecol. 9: 610–621. [DOI] [PubMed] [Google Scholar]
- Wasser, S., A. Shedlock, K. Comstock, E. Ostrander, B. Mutayoba et al., 2004. Assigning African elephants DNA to geographic region of origin: applications to the ivory trade. Proc. Natl. Acad. Sci. USA 101(41): 14847–14852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1943. Isolation by distance. Genetics 28: 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F., 1982. The Potts model. Rev. Mod. Phys. 54: 235–268. [Google Scholar]
- Zhang, Y., M. Brady and S. Smith, 2001. Segmentation of brain MR Images through a hidden Markov random field model and the Expectation-Maximization algorithm. IEEE Trans. Med. Imag. 20: 45–57. [DOI] [PubMed] [Google Scholar]