

Abstract
G-proteins play critical roles in many cellular processes and are regulated by accessory proteins that modulate the nucleotide-bound state. Such proteins, including eukaryotic translation elongation factor 1A (eEF1A), are frequently reactivated by guanine nucleotide exchange factors (GEFs). In the yeast Saccharomyces cerevisiae, only the catalytic subunit of the GEF complex, eEF1Bα, is essential for viability. The requirement for the TEF5 gene encoding eEF1Bα can be suppressed by the presence of excess substrate, eEF1A. These cells, however, have defects in growth and translation. Two independent unbiased screens performed to dissect the cause of these phenotypes yielded dominant suppressors that bypass the requirement for extra eEF1A. Surprisingly, all mutations are in the G-protein eEF1A and cluster in its GTP-binding domain. Five mutants were used to construct novel strains expressing only the eEF1A mutant at normal levels. These strains show no growth defects and little to no decreases in total translation, which raises questions as to the evolutionary expression of GEF complexity and other potential functions of this complex. The location of the mutations on the eEF1A-eEF1Bα structure suggests that their mechanism of suppression may depend on effects on the conserved G-protein elements: the P-loop and NKXD nucleotide-binding element.
MANY steps in the process of protein synthesis are regulated or stimulated by energy-requiring ATPases or GTPases. Several critical soluble translation factors are G-proteins, such as the eukaryotic initiation factor 2 (eIF2), the eukaryotic elongation factors 1A and 2 (eEF1A, formerly EF-1α and eEF2) and the release factor 3 (eRF3). Both eIF2 and eEF1A have identified guanine nucleotide exchange factors (GEFs), which help regulate the activity of these proteins to allow the delivery of aminoacyl-tRNAs (Met-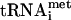 or all other aa-tRNAs, respectively) to the ribosome via the classic “molecular switch” used by many G-proteins (Bourne et al. 1991). While the mechanism of regulation is conserved, the structure and the sequence of GEFs themselves vary dramatically. The GEF for eIF2, eIF2B, consists of five subunits (reviewed in Hershey and Merrick 2000). While these include both catalytic and regulatory proteins, the C-terminal amino acids 544–704 of the eIF2Bε subunit define the minimal catalytic region of the GEF in vitro and in vivo (Gomez et al. 2002). The GEF for Saccharomyces cerevisiae eEF1A, eEF1B, is composed of two subunits (Anand et al. 2001). The eEF1Bα subunit (formerly EF-1β) is catalytic whereas the eEF1Bγ (formerly EF-1γ) subunit appears to regulate the activity of eEF1Bα. A third subunit, eEF1Bβ (formerly EF-1δ), is found only in metazoans and exhibits catalytic GEF activity, although its role in the cell is not well understood (van Damme et al. 1990). The minimal catalytic fragments of eIF2Bε and eEF1Bα show no conservation in sequence or structure (Andersen et al. 2000; Boesen et al. 2004).
or all other aa-tRNAs, respectively) to the ribosome via the classic “molecular switch” used by many G-proteins (Bourne et al. 1991). While the mechanism of regulation is conserved, the structure and the sequence of GEFs themselves vary dramatically. The GEF for eIF2, eIF2B, consists of five subunits (reviewed in Hershey and Merrick 2000). While these include both catalytic and regulatory proteins, the C-terminal amino acids 544–704 of the eIF2Bε subunit define the minimal catalytic region of the GEF in vitro and in vivo (Gomez et al. 2002). The GEF for Saccharomyces cerevisiae eEF1A, eEF1B, is composed of two subunits (Anand et al. 2001). The eEF1Bα subunit (formerly EF-1β) is catalytic whereas the eEF1Bγ (formerly EF-1γ) subunit appears to regulate the activity of eEF1Bα. A third subunit, eEF1Bβ (formerly EF-1δ), is found only in metazoans and exhibits catalytic GEF activity, although its role in the cell is not well understood (van Damme et al. 1990). The minimal catalytic fragments of eIF2Bε and eEF1Bα show no conservation in sequence or structure (Andersen et al. 2000; Boesen et al. 2004).
The inhibition of the nucleotide exchange reaction on eIF2 is a major regulatory step under cellular conditions of amino acid starvation, heme deficiency, endoplasmic reticulum stress, and viral infection (reviewed in Rabinow et al. 1993; Rodnina et al. 1995; Chen 2000; Hinnebusch 2000; Kaufman 2000; Ron and Harding 2000). While evidence indicates that eEF1Bα is a target for kinases (Janssen et al. 1988; Chang and Traugh 1997) and phosphatases (de Nadal et al. 2001), the effects of these modifications on gene expression are not well understood. In vitro dephosphorylation of Artemia salina eEF1Bα results in increased activity in nucleotide exchange (Janssen et al. 1988). In contrast, in the context of the full eEF1 complex in vivo, insulin treatment, S6 kinase activity, or in vitro protein kinase C treatment stimulates eEF1A, eEF1Bα, and eEF1Bβ (metazoan-specific) phosphorylation, nucleotide exchange, and total translation (Peters et al. 1995; Chang and Traugh 1997, 1998). Additionally, loss of eEF1Bγ or eEF1Bα in yeast results in resistance to oxidative stress (Olarewaju et al. 2004). Thus, much remains to be determined about the regulation of eEF1Bαγ activity and its subsequent effects on the activity of eEF1A.
Structural and functional studies in the S. cerevisiae system have illuminated some aspects of eEF1Bα function. Structural analysis of yeast eEF1A with the catalytic C terminus of eEF1Bα indicates that one face of eEF1Bα interacts with domain II of eEF1A while the other interacts with domain I, which contains the nucleotide-binding region (Andersen et al. 2000). This is a significant difference from interactions of the single subunit prokaryotic GEF EF-Ts with domains I and III of EF-Tu (Kawashima et al. 1996). Mutations in conserved residues of eEF1Bα that bind domain II of eEF1A cause conditional growth defects, reduce total translation, and enhance fidelity at nonsense codons (Carr-Schmid et al. 1999b; Andersen et al. 2000). K205 of eEF1Bα is in close proximity to the Mg+2-binding site and is proposed to displace the Mg+2 ion, thereby catalyzing nucleotide exchange. In addition, a K205A mutation causes lethality (Andersen et al. 2001). While eEF1Bα is normally essential for viability (Hiraga et al. 1993), cells can survive without the protein in the presence of excess substrate, eEF1A. Such an eEF1Bα-deficient strain, however, shows significant growth defects such as a 50% increase in doubling time, temperature (Ts−)- and cold (Cs−)-sensitive growth, reduced translational fidelity, and increased sensitivity to translation inhibitors (Kinzy and Woolford 1995). When some mutations in the G domain of eEF1A are provided as the extra copy of eEF1A, the Cs− growth defect is suppressed (Kinzy and Woolford 1995; Carr-Schmid et al. 1999a), indicating that it is possible to manipulate the system in vivo to more efficiently reduce the requirement for nucleotide exchange.
The result that overexpression of the G-protein substrate can bypass an essential GEF in vivo can be used as a genetic system to interpret the effect of GTPase function in the absence of regulation by its GEF. However, eEF1A overexpression also affects actin cytoskeleton organization (Munshi et al. 2001). Thus, a system lacking the requirement for eEF1A overexpression focuses the analysis strictly on the loss of GEF function. In the case of eEF1Bα, it can be also used as a model system to determine how the cell responds when the proposed rate-limiting step of translation elongation has been disrupted. We have created an eEF1Bα-deficient strain using suppressors of the requirement for this normally essential protein and normal levels of eEF1A. The screen was performed in strains with and without the [PSI+] prion, a form of the release factor eRF3. The screens yielded 4 and 7 mutations, respectively, 10 of which are dominant. While the screen was unbiased, and the strain contained three copies of the gene encoding eEF1A, all 9 unique mutations are located in one of the two genes encoding eEF1A. Interestingly, all mutations are within or in close proximity to the nucleotide-binding domains. One of the mutants, A117V, results in altered mobility of eEF1A in SDS–PAGE. Additionally, all 9 mutants are functional as the only form of eEF1A. A representative subset of mutants was utilized to prepare strains where these forms of eEF1A are the only copy in the cell and the gene encoding eEF1Bα is deleted. Thus, this allows us to determine the function and the necessity of eEF1Bα in the cell. Furthermore, the clustered locations of these mutations in eEF1A provide valuable information on the different roles of the P-loop and the NKXD regions of G-proteins.
MATERIALS AND METHODS
Strains and media:
Escherichia coli DH5α was used for plasmid preparation. S. cerevisiae strains used in these studies are listed in Table 1. Standard yeast genetic methods were employed (Mortimer and Hawthorne 1966; Sherman et al. 1986). Yeast cells were grown in either YEPD (1% Bacto yeast extract, 2% peptone, 2% dextrose) or defined synthetic complete media (C or C−) supplemented with 2% dextrose as a carbon source unless noted. Yeast were transformed by the lithium acetate method (Ito et al. 1983). Mating-type switching in yeast using the HO endonuclease was performed as described (Herskowitz and Jensen 1991). Strains lacking the chromosomal TEF1, TEF2, and TEF5 genes were constructed by PCR of the tef5∷TRP1 locus from TKY298, transformation into a strain bearing a plasmid-borne eEF1A mutant and tef1∷LEU2 tef2Δ deletions of the eEF1A genes, and selection on C-Trp. All strains were confirmed as lacking eEF1Bα by Western blot analysis.
TABLE 1.
S. cerevisiae strains
| Strain | Genotype | Source |
|---|---|---|
| MC213 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF2 TRP1 | Sandbaken and Culbertson (1988) |
| MC 214 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF2 URA3 | Sandbaken and Culbertson (1988) |
| JWY4231 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 pTEF5 URA3 | Carr-Schmid et al. (1999b) |
| TKY235 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF5 LEU2 | Carr-Schmid et al. (1999b) |
| TKY298 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF2 URA3 | This work |
| TKY299 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF2 LEU2 | This work |
| TKY352 | MATa ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF1 LEU2 | This work |
| TKY372 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 TEF1 SBD1 (TEF2-20 R164K) | This work |
| TKY373 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-his4-713 tef5∷TRP1 TEF2 sbd2 (TEF1-21 D156E) | This work |
| TKY374 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF1 SBD3 (TEF2-17 D156N) | This work |
| TKY375 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 TEF1 SBD4 (TEF2-22 T22S) | This work |
| TKY603 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF5 LEU2[psi−] | This work |
| TKY604 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 pTEF2 URA3[psi−] | This work |
| TKY 631 | MATα ura3-52 lys2-81 ade2-101trp1Δ63 his3Δ200leu2Δ1 tef5∷KanMX pTEF5 URA3 | This work |
| TKY646 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 SBD1 pTEF2 URA3 | This work |
| TKY647 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2- l his4-713 tef5∷TRP1 sbd2 pTEF2 URA3 | This work |
| TKY648 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 SBD3 pTEF2 URA3 | This work |
| TKY649 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 SBD4 pTEF2 URA3 | This work |
| TKY 726 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 pTEF1 LEU2 | This work |
| TKY767 | MATα ura3-52 lys2-81 ade2-101trp1Δ63his3Δ200 leu2Δ1 tef5∷KanMX pTEF1 TRP1 | This work |
| TKY781 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF2 SBD5 (TEF1-4 E122K) [psi−] | This work |
| TKY782 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF1 SBD6 (TEF2-17 D156N) [psi−] | This work |
| TKY783 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1his4-713 tef5∷TRP1 TEF2 SBD7 (TEF1-20 R164K) [psi−] | This work |
| TKY784 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF1 SBD8 (TEF2-23 A112T) [psi−] | This work |
| TKY785 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF2 SBD9 (TEF1-24 A117T) [psi−] | This work |
| TKY786 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF2 SBD10 (TEF1-25 A117V) [psi−] | This work |
| TKY787 | MATα ura3-52 trp1Δ101 lys2-801 leu2Δ1 met2-1 his4-713 tef5∷TRP1 TEF2 SBD11 (TEF1-26 A117V T172A) [psi−] | This work |
| TKY 789 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (R164K) | This work |
| TKY 791 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (A117V T172A) | This work |
| TKY 846 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (D156E) | This work |
| TKY 847 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (D156N) | This work |
| TKY 848 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (T22S) | This work |
| TKY 849 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (A112T) | This work |
| TKY 850 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (E122K) | This work |
| TKY 851 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (A117T) | This work |
| TKY 852 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ pTEF1 URA3 (A117V) | This work |
| TKY 961 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ tef5∷TRP1 pTEF1 URA3 (R164K) | This work |
| TKY 962 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ tef5∷TRP1 pTEF1 URA3 (D156N) | This work |
| TKY 963 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ tef5∷TRP1 pTEF1 URA3 (T22S) | This work |
| TKY 964 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ tef5∷TRP1 pTEF1 URA3 (A112T) | This work |
| TKY 965 | MATα ura3-52 leu2-3,112 trp1Δ1 lys2-20 met2-1 his4-713 tef1∷LEU2 tef2Δ tef5∷TRP1 pTEF1 URA3 (A117V) | This work |
DNA manipulations:
Recombinant DNA techniques were performed as described (Sambrook et al. 1989). Restriction endonucleases and DNA-modifying enzymes were obtained from Roche Biochemicals (Indianapolis). The TEF1 and TEF2 genes were recovered from the genome of SBD mutants by PCR and the coding region was directly sequenced. Plasmids expressing the identified mutant forms of eEF1A were prepared by either in vivo recombination of a pRS316 (URA3 CEN)-based plasmid or use of the Quikchange mutagenesis kit (Stratagene, La Jolla, CA) using pTKB754 as the template. The resulting plasmids were transformed into TKY767 and grown on C-Ura to confirm that the eEF1Bα-deficiency phenotype was suppressed. Each cloned gene containing an SBD mutant was transformed into MC214, loss of the wild-type eEF1A plasmid was monitored by growth on 5-fluoroanthranilic acid (5-FAA) (Toyn et al. 2000), and the recovered strain was analyzed for growth defects.
EMS mutagenesis and isolation of suppressors of the eEF1Bα-deficiency:
TKY298 (2 × 108 cells) or TKY604 cells (2 × 108) were mutagenized by a modification of standard procedures (Lawrence 1991). Cells were washed and resuspended in 2 ml of 50 mm potassium phosphate buffer. Resuspended cells (100 μl) were added to a 10% sodium thiosulfate solution and kept as a zero time point. Ethyl methanesulfonate (EMS; 40 μl) was added to the remaining cells and shaken at 30°. Aliquots of 600 μl of cells were transferred at 30, 60, and 90 min to a 10% sodium thiosulfate solution to neutralize the EMS, washed twice in sterile water, resuspended in 5 ml of 15% glycerol, and stored at −80°. The percentage of surviving cells was determined by plating a dilution of cells from each time point and counting the colonies after 2–3 days of incubation. For TKY298 [PSI+], ∼30,000 colonies from the 30-min time point (53% killing) and 70,000 colonies from the 60-min time point (61% killing) were screened by plating on YEPD at the nonpermissive temperature of 37°. Colonies that grew well at 37° were patched onto fresh YEPD plates, grown at 30°, and then confirmed for growth at 37°. For TKY604 [psi−], ∼7500 colonies from the 30-min time point (40% killing) were plated on YEPD at the permissive temperature of 30° to allow recovery and then replica plated to 5-FOA at 37° and 24°. Colonies that grew better on 5-FOA at 37° than at 24° were patched onto YEPD plates, grown at 30°, and then confirmed for growth at 37° by streaking and spotting of cells on YEPD.
Drug sensitivity and growth assay:
Two-milliliter cultures of each strain were grown at 30° in the appropriate media to midlog phase and independent colonies of each strain were assayed as previously described (Carr-Schmid et al. 1999b). The concentrations of drugs used were 2 mm cycloheximide, 25 mm hygromycin B, and 1.3 m (800 mg/ml) paromomycin. Sensitivity to each drug was measured by the radius of inhibition of growth around each disc in millimeters. Relative growth of wild-type, eEF1Bα-deficient, and suppressor strains was assayed by streaking or serial dilution spotting as previously described (Carr-Schmid et al. 1999b).
Western blot analysis:
Proteins were extracted from suppressor, wild-type, and eEF1Bα-deficient strains at an OD600 of 0.3–0.8. Cells were spun down for 5 min at 5000 rpm, resuspended in 0.4 ml of cold lysis buffer (100 mm Tris–HCl, pH 8, 20% glycerol, 1 mm DTT, and 1 mm PMSF), and lysed by vortexing with glass beads. Concentration of protein in the extract was determined using Bradford's reagent (Bio-Rad, Hercules, CA) and protein levels were analyzed by Western blot analysis using polyclonal antibodies to Rpa1p (provided by Steven Brill, Rutgers University) and yeast eEF1A (Carr-Schmid et al. 1999a).
Nonsense and missense suppression and total translation assays:
Nonsense suppression assays were performed on strains containing URA3-based CEN plasmids containing lacZ expressed from the PGK1 promoter with the PGK1 transcriptional terminator with either the wild-type sequence (pUKC815tail) or an in-frame UAA (pUKC817tail), UAG (pUKC818tail), or UGA (pUKC819tail) codon (Carr-Schmid et al. 1999b). The strains containing each plasmid were grown overnight at 30° in C-Ura to midlog phase. At least four samples for each strain were analyzed in duplicate using the ortho-nitrophenyl-β-galactoside assay as previously described (Dinman and Kinzy 1997) and standard deviations were calculated. Dual luciferase assays were performed with reporter systems to measure nonsense and missense suppression. Nonsense suppression assays were performed on strains containing URA3-based CEN plasmids containing renilla and firefly luciferase reporters expressed from the ADH1 promoter with the CYC1 terminator and either the wild-type sequence (AAA) or a stop codon (UAA) (Harger and Dinman 2003). Missense suppression assays were performed on strains containing URA3-based CEN plasmids containing a CAC (His)-to-CGC (Arg) mutation in firefly luciferase at codon 245 (Salas-Marco and Bedwell 2005). Strains containing each plasmid were grown overnight at 30° in C-Ura to midlog phase. Cells were harvested by centrifugation and washed twice with 0.5–1 ml of cold lysis buffer (1× PBS, pH 7.4, 1 mm PMSF). Cell suspensions were lysed with glass beads and resuspended in cold lysis buffer. At least three independent colonies of each strain were analyzed in triplicate using the Dual-Luciferase system (Promega, Madison, WI) and a microtiter plate luminometer (MTX Lab Systems) and standard deviations were calculated. In vivo [35S]methionine incorporation was performed as described (Carr-Schmid et al. 1999b).
RESULTS
An eEF1Bα-deficient strain shows altered translation phenotypes:
Prior work indicates a strain lacking eEF1Bα where viability is restored by excess eEF1A, is sensitive to translation inhibitors such as paromomycin, and shows suppression of the lys2-801 (UAG) nonsense mutation in vivo (Kinzy and Woolford 1995). The alteration in nonsense suppression was quantitated using a lacZ reporter construct. Wild-type (TKY235) and eEF1Bα-deficient (TKY299) strains were assayed for the production of β-galactosidase indicative of suppression of an in-frame UAA, UAG, or UGA codon. The eEF1Bα-deficient strain showed a 5- to 10-fold increase in nonsense suppression at all three stop codons (Figure 1A), indicating omnipotent nonsense suppression. Total translation was also monitored, and an eEF1Bα-deficient strain showed an ∼50% reduction in total protein synthesis as measured by 35Met incorporation (Figure 1B). Neither effect is due to the extra copy of the TEF2 gene encoding eEF1A, since a strain with the normal copies of eEF1A and eEF1Bα genes on the chromosome as well as a TEF2 plasmid shows wild-type nonsense suppression and total translation (Munshi et al. 2001; Figure 1B). Thus, while an eEF1Bα-deficient strain is viable, there are consequences to the lack of catalyzed nucleotide exchange and/or the presence of eEF1Bα, which could be illuminated by the analysis of suppressors of the defects of eEF1Bα-deficient strains.
Figure 1.—

(A) A wild-type eEF1Bα (TKY235, pTEF5 LEU2, solid bars) and an eEF1Bα-deficient (TKY299, TEF2 LEU2, shaded bars) strain were assayed for the ability to readthrough the three stop codons using a lacZ-based assay. Results show the percentage of readthrough of the indicated stop codon and represent the average of a minimum of four samples. The error bars represent the propagation of error calculated as the standard deviation of a minimum of three samples. (B) Strains, as in A, containing pRS316 (TKY235, squares and TKY299, triangles) or with a URA3 TEF2 plasmid (TKY235, circles) were grown to midlog phase in C-Ura-Met, diluted, and grown for varying times in C-Ura-Met with [35S]methionine and total protein synthesis measured by TCA precipitation. Data are represented as counts per minute per A600 unit.
Isolation of mutations that suppress the conditional growth defect of a cell lacking eEF1Bα:
To identify mutations in genes that suppress the defects of a cell lacking eEF1Bα, yeast strain TKY298 bearing two chromosomal and one plasmid-borne gene encoding eEF1A as well as a chromosomal deletion of the TEF5 gene encoding eEF1Bα were mutagenized with EMS to 53–61% lethality. Approximately 100,000 colonies were screened for growth at the restrictive temperature of 37°, yielding four independent colonies. To determine if the putative suppressor mutations were a result of a mutation in the plasmid-borne TEF2 gene, all four strains were transformed with a TEF1 LEU2 plasmid (pTKB168) and loss of the TEF2 URA3 plasmid was monitored by growth on 5-FOA (Boeke et al. 1987). The resulting strains still demonstrated wild-type growth at 37° (data not shown), indicating that the four suppressor mutations reside in the chromosomal DNA. Surprisingly, all four strains also grew on 5-FOA when an empty LEU2 plasmid was present, exhibiting suppression of not only the Ts− defect of the eEF1Bα-deficient strain, but also the requirement for excess eEF1A (Figure 2A). The suppressor strains were termed SBD for suppressor of an eEF1Bα-deficiency. To determine if the mutations were dominant or recessive, each of the SBD mutant strains with the TEF2 URA3 plasmid (TKY646-649) was mated to the eEF1Bα-deficient strain TKY352 and diploids were selected. The TEF1 LEU2 plasmid was lost by nonselective growth and the diploids were streaked on 5-FOA. Loss of the suppressor phenotype, 5-FOA resistance, was seen for TKY647 (sbd2), indicating a recessive mutation (Figure 2B). However, diploids containing the suppressor mutation from TKY646, TKY648, and TKY649 (SBD1, -3, and -4) allowed the strains to survive without excess eEF1A and are thus dominant (Figure 2B).
Figure 2.—

(A). Wild-type eEF1Bα (TKY235), the parental eEF1Bα-deficient (TKY298), and the SBD1–4 (TKY646-649) strains were grown on 5-FOA for 3 days at 30°. (B) Strains, as in A, were mated to the eEF1Bα-deficient strain TKY352, diploids were selected, streaked onto 5-FOA, and grown for 3 days at 30°. (C) Wild type (JWY4231, pTEF5 URA3), the parental eEF1Bα-deficient (TKY298), and SBD1–4 lacking the TEF2 URA3 plasmid (TKY372-375) strains were grown in YEPD and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted and grown at 13,° 24,° 30,° and 37° for 2–8 days. (D) Wild-type (TKY235), eEF1Bα-deficient (TKY299), or SBD1–4 strains (TKY372-375) were assayed for the ability to readthrough a UAA stop codon using a lacZ-based assay. Results show the percentage of readthrough of the stop codon and represent the average of a minimum of four samples. The error bars represent the propagation of error calculated as the standard deviation for a minimum of four samples. (E) Equal amounts of total yeast proteins from wild-type (TKY235), eEF1Bα-deficient (TKY298), and SBD1–4 (TKY372-375) strains, as determined by Bio-Rad assay, were analyzed by SDS–PAGE and Western blot analysis with polyclonal antibodies to yeast eEF1A and yeast Rpa1p (as a loading control).
Growth was assayed for the four mutants without an extra eEF1A encoding plasmid. At 37°, growth was essentially as for wild type for the suppressor strains; at 24°, SBD1 and SBD3 grew slightly better than sbd2 and SBD4 strains, while, at 13°, all the suppressor strains resemble the eEF1Bα-deficient strain (Figure 2C). The SBD strains recovered the nonsense suppression phenotype of the eEF1Bα-deficient strain as monitored by a reduction in the level of β-galactosidase activity back to the wild-type levels (Figure 2D). To confirm that the strain had lost the extra copy of eEF1A, Western blot analysis was performed. All four suppressor strains show wild-type levels of eEF1A equivalent to two chromosomal genes encoding eEF1A, and not the excess protein seen in the eEF1Bα-deficient strain (Figure 2E). These are the first strains shown to be able to bypass the need for the nucleotide exchange factor eEF1Bα without the presence of excess eEF1A.
Isolation of mutations that bypass eEF1Bα in a [psi−] background:
In the course of analysis of the mutants, it was determined that the parent of the suppressor strains also contains the yeast prion [PSI+]. The presence of [PSI+], a form of the eukaryotic release factor 3 (eRF3), could alter the translational status of the cell and bias the screen results. Thus, a similar screen was performed specifically for bypass suppressors of the eEF1Bα deficiency using the [psi−] derivative of TKY298, TKY604. Since all the mutants in the first screen could lose the extra eEF1A-encoding URA3 plasmid, we identified strains that, following mutagenesis, could grow on 5-FOA at 37° better than at 25°. From 7500 colonies, 13 mutants were identified. Growth assays of the cells on YEPD indicated that the suppressors grew faster than the parent eEF1Bα-deficient strain and in many cases grew similarly to a wild-type strain (Figure 3A; data not shown). Diploids were prepared by mating each mutant back to an eEF1Bα-deficient parent strain of the opposite mating type (TKY726). All 13 diploids grew on 5-FOA as for the first screen and were thus dominant. Western analysis of equalized protein extracts from all suppressor, wild-type, and eEF1Bα-deficient strains showed that eEF1A protein levels for all SBD suppressors approximated wild-type levels and were qualitatively much less than the eEF1Bα-deficient strain (Figure 3B; data not shown). Four suppressor strains show heterogeneity as a doublet in the eEF1A band by Western analysis (Figure 3B; data not shown). This is not observed in the wild-type strain, because the protein sequences of the TEF1 and TEF2 open reading frames are 100% identical. This doublet suggests that, in these mutants, either TEF1 or TEF2 is changed in some way that causes a shift in mobility on an SDS–PAGE gel.
Figure 3.—

(A) The [psi−] wild-type (TKY603), parental eEF1Bα-deficient (TKY604), or SBD5-11 (TKY781-787) strains were grown in YEPD and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted and grown at 13°, 24°, 30,° and 37° for 2–8 days. (B) Equal amounts of total yeast proteins from strains, as in A, as determined by Bio-Rad assay, were analyzed by SDS–PAGE and Western blot analysis with polyclonal antibodies to yeast eEF1A and yeast Rpa1p (as a loading control).
All SBD mutants are in a chromosomal eEF1A-encoding gene:
On the basis of prior results of some eEF1A mutations permitting enhanced viability in the absence of eEF1Bα, the dominant phenotype of the 16 mutants from the two screens, and the heterogeneity of the eEF1A band in Western blot analysis, the genes encoding eEF1A were analyzed from the mutant strains. Initially, the TEF1 and TEF2 genes were cloned from the SBD7 and SDB11 mutant strains by in vivo recombination into a pRS316 plasmid (URA3 CEN). The resulting plasmids were transformed into the eEF1Bα-deficient strain TKY767, plated on 5-FAA to monitor the loss of the TEF1 TRP1 plasmid, recovered on YEPD, and spotted at various temperatures. The plasmids encoding TEF1, but not TEF2, from either suppressor allowed enhanced growth at 24°, 30°, and 37°, indicating the SBD phenotype (Figure 4A). The genes were sequenced and mutations of R164K (SBD7) and A117V T172A (SBD11) were identified in the TEF1 gene; however, the TEF2 sequences were wild type. On the basis of these results TEF1 and TEF2 were amplified from the genomic DNA from the remaining 11 mutants identified in the [psi−] screen. A total of seven different mutations were identified, three of them appearing more than once. Mutants that maintained a single eEF1A band by Western-contained mutations of E112K (SBD5, three isolates), D156N (SBD6), R164K (SBD7, three isolates), A112T (SBD8), and A117T (SBD9). Mutants with the doublet by Western analysis contained mutations of A117V (SBD10, three isolates) and A117V T172A (SBD11). Sequencing of the TEF1 and TEF2 genes from SBD1-4 mutants demonstrated eEF1A mutants of R164K (SBD1), D156E (sbd2), D156N (SBD3), and T22S (SBD4). Thus, two mutants, R164K and D156N, were found in both screens. Interestingly, E122K and D156N were previously identified as allowing better growth at low temperatures of a strain lacking eEF1Bα when present as the third copy of eEF1A (Kinzy and Woolford 1995; Carr-Schmid et al. 1999a). To confirm the results from the genomic DNA sequencing, site-directed mutagenesis was utilized to produce each mutation in a TEF1 URA3 CEN plasmid. All constructs were transformed into the eEF1Bα-deficient strain, as in Figure 4A, and promoted enhanced growth (Figure 4B), confirming that they can cause the SBD phenotype. Interestingly, growth of these strains with the three eEF1A-encoding genes was better suppressed at lower temperatures, even though the original SBD strains with the normal two eEF1A-encoding genes showed better growth at higher temperatures (Figures 2 and 3).
Figure 4.—

(A) Wild type (JWY235), eEF1Bα-deficient (JWY767, pTEF1 TRP1) with pRS316, and JWY767 transformed with the plasmids containing the TEF1 or TEF2 gene cloned from TKY789 (SBD7) and TKY791 (SBD11) strains were grown in C-Ura and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted onto C-Ura medium and grown at 24°, 30°, and 37° for 2–8 days. (B) Wild-type (TKY631, pTEF5 URA), eEF1Bα-deficient (TKY767) with pRS316, and TKY767 transformed with a TEF1 (SBD mutant) URA3 plasmid strains were grown and assayed as in A.
To look more closely at translation effects, sensitivity to translation inhibitors was monitored for all 11 SBD mutant strains, which lack eEF1Bα and have one wild type and one mutant copy of a chromosomal eEF1A gene. The results show that while there was a modest difference in the sensitivity to the drugs cycloheximide and hygromycin B, some SBD mutants partially suppressed the severe paromomycin-sensitive phenotype of the eEF1Bα-deficient strain. However, all strains expressing SBDs remained hypersensitive to paromomycin compared to the isogenic wild-type strain (Tables 2 and 3). To determine if any secondary phenotypes are conferred by the suppressor mutations when eEF1Bα is restored, the strains containing SBD1-11 were transformed with a TEF5 URA3 plasmid. None of the strains showed conditional or slow-growth phenotypes with eEF1Bα present (data not shown).
TABLE 2.
Drug sensitivity phenotypes of mutant strains isolated from a [PSI+] eEF1Bα-deficient mutant screen
| Strains | Hygro (25 mm) | Cyclo (2 mm) | Paromo (1.3 m) | |
|---|---|---|---|---|
| TKY235 | WT | 2.5a | 12.8 | 3.3 |
| eEF1Bα | ||||
| TKY298 | eEF1Bα deficient | 3.0 | 16.3 | 9.3 |
| TKY372 | SBD1 | 2.3 | 15.5 | 4.0 |
| TEF2 | ||||
| R164K | ||||
| TKY373 | sbd2 | 1.3 | 12.2 | 4.5 |
| TEF1 | ||||
| D156E | ||||
| TKY374 | SBD3 | 3.5 | 15.2 | 8.0 |
| TEF2 | ||||
| D156N | ||||
| TKY375 | SBD4 | 2.5 | 13.2 | 6.0 |
| TEF2 | ||||
| T22S |
Radius of inhibition of growth around the drug-containing filter in millimeters on YEPD.
TABLE 3.
Drug sensitivity phenotypes of mutant strains isolated from a [psi−] eEF1Bα-deficient mutant screen
| −eEF1Bα
|
||||
|---|---|---|---|---|
| Hygro (25 mm) | Cyclo (2 mm) | Paromo (1.3 m) | ||
| TKY603 | Wild type | 2.8a | 9.0 | 1.0 |
| eEF1Bα | ||||
| TKY604 | eEF1Bα deficient | 4.0 | 14.8 | 9.3 |
| TKY781 | SBD5 | 4.3 | 13.8 | 5.8 |
| TEF1 | ||||
| E122K | ||||
| TKY782 | SBD6 | 3.5 | 11.8 | 4.3 |
| TEF2 | ||||
| D156N | ||||
| TKY783 | SBD7 | 3.3 | 13.3 | 2.5 |
| TEF1 | ||||
| R164K | ||||
| TKY784 | SBD8 | 2.8 | 11.8 | 2.0 |
| TEF2 | ||||
| A112T | ||||
| TKY785 | SBD9 | 3.5 | 14.3 | 4.5 |
| TEF1 | ||||
| A117T | ||||
| TKY786 | SBD10 | 3.5 | 12.5 | 4.5 |
| TEF1 | ||||
| A117V | ||||
| TKY787 | SBD11 | 4.5 | 12.5 | 3.5 |
| TEF1 | ||||
| A117V | ||||
| T172A | ||||
Radius of inhibition of growth around the drug-containing filter in millimeters on YEPD.
All SBD mutants in eEF1A map to the nucleotide-binding domain:
SBD mutations that allow the eEF1A protein to function without its normally essential exchange factor are all located in domain I, the nucleotide-binding domain (Figure 5). Each mutation is located in the nucleotide-binding domain or in a very close proximity to at least one of the conserved sequence elements of the G-proteins. The mutations interact directly with either the nucleotide or the motifs that bind and stabilize it.
Figure 5.—

X-ray crystal structure of S. cerevisiae eEF1A (shading) shows that the SBD mutations (arrows) T22 (S, magenta), A112 (T, cyan), A117 (V, T, purple), E122 (K, yellow), D156 (E, N, orange), and R164 (K, pink) that suppress the requirement for the eEF1Bα (brown) cluster in the GTP-binding domain (Andersen et al. 2000). GDP is shown in green.
eEF1A mutants that suppress the eEF1Bα requirement are viable as the only copy of eEF1A:
Strains that express only the SBD form of eEF1A were prepared by plasmid shuffling in a tef1∷LEU2 tef2Δ strain. All mutants allowed viability with no growth defects at permissive temperatures compared to wild-type cells (Figure 6). A112T, and to a lesser extent D156E, show a cold-sensitive temperature defect. To assess effects on protein synthesis, sensitivity to different translation inhibitors was monitored (Table 4, first three columns). Interestingly, all the SBD mutant strains show at least a slight increase in hygromycin sensitivity. Cycloheximide sensitivity was unaffected in these strains. Strains harboring the R164K or A117V mutants showed paromomycin resistance, whereas T22S and A112T strains showed the greatest sensitivity to the drug.
Figure 6.—

Strains expressing wild-type eEF1A (MC213 pTEF1 TRP1) or SBD mutants (TKY789, 791, 846-852) from a plasmid as the only form were grown in YEPD and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted onto YEPD plates and grown at 13°, 24°, 30°, and 37° for 2–8 days.
TABLE 4.
Drug sensitivity phenotypes of the strains expressing SBD mutants as the only form of eEF1A
| +eEF1Bα
|
−eEF1Bα:
|
||||
|---|---|---|---|---|---|
| Strains | Hygro (50 mm) | Cyclo (2 mm) | Paromo (1.3 m) | Paromo (1.3 m) | |
| TKY102 | Wild type | 15a | 33 | 14.7 | NA |
| eEF1Bα | |||||
| TKY789 | SBD1/SBD7 | 17.3 | 31 | 9.7 | 20 |
| TEF1 R164K | |||||
| TKY846 | sbd2 | 18.7 | 27.7 | 14.7 | ND |
| TEF1 D156E | |||||
| TKY847 | SBD3/SBD6 | 18.7 | 31.7 | 16.7 | 22.3 |
| TEF2 D156N | |||||
| TKY848 | SBD4 | 19 | 30 | 18.7 | 21 |
| TEF2 T22S | |||||
| TKY850 | SBD5 | 18 | 30 | 16.3 | ND |
| TEF1 E122K | |||||
| TKY849 | SBD8 | 21.3 | 33.7 | 22.3 | 22.3 |
| TEF2 A112T | |||||
| TKY851 | SBD9 | 18 | 29.7 | 14 | ND |
| TEF1 A117T | |||||
| TKY852 | SBD10 | 18 | 33.3 | 12 | 16.7 |
| TEF1 A117V | |||||
| TKY791 | SBD11 | 18.3 | 31 | 17 | ND |
| TEF1 | |||||
| A117V/T172A | |||||
NA; not applicable. ND; not determined.
Radius of inhibition of growth around the drug-containing filter in millimeters on YEPD.
SBD mutations do not have additive effects on suppression:
To determine if the eEF1A mutations conferring the SBD phenotype function through the same pathway, four double mutations were prepared. The T22S/A117V, R164K/A117V, A117V/A112T, and E122K/A112T double mutants were chosen on the basis of the distance between the side chains in the structure or the orientation of the side groups relative to each other (Figure 5). A strain with the double mutants present as a plasmid-borne extra copy and lacking eEF1Bα was assayed for growth at 24°, 30°, and 37°. The T22S/A117V mutant showed modest suppression of the loss of eEF1Bα, albeit at a lower level than seen for the single mutants. The R164K/A117V mutant showed little to no growth suppression. Interestingly, the A117V/A112T and E122K/A112T double mutants not only fail to suppress the requirement for eEF1Bα, but also show a dominant negative effect on cell growth (Figure 7). None of the double mutants were viable as the only form of eEF1A in a cell containing eEF1Bα (data not shown), indicating that they compromise functions beyond that tolerated by the cell.
Figure 7.—

Wild type (TKY631, pTEF5 URA3, eEF1Bα-deficient (TKY767) with pRS316 and TKY767 expressing the indicated TEF2 SBD double-mutant plasmid strains were grown in C-Ura and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted onto C-Ura plates and grown at 24°, 30°, and 37° for 2–8 days.
SBD mutations can support viability as the only form of eEF1A in the absence of eEF1Bα:
To establish the consequence of the full loss of eEF1Bα and a mutant form of eEF1A less dependent on the normally essential activity of this exchange factor, strains were constructed lacking the chromosomal eEF1A and eEF1Bα genes. Strains were prepared by recombination of a tef5∷TRP1 deletion cassette in the strains bearing the R164K, A117V, T22S, A112T, or D156N SBD mutations as the only form of eEF1A on a plasmid. All the strains produced viable cells that lack the eEF1Bα-encoding gene as detected by DNA and protein analysis. The strains showed no growth defect compared to the wild-type strain (Figure 8) or to the strains harboring the same mutations as the only form but with eEF1Bα present (compare to Figure 6).
Figure 8.—

A wild-type strain (MC213 pTEF1 TRP1) and strains TKY961–965 obtained by transforming the tef5∷TRP1 fragment into TKY789, 847–849, and 852 and selected by growth on C-Trp were grown in YEPD and diluted to an A600 of 1.0. Ten-fold serial dilutions were spotted onto YEPD plates and grown at 13°, 24°, 30°, and 37° for 2–8 days.
To determine the effects of these strains on translation, in vivo [35S]methionine labeling assays were performed. A112T and T22S strains showed a 25 or 35% reduction, respectively, in total translation both in the presence and in the absence of eEF1Bα. However, R164K and A117V strains translated as efficiently as wild type in the presence of eEF1Bα. In the absence of eEF1Bα, A117V showed a 25% decrease in total protein synthesis, while R164K showed a 25% increase (Table 5). Drug sensitivity assays were performed to determine the paromomycin sensitivity of these strains. In the absence of eEF1Bα, all the mutant strains except A112T were more sensitive to paromomycin compared to the presence of eEF1Bα (Table 4). To measure the effect of these strains on the accuracy of translation, we performed in vivo dual luciferase assays. Previously used lacZ reporters were compared to dual luciferase reporters for the analysis eEF1A mutants and all mutants reproduced same trends in nonsense or frameshift suppression (data not shown). While A112T and T22S strains showed higher nonsense suppression rates by 2.5- to 2.7-fold compared to wild type, R164K and A117V strains showed no effect (Table 5). No mutation affected missense suppression (Table 5).
TABLE 5.
Total translation and nonsense and missense effects of the SBD mutant strains in the presence and the absence of eEF1Bα
| Total translation relative to wild type (% change) | Nonsense suppression (UAA) (Rtest/Rcon)a | Missense suppression (Rtest/Rcon)a | |
|---|---|---|---|
| Wild type | 100 | 0.029 ± 0.01 | 0.01 ± 0.004 |
| R164K + eEF1Bα | 100 | ND | ND |
| R164K − eEF1Bα | 125 | 0.034 ± 0.01 | 0.012 ± 0.004 |
| A117V + eEF1Bα | 100 | ND | ND |
| A117V − eEF1Bα | 75 | 0.028 ± 0.01 | 0.008 ± 0.003 |
| T22S + eEF1Bα | 65 | ND | ND |
| T22S − eEF1Bα | 65 | 0.073 ± 0.02 | 0.01 ± 0.004 |
| A112T + eEF1Bα | 75 | ND | ND |
| A112T − eEF1Bα | 75 | 0.079 ± 0.02 | 0.012 ± 0.003 |
| D156N + eEF1Bα | 125 | ND | ND |
| D156N − eEF1Bα | 100 | ND | ND |
ND, not determined.
Rtest/Rcon: the activity ratio derived from lysates expressing test cassettes divided by the activity ratio of the control reporter. Standard deviations were calculated using a minimum of four samples.
DISCUSSION
While the guanine nucleotide exchange factor of eEF1A, eEF1Bα, is essential for cell viability, recent studies showed that eEF1A is able to dissociate GDP without its GEF although with a 700-fold slower rate (Pittman et al. 2006). This slow rate of exchange is not sufficient for cell viability but can be partially compensated for by an extra copy of eEF1A (Kinzy and Woolford 1995). Cells lacking eEF1Bα with an extra copy of wild-type eEF1A exhibit slower-growth phenotypes, a 50% reduction in total translation, and reduced translational fidelity. Interestingly, SBD mutations in eEF1A likely cause a more favorable conformation of eEF1A for nucleotide exchange, allowing it to function efficiently without eEF1Bα and thus suppress most of the eEF1Bα deficiency phenotypes. Two genetic screens were performed to isolate the suppressors of the eEF1Bα deficiency, and although the screens were developed specifically to avoid eEF1A mutations by the presence of three eEF1A-encoding genes, both screens exclusively yielded eEF1A mutations. Since the same mutation was selected more than once on multiple occasions, it appears that eEF1A mutations are the major, if not the sole, suppressors of the lethal effects of the loss of eEF1Bα.
An analysis of three different strains containing the SBD mutants provided unique insights into the requirement for eEF1Bα. The initial eEF1Bα-deficient strains isolated had one wild type and one SBD mutant eEF1A-encoding gene and thus normal eEF1A protein levels. Previous studies of suppression of the requirement for eEF1Bα used strains with the two chromosomal wild-type eEF1A-encoding genes and an eEF1A mutant on a plasmid (Kinzy and Woolford 1995) and showed a significant difference in the temperature effects. While the SBD mutant strains with two eEF1A genes suppress growth defects at 37° (Figures 2 and 3), eEF1Bα-deficient strains from this (Figure 4) or prior work (Kinzy and Woolford 1995; Carr-Schmid et al. 1999a) with three eEF1A genes show enhanced growth at low temperatures. This likely relates to the effects of excess eEF1A on the actin cytoskeleton, which results in reduced growth at elevated temperatures (Munshi et al. 2001). The new strains lacking eEF1Bα and both chromosomal eEF1A genes and thus expressing only the SBD form of eEF1A indicate that it is in fact possible to suppress all the deficiencies of the loss of the nucleotide exchange factor with little to no effect on growth (Figure 8).
The slow-growth phenotype of an eEF1Bα-deficient strain is suppressed in the presence of SBD mutants of eEF1A. However, while SBD strains with one wild-type and one SBD mutant eEF1A gene exhibit modest differences in the sensitivity to the drugs cycloheximide and hygromycin B, some SBD mutants only partially suppress the severe paromomycin-sensitive phenotype of the eEF1Bα-deficient strain. These results indicate that SBD mutations cannot completely recover the paromomycin sensitivity phenotype, suggesting reduced A-site fidelity. Strains expressing only the SBD form of eEF1A with eEF1Bα show widely varying sensitivity or resistance to paromomycin compared to the isogenic wild-type strain. In the absence of eEF1Bα, however, all the eEF1A mutant strains show higher sensitivity to paromomycin than a wild-type strain with eEF1Bα. A112T and T22S mutants exhibit translational readthrough of a UAA stop codon, correlating with their higher paromomycin sensitivity and reduced translation rates. The fact that not all phenotypes are suppressed underscores the additional translational roles of eEF1Bα other than nucleotide exchange, especially translational fidelity and, potentially, the proposed channeling of aa-tRNA to eEF1A (Andersen et al. 2000).
Examination of the crystal structure of the eEF1A:eEF1Bα complex revealed a series of important hints for understanding the suppression of the GEF requirement as well as the mechanism of guanine nucleotide exchange. The established role of eEF1Bα is to accelerate the rate of GDP release following GTP binding to eEF1A. Structurally, eEF1Bα is suggested to displace the Mg+2 ion associated with the nucleotide, stimulating GDP release (Andersen et al. 2001; Pittman et al. 2006). The SBD forms of eEF1A are located in the nucleotide-binding domain. Analysis of the SBD mutations mapped onto the structure of eEF1A indicate that they are near the P-loop or the NKXD motif. The P-loop is essential for phosphate binding of the nucleotide while the NKXD motif binds to the base of the nucleotide. Together with the functional data, two classes of mutants were identified: class I mutants A112T and T22S and class II mutants R164K and A117V.
For the class I mutants, T22 of the P-loop directly interacts with the α-phosphate of GDP via an amide group while A112 is located on the β-sheet between a β-sheet that connects to the P-loop and a second that connects to the NKXD element. Both A112 and T22 are conserved in E. coli and Thermus aquaticus EF-Tu and human eEF1A, pointing out their importance in the G-domain. In the absence of its GEF, the spontaneous GDP dissociation of eEF1A is 85 times more rapid than that of EF-Tu (Gromadski et al. 2002; Pittman et al. 2006); this higher GDP dissociation rate of eukaryotic eEF1A may be what allows SBD mutants to be functional as the only form of eEF1A. Class I mutant strains show a 2.5- to 2.7-fold increase in nonsense suppression while class II mutants do not affect suppression. The P-loop is essential for the molecular switch between active and inactive forms of the proteins and the disturbance of the P-loop is suggested to be the major reason for the decreased affinity of GDP (Vetter and Wittenghofer 2001). Class I mutations also showed a 25–35% decrease in total translation rates with or without eEF1Bα. The presence of eEF1Bα thus does not alter translation efficiency.
A117 of class II is in the upper tip of the same β-sheet containing A112. The observation that A117V shows a doublet on SDS–PAGE gel may suggest a conformational change. This is consistent with the effect of the known nucleotide-dependent changes in conformations of G-proteins (Vetter and Wittenghofer 2001; Spoerner et al. 2005). A117 is conserved in E. coli and T. aquaticus EF-Tu and in human eEF1A, whereas R164 is conserved only in yeast and in human eEF1A. In prokaryotic systems, this residue is a leucine, which may indicate the importance of a charged residue in this position for the eukaryotic systems.
eEFSec is the Sec-specific counterpart of eEF1A that incorporates selenocysteine into the protein. eEFSec is predicted to function independently of a guanine nucleotide exchange factor (Copeland 2003). While the residues of the both class I and class II mutations are identical in prokaryotic elongation factor SelB, none of the residues are conserved in eEFSec. This may indicate the importance of these residues such that eEFSec does not require exchange activity.
Double mutants that altered amino acids near both the P-loop and the guanine base were not viable as the only form of eEF1A and did not function as efficient suppressors of the requirement for eEF1Bα. This suggests that the effects on nucleotide release or binding surpass the threshold needed for cell survival. This finding also indicates that the suppression of the exchange factor is the common result of all SBDs but likely is achieved through separate alterations of the consensus elements.
This study presents the eEF1Bα function as dispensable for the cell, likely as long as nucleotide exchange or GDP release rates can be maintained above a certain threshold. Some of the G-proteins, including translation factors eRF3 and eEF2, do not depend on separate nucleotide exchange factors. However, some studies suggest that the ribosome acts as a GEF for prokaryotic RF3 and EF-G (Zavialov et al. 2005). Further determination of the GDP release and nucleotide exchange rates will also identify the step of the suppression during the G-protein cycle and provide more information on this novel exchange mechanism and its relationship to other G-proteins that lack a GEF.
Although eEF1B has two subunits in S. cerevisiae, only eEF1Bα has guanine nucleotide exchange activity. Furthermore, comparison of the crystal structure of the eEF1A:eEF1Bα complex to the prokaryotic EF-Tu-GDPNP-aa-tRNA structure has led to the suggestion that eEF1Bα may help to channel aa-tRNA to eEF1A (Andersen et al. 2000). The eEF1Bαγ complex is also proposed to have other regulatory functions in yeast, as both subunits are suggested to play a role in the oxidative stress response pathway (Olarewaju et al. 2004). The resulting phenotypes of the SBD strains may also be caused by the impairment of the additional functions of the eEF1Bα. These results suggest that GEFs for the elongation factors have gained more complexity and perhaps more functions in the cell throughout evolution. Further studies will determine the additional functions of these proteins.
Acknowledgments
We thank Joanna Slusky for initial contributions to the work; Andrew Vershon, Jonathan Dinman, Paul Copeland, and members of the Kinzy Lab for helpful comments; and the Robert Wood Johnson Medical School DNA facility for DNA sequencing. This work was supported by National Institutes of Health (NIH) grant RO1 GM57483 to T.G.K. M.R.V. was supported in part by NIH grant T25 GM55145.
References
- Anand, M., L. Valente, A. Carr-Schmid, R. Munshi, O. Olarewaju et al., 2001. Functions of the translation elongation factor 1 in the yeast Saccharomyces cerevisiae. Symp. Quant. Biol. 66: 439–448. [DOI] [PubMed] [Google Scholar]
- Andersen, G. R., L. Pedersen, L. Valente, I. Chatterjee, T. G. Kinzy et al., 2000. Structural basis for nucleotide exchange and competition with tRNA in the yeast elongation factor complex eEF1A:eEF1Bα. Mol. Cell 6: 1261–1266. [DOI] [PubMed] [Google Scholar]
- Andersen, G. R., L. Valente, L. Pedersen, T. G. Kinzy and J. Nyborg, 2001. Crystal structures of nucleotide exchange intermediates in the eEF1A-eEF1Balpha complex. Nat. Struct. Biol. 8: 531–534. [DOI] [PubMed] [Google Scholar]
- Boeke, J. D., J. Trueheart, G. Natsoulis and G. R. Fink, 1987. 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154: 164–175. [DOI] [PubMed] [Google Scholar]
- Boesen, T., S. S. Mohammad, G. D. Pavitt and G. R. Andersen, 2004. Structure of the catalytic fragment of translation initiation factor 2B and identification of a critically important catalytic residue. J. Biol. Chem. 279: 10584–10592. [DOI] [PubMed] [Google Scholar]
- Bourne, H. R., D. A. Sanders and F. McCormick, 1991. The GTPase superfamily: conserved structure and molecular mechanism. Nature 349: 117–127. [DOI] [PubMed] [Google Scholar]
- Carr-Schmid, A., N. Durko, J. Cavallius, W. C. Merrick and T. G. Kinzy, 1999. a Mutations in a GTP-binding motif of eEF1A reduce both translational fidelity and the requirement for nucleotide exchange. J. Biol. Chem. 274: 30297–30302. [DOI] [PubMed] [Google Scholar]
- Carr-Schmid, A., L. Valente, V. I. Loik, T. Williams, L. M. Starita et al., 1999. b Mutations in elongation factor 1β, a guanine nucleotide exchange factor, enhance translational fidelity. Mol. Cell. Biol. 19: 5257–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Y. W., and J. A. Traugh, 1997. Phosphorylation of elongation factor 1 and ribosomal protein S6 by multipotential S6 kinase and insulin stimulation of translational elongation. J. Biol. Chem. 272: 28252–28257. [DOI] [PubMed] [Google Scholar]
- Chang, Y. W., and J. A. Traugh, 1998. Insulin stimulation of phosphorylation of elongation factor 1 (eEF-1) enhances elongation activity. Eur. J. Biochem. 251: 201–207. [DOI] [PubMed] [Google Scholar]
- Chen, J. J., 2000. Heme-regualted eIF2α kinase, pp. 529–546 in Translational Control of Gene Expression, edited by N. Sonenberg, J. W. B. Hershey and M. B. Mathews. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Copeland, P. R., 2003. Regulation of gene expression by stop codon recoding: selenocysteine. Gene 312: 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nadal, E., R. P. Fadden, A. Ruiz, T. Haystead and J. Arino, 2001. A role for the ppz ser/thr protein phosphatases in the regulation of translation elongation factor 1balpha. J. Biol. Chem. 276: 14829–14834. [DOI] [PubMed] [Google Scholar]
- Dinman, J. D., and T. G. Kinzy, 1997. Translational misreading: mutations in translation elongation factor 1α differentially affect programmed ribosomal frameshifting and drug sensitivity. RNA 3: 870–881. [PMC free article] [PubMed] [Google Scholar]
- Gomez, E., S. S. Mohammad and G. D. Pavitt, 2002. Characterization of the minimal catalytic domain within eIF2B: the guanine-nucleotide exchange factor for translation initiation. EMBO J. 21: 5292–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski, K., H. Wieden and M. Rodnina, 2002. Kinetic mechanism of elongation factor Ts-catalyzed nucleotide exchange in elongation factor Tu. Biochemistry 41: 162–169. [DOI] [PubMed] [Google Scholar]
- Harger, J. W., and J. D. Dinman, 2003. An in vivo dual-luciferase assay system for studying translational recoding in the yeast Saccharomyces cerevisiae. RNA 9: 1019–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey, J. W. B., and W. C. Merrick, 2000. The pathway and mechanism of initiation of protein synthesis, pp. 33–88 in Translational Control of Gene Expression, edited by N. Sonenberg, J. W. B. Hershey and M. B. Mathews. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Herskowitz, I., and R. E. Jensen, 1991. Putting the HO gene to work: practical uses for mating-type switching. Methods Enzymol. 194: 132–146. [DOI] [PubMed] [Google Scholar]
- Hinnebusch, A. G., 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes, pp. 185–244 in Translational Control of Gene Expression, edited by N. Sonenberg, J. W. B. Hershey and M. B. Mathews. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Hiraga, K., K. Suzuki, E. Tsuchiya and T. Miyakawa, 1993. Cloning and characterization of the elongation factor EF-1β homologue of Saccharomyces cerevisiae. EF-1β is essential for growth. FEBS Lett. 316: 165–169. [DOI] [PubMed] [Google Scholar]
- Ito, H., Y. Fukuda, K. Murata and A. Kimura, 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153: 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen, G. M. C., G. D. F. Maessen, R. Amons and W. Moller, 1988. Phosphorylation of elongation factor 1β by an exogenous kinase affects its catalytic nucleotide exchange activity. J. Biol. Chem. 263: 11063–11066. [PubMed] [Google Scholar]
- Kaufman, R. J., 2000. Double-stranded RNA-activated protein kinase PKR, pp. 503–528 in Translational Control of Gene Expression, edited by N. Sonenberg, J. W. B. Hershey and M. B. Mathews. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Kawashima, T., C. Berthet-Colominas, M. Wulff, S. Cusack and R. Leberman, 1996. The structure of the Escherichia coli EF-Tu · EF-Ts complex at 2.5Å resolution. Nature 379: 511–518. [DOI] [PubMed] [Google Scholar]
- Kinzy, T. G., and J. L. Woolford, Jr., 1995. Increased expression of Saccharomyces cerevisiae translation elongation factor EF-1α bypasses the lethality of a TEF5 null allele encoding EF-1β. Genetics 141: 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, C. W., 1991. Classical mutagenesis techniques. Methods Enzymol. 194: 273–281. [DOI] [PubMed] [Google Scholar]
- Mortimer, R. K., and D. C. Hawthorne, 1966. Genetic mapping in Saccharomyces. Genetics 53: 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi, R., K. A. Kandl, A. Carr-Schmid, J. L. Whitacre, A. E. Adams et al., 2001. Overexpression of translation elongation factor 1α affects the organization and function of the actin cytoskeleton in yeast. Genetics 157: 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olarewaju, O., P. A. Ortiz, W. Chowdhury, I. Chatterjee and T. G. Kinzy, 2004. The translation elongation factor, eEF1B, plays a role in the oxidative stress response pathway. RNA Biol. 1: 12–17. [DOI] [PubMed] [Google Scholar]
- Peters, H. I., Y.-W. E. Chang and J. A. Traugh, 1995. Phosphorylation of elongation factor 1 (EF-1) by protein kinase C stimulates GDP/GTP-exchange activity. Eur. J. Biochem. 234: 550–556. [DOI] [PubMed] [Google Scholar]
- Pittman, Y., L. Valente, G. R. Jeppesen, G. R. Andersen and S. Patel, 2006. Mg+2 and key lysine modulate exchange activity of eukaryotic translation elongation factor 1Bα. J. Biol. Chem. 281: 19457–19468. [DOI] [PubMed] [Google Scholar]
- Rabinow, L., S. L. Chiang and J. A. Birchler, 1993. Mutations at the Darkener of apricot locus modulate transcript levels of copia and copia-induced mutations in Drosophila melanogaster. Genetics 134: 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina, M. V., R. Fricke, L. Kuhn and W. Wintermeyer, 1995. Codon-dependent conformational change of elongation factor Tu preceding GTP hydrolysis on the ribosome. EMBO J. 14: 2613–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron, D., and H. P. Harding, 2000. PERK and translational control by stress in the endoplasmic reticulum, pp. 547–560 in Translational Control of Gene Expression, edited by N. Sonenberg, J. W. B. Hershey and M. B. Mathews. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Salas-Marco, J., and D. M. Bedwell, 2005. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 348: 801–815. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E. F. Fritsch and T. Maniatis, 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sandbaken, M. G., and M. R. Culbertson, 1988. Mutations in elongation factor EF-1α affect the frequency of frameshifting and amino acid misincorporation in Saccharomyces cerevisiae. Genetics 120: 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F., G. R. Fink and J. B. Hicks, 1986. Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Spoerner, M., A. Nuehs, P. Ganser, C. Herrman, A. Wittinghofer et al., 2005. Conformational states of Ras complexed with the GTP analogue GppNHp or GppCH2p: implications for the interaction with effector proteins. Biochemistry 44: 2225–2236. [DOI] [PubMed] [Google Scholar]
- Toyn, J. H., P. L. Gunyuzlu, W. H. White, L. A. Thompson and G. F. Hollis, 2000. A counterselection for the tryptophan pathway in yeast: 5-fluoroanthranilic acid resistance. Yeast 16: 553–560. [DOI] [PubMed] [Google Scholar]
- van Damme, H. T. F., R. Karssies, C. J. Timmers, G. M. C. Janssen and W. Moller, 1990. Elongation factor 1β of artemia: localization of functional sites and homology to elongation factor 1δ. Biochim. Biophys. Acta 1050: 241–247. [DOI] [PubMed] [Google Scholar]
- Vetter, I. R., and A. Wittenghofer, 2001. The guanine nucleotide-binding switch in three dimensions. Science 294: 1299–1304. [DOI] [PubMed] [Google Scholar]
- Zavialov, A. V., V.V. Hauryliuk and M. Ehrenberg, 2005. Guanine-nucleotide exchange on ribosome-bound elongation factor G initiates the translocation of tRNAs. J. Biol. 4: 9.. [DOI] [PMC free article] [PubMed] [Google Scholar]