

Abstract
Elucidating genetic influences on bison growth and body composition is of interest, not only because bison are important for historical, cultural, and agricultural reasons, but also because their unusual population history makes them valuable models for finding influential loci in both domestic cattle and humans. We tested for trait loci associated with body weight, height, and bison mass index (BMI) while controlling for estimated ancestry to reduce potential confounding effects due to population admixture in 1316 bison sampled from four U.S. herds. We used 60 microsatellite markers to model each phenotype as a function of herd, sex, age, marker genotypes, and individual ancestry estimates. Statistical significance for genotype and its interaction with ancestry was evaluated using the adaptive false discovery rate. Of the four herds, two appeared to be admixed and two were nonadmixed. Although none of the main effects of the loci were significant, estimated ancestry and its interaction with marker loci were significantly associated with the phenotypes, illustrating the importance of including ancestry in the models and the dependence of genotype–phenotype associations on background ancestry. Individual loci contributed ∼2.0% of variation in weight, height, and BMI, which confirms the utility and potential importance of adjusting for population stratification.
INTENSIVE linkage analysis in livestock species over the past two decades has led to the mapping of loci for economically important traits. Numerous regions of the domestic cattle genome, for example, have been linked to quantitative trait loci (QTL) for body weight and carcass characteristics (Casas et al. 1998, 2000, 2001; Elo et al. 1999; Stone et al. 1999; Grosz and MacNeil 2001; Kim et al. 2003), increasing the potential for using marker-assisted selection to improve traits of economic importance. North American bison (Bison bison) have recently become an important alternative and complementary meat source to beef cattle; >90% of bison are maintained on private ranches (Halbert 2003), where they are raised primarily for meat production. Although bison and domestic cattle are classified into different genera (Bison and Bos) and are estimated to have diverged 1.0–1.5 million years ago (Hartl et al. 1988; Wall et al. 1992; Ritz et al. 2000), they still have the same number of chromosomes (m = 30), the same chromosome banding patterns (Basrur and Moon 1967; Ying and Peden 1977), and a highly similar autosomal gene content and order (Schnabel et al. 2003). Interbreeding between bison and domestic cattle can result in fertile offspring, and modern technologies have been used to detect domestic cattle genetic introgression in many extant bison populations (Polziehn et al. 1995; Ward et al. 1999; Ward 2000; Halbert et al. 2005).
Although the recently enriched bovine linkage map (Ihara et al. 2004) has become a critical resource in the dissection of bovine quantitative traits including those of bison, only a few studies have reported microsatellite variation in North American bison (Momens et al. 1998; Wilson and Strobeck 1999; Ward 2000; Halbert 2003; Schnabel et al. 2003; Halbert et al. 2004). Moreover, only one QTL genome scan has so far been reported in bison (Schnabel et al. 2003); these investigators used a linkage map of 292 microsatellite markers spanning all 29 autosomes. A single quantitative trait locus was found to be significant for growth characteristics (17-month weight) and was located in the same region identified by Kim et al. (2003) that houses a locus for hot carcass weight in domestic cattle. More work remains to be done for bison to fully benefit from the identification of economically important genes through the dense bovine linkage map.
When feasible, the inbred line cross is a nearly ideal research design for initial detection of QTL by marker–trait associations because of the substantial long-range linkage disequilibrium (LD) it creates (Lynch and Walsh 1998). On the contrary, commercial livestock populations are predominantly outbred such that QTL segregate within families, and thus we must rely on analysis of relatives to provide pedigree information and the LD necessary for QTL detection (Lynch and Walsh 1998). Use of variances to estimate within-family variation leads to decreased power of QTL detection because variances are estimated less precisely than the means used in the inbred line crosses. The use of variances and the incomplete LD resulting from a lack of informative markers are the two major disadvantages of within-family linkage analysis. Herein, we apply an association analysis approach to map QTL in bison populations, which has distinct advantages over other methods: the potential to detect alleles with minor or modest phenotypic effects (Risch and Merikangas 1996) without pedigree information and the capacity to account for population stratification. Moreover, using admixed populations to detect QTL fills an important niche between intercross or family-based linkage studies and association studies among unrelated individuals in panmictic populations, since admixed populations have less disequilibrium than the former, but more than the latter (McKeigue 2005).
Recently created admixture between genetically differentiated populations provides high levels of LD for loci located as far apart as 10–20 cM (Chakraborty and Weiss 1988; McKeigue 1997; Shriver et al. 2003) and genetic substructure effects (interindividual variation in admixture proportions), requisite for greater statistical power to detect QTL. Variations in individual ancestry, which will generally be highly correlated with variations in individual admixture, create confounding effects and may lead to increased false positive associations (Parra et al. 1998; Lautenberger et al. 2000; Pfaff et al. 2001; Nordborg and Tavaré 2002; Gauderman 2003; Hoggart et al. 2004), thereby necessitating adjustment for ancestry effects a priori.
The potential for admixture effects in bison populations is significant for several reasons. First, within-population substructure has been detected in the Yellowstone National Park bison population (Halbert 2003) and remains to be examined in other populations. Additionally, nearly all extant bison populations were derived from a few founding lineages within the past 120 years (Coder 1975; Halbert 2003). Finally, evidence of domestic cattle nuclear introgression is present in most bison populations evaluated to date (Halbert 2003; Halbert et al. 2005). These three potential sources of admixture necessitate appropriate measures of and corrections for substructure effects in analysis of QTL in bison populations. Herein, we present the first study that tests for association and evaluate the contribution of nuclear microsatellite markers and population stratification to body size (weight and height) and relative body mass in U.S. federal bison herds.
MATERIALS AND METHODS
Sample and data collection:
Bison from the following U.S. federal herds were sampled for this study: Badlands National Park (BNP, n = 495); Fort Niobrara National Wildlife Refuge (FN, n = 75); Theodore Roosevelt National Park, South Unit (TRS, n = 371); and Wind Cave National Park (WC, n = 375). In total there were 1316 bison with ∼60% females and 40% males in each herd except FN (Table 1).
TABLE 1.
Mean ± standard deviation of weight, height, age, and mass index in the bison samples
| Herd | Sex | N | Weight (kg) | Height (m) | Age (yr) | Bison mass index | Exponent of height |
|---|---|---|---|---|---|---|---|
| BNP | Female | 296 | 432.5 ± 96.55 | 1.36 ± 0.10 | 4.21 ± 3.10 | 162.7 ± 5.57 | 3.09 ± 0.10 |
| Male | 199 | 537.2 ± 189.90 | 1.46 ± 0.16 | 3.55 ± 2.31 | 143.5 ± 5.91 | 3.40 ± 0.09 | |
| FN | Female | 37 | 210.9 ± 70.85 | — | 0.67 ± 0.24 | — | — |
| Male | 38 | 219.4 ± 77.88 | — | 0.67 ± 0.24 | — | — | |
| TRS | Female | 230 | 370.1 ± 127.84 | 1.34 ± 0.07 | 3.07 ± 3.21 | 220.1 ± 15.5 | 3.28 ± 0.23 |
| Male | 141 | 388.8 ± 196.39 | 1.39 ± 0.11 | 2.19 ± 1.98 | 158.6 ± 12.3 | 3.33 ± 0.21 | |
| WC | Female | 223 | 397.2 ± 70.29 | 1.39 ± 0.08 | 5.94 ± 6.22 | 200.2 ± 12.3 | 2.05 ± 0.18 |
| Male | 152 | 438.0 ± 180.57 | 1.43 ± 0.14 | 1.72 ± 2.03 | 107.3 ± 10.1 | 3.78 ± 0.22 |
There is a well-known relationship between body weight and height that in humans is often expressed as body mass index (kilograms per square meter) as a proxy measure of adiposity. Using the so-called Benn index approach (Garn and Pesick 1982), we derived a similar index for bison. We used a nonlinear regression (weight = τ × heightλ), where τ and λ are coefficients to be estimated. A bison mass index (BMI) was then calculated for each animal as 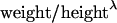 , where weight and height were expressed in kilograms and meters, respectively. Sex-specific values of τ and λ were estimated from the data for the entire sampled bison population and then for each herd (Table 1).
, where weight and height were expressed in kilograms and meters, respectively. Sex-specific values of τ and λ were estimated from the data for the entire sampled bison population and then for each herd (Table 1).
A summary of the relationships among the bison herds sampled on the basis of historical records is shown in Figure 1. Tail hair samples, jugular or tail vein blood samples, and body size measurements (weight and height) were collected by park personnel during annual bison roundups. Total body weight was measured using standard electronic livestock scales; height was calculated as the vertical distance from the chute platform to the highest point of the hump. Weight and height data were collected in English customary units (pounds, feet) and converted to metric units (kilograms, meters). Bison sampled from the FN herd included only calves from the 2002 roundup, from which height data are not available. DNA extraction from hair and whole blood samples, multiplex polymerase chain reaction design, and microsatellite analyses were performed as previously described (Halbert 2003; Halbert et al. 2004). Sixty microsatellite marker loci distributed throughout the bison genome were scored for each sample. Alleles previously identified as of domestic cattle origin (Halbert et al. 2005) were detected for the following markers: BM4307 (BNP, FN, TRS), BMS4017 (BNP, FN, TRS), and BMS2270 (BNP). The effects of these markers on bison weight and height were evaluated.
Figure 1.—

Establishment of the bison herds in this study, adapted from Coder (1975) and Halbert (2003). Parentheses indicate the year animals were transferred from one herd to another, as denoted by arrows. NM, national monument; NP, national park; NWR, national wildlife refuge; NY, New York; SP, state park.
Association of markers with weight, height, and BMI:
Before polymorphic microsatellite markers were used to evaluate their association with weight, height, and BMI, they were first used along with herd classification to estimate ancestry, using STRUCTURE software developed by Pritchard et al. (2000). We tested the association between marker genotype and estimated ancestry with weight and height by multivariate analysis of covariance and with BMI using analysis of variance (Huberty and Morris 1989). The proportion of variance contributed by genotype and estimated ancestry in the phenotypes was measured by the change in R2 between full and reduced models. The full model included the effects of genotype and genotype-by-estimated ancestry whereas the reduced model excluded these effects.
STRUCTURE was used to resolve bison population stratification and to assign individual animals to clusters (subpopulations) on the basis of their allele frequencies at multiple loci (Figure 2). STRUCTURE is a model-based clustering approach; it assumes that K latent subpopulations exist in the sample and assigns each sampled individual to them probabilistically. For example, an admixed population is assigned jointly to two or more subpopulations depending on the degree to which the genome of the admixed individual is composed of DNA segments that descended from one particular parental population relative to the others. The Markov chain Monte Carlo (MCMC) procedure is used to estimate populations of origin, populationwide marker allele frequencies, and proportions of individual admixture conditional on the observed variables, i.e., the marker genotype matrix denoted by X. Estimates of allele frequencies obtained are ultimately used to compute the likelihood of population origins given the genotype [i.e., assuming K populations, the probability of K given X was inferred: P(K | X)].
Figure 2.—

Summary of clustering results assuming five populations. Each point shows the mean estimated ancestry for several animals in the sample. The animals were labeled a posteriori according to herd of origin (color coded). Groups represented by the dots labeled 1, 2, and 3 likely reflect the historic relationship between FN and WC due to transfers of bison from Yellowstone National Park (Figure 1).
The most plausible number of subpopulations or clusters and the proportion of membership in the K clusters were obtained using log probability of the data (Log P(D)), which is used to estimate the posterior probability of data for a given K, Pr(X | K) (Pritchard and Wen 2003). We ran an MCMC scheme for K-values between 2 and 7, where each burn-in and MCMC length was 10,000 (Evanno et al. 2005). Twenty runs for each K were then carried out to quantify the amount of variation of likelihood values for the data conditioned on K. The real K-value was arrived at by examining the distribution of likelihood values and variance between runs across K. As recommended by Pritchard and Wen (2003), we plotted these distributions for each K (Figures 3 and 4). The average change in likelihood values tended to plateau or increase only slightly after the correct value of K is reached (Figure 3), while the variance between runs increased substantially (Figure 4). Coupled with information provided by predefined classification of bison into herds, we chose K = 5 for this data set. There is currently no accurate method for estimating K. In fact, according to Falush et al. (2003), the probability of alleles given K is maximized at high values of K. After determining the value of K for the data set, we estimated individual admixture for all animals in the data and the proportion of membership to the K classes from collecting data from 600,000 MCMC iterations after a burn-in period of 20,000 iterations (Pritchard and Wen 2003). The admixture estimates obtained were used as covariates in the subsequent association study.
Figure 3.—

The distribution of likelihood values for data conditioned on K. The trend increases distinctly from K = 2 to 5 and then plateaus off, which is consistent with a signal that the real value of K has been attained.
Figure 4.—

The distribution of between-runs (20 runs) variance of likelihood values for each K. After K = 5, the among-runs variance for K ≥ 6 increased tremendously.
We computed marker information content for ancestry (f) using the formula proposed by McKeigue (1998). Markers with large differences in allele frequencies among the bison herds explained stratification results appropriately. The average of this measure (i.e., f-value) is directly related to the fixation index between two populations (FST), often used to measure genetic distance between any two populations (Wright 1951).
Statistical model:
Data analysis proceeded in stages: descriptive statistics, significance tests, and estimates of effects (i.e., contribution of genotype, estimated ancestry, and genotype-by-estimated ancestry interaction). Analyses were implemented using SAS version 9.1. Because of the high correlation between weight and height at each locus in the data set (>0.75), tests of association with predictors (herd, sex, age, genotype, probability of inferred ancestry, genotype-by-estimated ancestry interaction) and their contribution were performed assuming a multivariate analysis model (Huberty and Morris 1989). Lack of records for height in the FN herd led to its exclusion from the analysis, although we used its genotypic information to estimate coefficients of ancestry.
Three models were implemented: the genotype model (tested the significance of genotype), the ancestry model (tested the effect of genotype and ancestry), and the interaction model (tested the effect of genotype, estimated ancestry, and genotype-by-estimated ancestry interaction). The overall model can be represented in matrix notation as 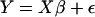 , where Y is an n × p matrix (n = number of observations and p = number of dependent variables, in this case, p = 2 or 1, when considering weight and height or BMI) and X is n × k matrix (k = number of predictors of the dependent variables). The number of predictors depended on the statistical model: the reduced model consisted primarily of the herd, sex, and age effects, where the age effect was included in both linear and quadratic terms. The full model—genotype, ancestry, and interaction models—consisted of genotype, estimated ancestry, and genotype-by-estimated ancestry interaction, respectively, in addition to the effects in the reduced model (i.e., herd, sex, and age). The genotype model tested for the effects of genotype on weight, height, and BMI in the absence of ancestry whereas the ancestry model tested for the effect of genotype and ancestry on weight, height, and BMI. Genotype-by-estimated ancestry effects on weight, height, and BMI were tested in the interaction model. The models afforded an opportunity to test the effect of marker genotype on weight and height in the presence and absence of ancestry.
, where Y is an n × p matrix (n = number of observations and p = number of dependent variables, in this case, p = 2 or 1, when considering weight and height or BMI) and X is n × k matrix (k = number of predictors of the dependent variables). The number of predictors depended on the statistical model: the reduced model consisted primarily of the herd, sex, and age effects, where the age effect was included in both linear and quadratic terms. The full model—genotype, ancestry, and interaction models—consisted of genotype, estimated ancestry, and genotype-by-estimated ancestry interaction, respectively, in addition to the effects in the reduced model (i.e., herd, sex, and age). The genotype model tested for the effects of genotype on weight, height, and BMI in the absence of ancestry whereas the ancestry model tested for the effect of genotype and ancestry on weight, height, and BMI. Genotype-by-estimated ancestry effects on weight, height, and BMI were tested in the interaction model. The models afforded an opportunity to test the effect of marker genotype on weight and height in the presence and absence of ancestry.
Wilk's λ-test statistic was used to test for significance at the 0.05 α-level. To control for potential type I error rate inflation due to multiple testing resulting from testing 60 marker loci, we used Benjamini and Hochberg's (2000) adaptive false discovery rate (FDR) procedure and reported marker genotypes significantly associated with the phenotypes, estimated ancestry, and genotype-by-estimated ancestry interactions. To investigate significant associations further, we examined solutions from generalized linear model analyses to ascertain specific genotypes responsible for the significant effects.
RESULTS
Descriptive statistics:
Each of the 60 loci evaluated contained 3–10 alleles (Halbert 2003). Table 1 summarizes the means (± standard deviation) of weight, height, BMI, and age relative to the herds and sex of the sampled bison population. More females were sampled in each herd than males. The BNP herd had the highest mean weight for both males and females followed by WC, TRS, and FN. Mean heights in BNP and WC were comparable. Males were on average heavier, taller, and younger than females; their weights showed more variability than those of females as indicated by larger standard deviations (Table 1). Heteroscedasticity testing using Levene's test (Levene 1960) revealed significant (P < 0.001) variance heterogeneity in weight between males and females. These results suggest that significant difference in herd, age, and sex may partly explain the differences in mean weight, height, and BMI of sampled animals. However, within-sex and between-herd differences in mean weight and BMI may largely be due to population stratification, as is evident from the results of the population substructure analysis below, suggesting a genetic basis for the notable significant differences between herds.
The populationwide BMI (± SE) and height exponent (± SE) were 192.7 ± 5.82 and 2.49 ± 0.09 for females and 143.4 ± 5.22 and 3.38 ± 0.09 for males, respectively. Herd-specific values suggest that female bison from TRS and WC herds had higher BMIs (>200) than those from the BNP herd. For males, the TRS herd had a higher BMI than both the BNP and the WC herds. Except for females from the WC herd, the exponent for height was slightly >3 in all herds. These results suggest that BMI is a ratio of body weight to the third order of height (BMI = weight/height3).
Population substructure:
Analysis of the four bison herds revealed that the FN and WC herds were homogenous (nonadmixed) whereas the BNP and TRS herds were admixed. Using estimated allele frequencies at the 60 marker loci, we assigned each individual a proportion of membership to each of the five clusters (1–5). Bison from the FN and WC herds were primarily assigned membership to clusters 2 and 4, respectively, with probabilities of 91 and 98%, respectively, suggesting that their genomes arose from these clusters. On the other hand, the BNP and TRS herds were assigned to two different populations each (Table 2), indicating some level of population subdivision within each of these herds. The BNP herd was assigned to clusters 3 and 4 with probabilities of 62 and 23%, while the TRS herd was assigned to clusters 5 and 1 with probabilities of 71 and 26%, respectively. These results imply a genetic connection between bison in the BNP and FN herds, which is consistent with the history of BNP herd establishment from FN bison (Figure 1). Analysis of results presented in Table 2, however, suggests a lack of genetic connection between the TRS and FN herds, contrary to the available historical records (Figure 1).
TABLE 2.
Proportion of membership of each predefined population in each of the five clusters
| Cluster (estimated ancestry)
|
||||||
|---|---|---|---|---|---|---|
| Herd | Total (N) | 1 | 2 | 3 | 4 | 5 |
| BNP | 495 | 0.063 | 0.017 | 0.629 | 0.225 | 0.066 |
| FN | 75 | 0.021 | 0.020 | 0.019 | 0.911 | 0.029 |
| TRS | 371 | 0.258 | 0.003 | 0.024 | 0.005 | 0.710 |
| WC | 375 | 0.006 | 0.979 | 0.005 | 0.004 | 0.005 |
Figure 2 shows a plot of clustering results for the bison in the sample, assuming that five subpopulations exist, as inferred above. The clustering results are largely consistent with the proportion of membership results shown in Table 2 except for the TRS herd, where the clustering and the probability of membership results somewhat disagree. The WC herd is connected to the FN and BNP herds as demonstrated by the labeled animals in Figure 2. The indirect connection between the WC and TRS herds through the FN herd is also implied from Figure 2 through animal 1.
In an attempt to explore further the apparent lack of genetic connection between TRS and FN herds, despite contrary evidence provided in the historical records detailing the establishment of each herd (Figure 1), an f-value was computed between all pairs of clusters at each locus. Table 3 summarizes the results on loci with high f-values (>20%) and reveals that overall, cluster 2 (equivalent to the WC herd) stood out as genetically distant from all other clusters. This suggests that the WC herd, which is a homogenous herd with a membership probability of 98%, is genetically distant from all other herds. A paired cluster 3 and 5, representing BNP and TRS, also had a high f-value for the 155 allele at BM4017 (Table 3). While the BNP herd was originally established with bison from the TRS herd, the noted difference between these populations is not surprising given the introduction of bison into BNP from an independent, and likely genetically distinct, herd in 1983 (Figure 1).
TABLE 3.
Summary of ancestry informative markers
| Locus | Alleles | Cluster combination (i and j) | fi and j |
|---|---|---|---|
| BM4028 | 114 | 2 and 3 | 0.30 |
| 116 | 1 and 2 | 0.24 | |
| 2 and 3 | 0.36 | ||
| 2 and 4 | 0.41 | ||
| 2 and 5 | 0.28 | ||
| 118 | 1 and 2 | 0.24 | |
| BM720 | 229 | 1 and 2 | 0.22 |
| BM4017 | 155 | 3 and 5 | 0.23 |
| TGLA227 | 72 | 1 and 2 | 0.22 |
| 73 | 1 and 2 | 0.21 | |
| URB011 | 153 | 1 and 2 | 0.22 |
| 2 and 4 | 0.25 |
Microsatellite marker locus BM4028 was the most informative marker of all 60 markers. From Table 3, the difference in frequency of allele 116 bp between cluster 2 (WC herd) and other clusters was notably >20%. Two other alleles at this locus (114 bp and 118 bp) showed similar trends.
Significant tests of herd, age, and sex:
In all cases, the overall regression model was significant (P ≤ 1.0 × 10−4), as were the effects of herd, sex, age, and age squared (P < 1.0 × 10−4). These results are consistent with our expectations based on the different management practices used within each herd, especially with regard to nutrition, disease control, and breeding structure (Berger and Peacock 1988; Halbert 2003).
Association of weight, height, and BMI with genotype and estimated ancestry:
Tests of association between traits and genotypes of polymorphic marker loci yielded nonsignificant results. However, estimated ancestry and its interaction with marker loci had significant associations with weight and height for some marker loci as summarized in Table 3. We used adaptive false discovery rate levels of 8.0 × 10−4, 4.3 × 10−3, 8.0 × 10−4, and 6.0 × 10−4, respectively, as thresholds to declare significance in the tests for the association of coefficients of ancestry for clusters 1, 2, 3, and 4 (q1, q2, q3, and q4, respectively) with the phenotypes. The corresponding threshold values for genotype-by-estimated ancestry interaction were 1.0 × 10−4, 2.1 × 10−3, 4 × 10−4, and 1.0 × 10−4. Genotypes in six marker loci, BM1905, BM4028, BM4513, BM7145, SPS113, and CSSM42, were significantly associated with q2. Genotypes in four marker loci, BM4028, CSSM42, BM4307, and BM7145, were significantly associated with q3. Only one marker locus each was significantly associated with q1 and q4: BM4028 and BM1905, respectively.
Table 4 summarizes significant genotype-by-estimated ancestry interaction effects on weight and height. Significant estimated ancestry effects at the BM4307 locus were particularly interesting because of the presence of domestic cattle-derived 197-bp alleles in some populations (Halbert 2003; Halbert et al. 2005). We observed significant (P ≤ 0.03) differences among means of 185/185 (coded 2), 185/197 (coded 1), and 197/197 (coded 0). Other marker loci suspected to carry alleles derived from domestic cattle included BM4017 and BMS2270, but because of the low frequency of their alleles in the sampled bison population, they were excluded from further analysis.
TABLE 4.
Significant interaction effects of estimated ancestry and marker genotype
| Marker locus | Chromosome | Positiona (cM) | Significant effectb | Significant genotype | P-value |
|---|---|---|---|---|---|
| BM4307 | 1 | 35.2 | q3 | <0.0001 | |
| BM4028 | 12 | 78.7 | q2 and q3 | 0.0008 | |
| BM711 | 8 | 83.6 | Genotype × q1 | 0.0001 | |
| BM2113 | 2 | 106.2 | Genotype × q2 | 127/153 | 0.0004 |
| Genotype × q4 | 143/153 | <0.0001 | |||
| TGLA227 | 21 | 67.3 | q2 | 0.0001 | |
| Genotype × q3 | 72/73 (weight) | 0.0004 | |||
| CSSM42 | 2 | 34.4 | q2 and q3 | <0.0001 | |
| q4 | 0.0006 | ||||
| BM7145 | 1 | 69.2 | q2 | 0.0016 | |
| q3 | <0.0001 | ||||
| BM1905 | 23 | 64.3 | Genotype × q4 | 172/176, 172/182, 172/184 | <0.0001 |
| SPS113 | 10 | 29.2 | q2 | 0.0003 | |
| BM4513 | 14 | 62.5 | q4 | 0.0043 | |
| Genotype × q4 | 0.0021 | ||||
| BMS2258 | 7 | 75.0 | Genotype × q4 | 127/142, 127/138, 138/142, 138/140, 138/142, 138/146, 140/146, 142/146, 146/148 | 0.0015 |
Chromosomal positions (centimorgans) as reported in the U.S. Department of Agriculture cattle gene-mapping database (http://www.animalgenome.org/cattle/).
Significant after adjusting for multiple testing.
Genotype-by-estimated ancestry interaction significantly influenced BMI at the RM372 and BM1905 marker loci in clusters 3 (q3) and 4 (q4). Coefficient of ancestry (q4) significantly influenced BMI at the BM1905 locus. Corresponding P-values for genotype × q3, genotype × q4, and q4 were 3.0 × 10−4, 2.8 × 10−4, and 2.0 × 10−4. We observed a total of four alleles at the BM1905 locus: two (172 and 176 bp) were common in three bison (BNP, TRS, and WC) herds and a similar number (182 and 184 bp) were present in the TRS herd only. Eight alleles were observed at the RM372 locus (114, 118, 128, 130, 132, 134, 136, and 138). The alleles combined variedly to constitute common genotypes in the sample.
Contribution of ancestry:
In addition to tests of association between genetic markers and the phenotypes under study, we also attempted to quantify the contribution of estimated ancestry to the linear model. Adjusting for variation of estimated ancestry in the model resulted in increased R2 as shown in Tables 5, 6, and 7, respectively, for weight, height, and BMI. The contribution of ancestry varied with marker loci, and summarized in the tables are results on loci where the change in R2 exceeded 5%.
TABLE 5.
The proportion of variance in body weight explained by polymorphic markers
| Rank | Locus | Marker genotype | Estimated ancestry | Full model |
|---|---|---|---|---|
| 1 | BMS2258 | 0.019 | 0.071 | 0.090 |
| 2 | BMS1857 | 0.016 | 0.067 | 0.083 |
| 3 | BM2113 | 0.018 | 0.064 | 0.082 |
| 4 | BM2830 | 0.015 | 0.066 | 0.081 |
| 5 | BMS1862 | 0.016 | 0.058 | 0.074 |
| 6 | BMS4017 | 0.009 | 0.064 | 0.073 |
| 7 | BM4311 | 0.017 | 0.052 | 0.069 |
| 8 | IL4 | 0.008 | 0.054 | 0.066 |
| 9 | BM720 | 0.017 | 0.049 | 0.066 |
| 10 | RM372 | 0.014 | 0.048 | 0.062 |
| 11 | URB011 | 0.018 | 0.041 | 0.060 |
| 12 | BMS812 | 0.013 | 0.047 | 0.060 |
| 13 | BM1225 | 0.015 | 0.044 | 0.059 |
| 14 | BMS527 | 0.016 | 0.043 | 0.059 |
TABLE 6.
The proportion of variance in height explained by polymorphic markers
| Rank | Locus | Genotype | Estimated ancestry | Full model |
|---|---|---|---|---|
| 1 | BMS4017 | 0.010 | 0.108 | 0.118 |
| 2 | BM2113 | 0.019 | 0.082 | 0.101 |
| 3 | BMS1862 | 0.022 | 0.070 | 0.092 |
| 4 | RM372 | 0.018 | 0.070 | 0.088 |
| 5 | BM720 | 0.015 | 0.072 | 0.087 |
| 6 | IL4 | 0.019 | 0.067 | 0.086 |
| 7 | BMS1857 | 0.010 | 0.070 | 0.080 |
| 8 | BMS2258 | 0.022 | 0.059 | 0.080 |
| 9 | BM2830 | 0.018 | 0.062 | 0.080 |
| 10 | BM188 | 0.014 | 0.062 | 0.076 |
| 11 | BM4311 | 0.026 | 0.048 | 0.074 |
| 12 | CSSM42 | 0.008 | 0.063 | 0.071 |
| 13 | BMS812 | 0.012 | 0.060 | 0.071 |
| 14 | BM4307 | 0.001 | 0.070 | 0.071 |
| 15 | BM1225 | 0.009 | 0.058 | 0.067 |
| 16 | BMS2270 | 0.000 | 0.066 | 0.065 |
| 17 | URB011 | 0.020 | 0.045 | 0.065 |
| 18 | ILSTS102 | 0.012 | 0.050 | 0.062 |
| 19 | BMS2639 | 0.011 | 0.049 | 0.061 |
TABLE 7.
The proportion of variance in relative body mass (BMI) explained by polymorphic markers
| Rank | Locus | Genotype | Estimated ancestry | Full |
|---|---|---|---|---|
| 1 | BM2830 | 0.013 | 0.076 | 0.089 |
| 2 | BMS2258 | 0.012 | 0.074 | 0.087 |
| 3 | RM372 | 0.009 | 0.064 | 0.072 |
| 4 | BM2113 | 0.013 | 0.059 | 0.071 |
| 5 | BMS1862 | 0.013 | 0.050 | 0.064 |
| 6 | URB011 | 0.022 | 0.041 | 0.063 |
| 7 | IL4 | 0.012 | 0.047 | 0.059 |
| 8 | BMS1857 | 0.012 | 0.045 | 0.058 |
| 9 | BMS2639 | 0.009 | 0.047 | 0.057 |
| 10 | BM4440 | 0.013 | 0.044 | 0.056 |
| 11 | BM720 | 0.014 | 0.037 | 0.051 |
| 12 | BMS4017 | 0.012 | 0.039 | 0.051 |
| 13 | BM4311 | 0.007 | 0.044 | 0.051 |
Estimated ancestry contributed more to the variation in the traits than genotype. In weight, for example, genotype alone contributed <2% of the variation whereas the inclusion of estimated ancestry led to an additional contribution in variation of weight (Table 5). In total, the full model accounted for up to 9% of variation in weight. The trend was similar in the case of height and BMI. At the same loci, albeit ranked differently, were contributions of estimated ancestry exceeding 5%. We observed more loci (21) with a change in R2 exceeding 5% for height than for weight (13) or BMI (14). Adjustment for estimated ancestry disqualified some marker loci that were otherwise significant to be nonsignificant (results not shown).
DISCUSSION
Recent genetic association studies have recommended adjusting for population stratification as a strategy for minimizing spurious association (Ewens and Spielman 1995; Parra et al. 1998; Lautenberger et al. 2000; Pfaff et al. 2001; Nordborg and Tavaré 2002; Hoggart et al. 2004). Failure to correct for even modest degrees of population stratification can result in unacceptably high type I error rates (Gauderman 2003) and reduced statistical power. Population stratification forms a crucial first step in defining a set of sampled populations otherwise predefined subjectively on the basis of nongenetic parameters (e.g., geographic location). For association mapping, this is important for confirming that the subjective classifications are consistent with genetic information and hence appropriate for studying the question of interest (Pritchard et al. 2000). In this study, data on weight, height, BMI, age, sex, and 60 microsatellite marker genotypes for individual bison from four U.S. populations were used to examine the relationship between marker genotypes and body size and relative body mass, identify population stratification, and assess the contribution of marker genotypes and population stratification to variation in body size traits. The model-based structured approach for analyzing population stratification as proposed by Pritchard et al. (2000) capitalizes on information provided by multilocus marker genotypes, large sample sizes, and prior classification to make accurate inferences about population substructure and individual probabilities of inferred ancestry.
The four bison subpopulations studied here have been distinguished by both geographical location and management practices for between 13 and 30 generations (Figure 1; generation time of 3 years) (Berger and Cunningham 1994). STRUCTURE not only succeeded in clustering the sampled population accurately, but also corroborated the known direct historical connections among subpopulations, particularly the direct genetic connection of FN with BNP and TRS and genetic distinctness of WC in relation to the other herds. Strikingly, indirect genetic connections were detected between WC and the other herds, a remnant of transfers of bison from Yellowstone National Park >90 years ago (Figure 1). Furthermore, within-subpopulation stratification was also identified for the BNP and TRS herds. Stratification within the BNP subpopulation was expected given the relatively recent introduction of bison from the Colorado National Monument (Figure 1). The somewhat unexpected stratification identified in the TRS herd may be a consequence of genetic drift, assortative mating, or other factors. Overall, the findings of this study offer a unique contribution to the increasing empirical evidence supporting the efficiency of STRUCTURE in assigning individuals to their populations of origin, despite the lack of formal procedures for estimating K (e.g., Pritchard and Donnelly 2001; Rosenberg et al. 2001; Manel et al. 2002; Turakulov and Easteal 2003; Evanno et al. 2005). That most of these studies are based on simulation places the current study's findings in the unique position of demonstrating the application of the method to real data. We conclude here that, on the basis of these findings, the pointers suggested by Pritchard and Wen (2003) seem to work reasonably for at least some real data as well.
STRUCTURE failed to capture the connection between TRS and FN because of lack of ancestry-informative markers. The genetic relationship between cluster 4 (FN herd) and cluster 5 (TRS herd) is so close that large differences in frequencies of marker alleles studied were nonexistent. This is supported by the small observed f-values (McKeigue 2005). Optimal marker loci for estimating proportions of ancestry should have different alleles fixed in each of the parental populations. In the absence of such loci, markers that demonstrate a large frequency difference (>20%) between the two parental populations are preferred. As shown in Table 3, none of the markers studied demonstrated frequency differences >20% between the TRS and FN herds.
Significant interaction between genotype and estimated ancestry provides insight into the genetic influence of the phenotypes studied. Marker loci that displayed significant interaction effects have been reported to be at putative regions for growth traits, including CSSM42 (BTA 2), BM4513 (BTA 1), and SPS113 (BTA 10) (Stone et al. 1999; Casas et al. 2001; Schnabel et al. 2003). In a sample of two private bison pedigrees, Schnabel et al. (2003) identified BM4513 as significantly associated with 17-month weight; Kim et al. (2003) also reported a putative QTL in this region for hot carcass weight in cattle. Additionally, SPS113 was reported by Casas et al. (2001) to be located in a region that may harbor a QTL associated with marbling score in cattle crosses. The significant interaction effects we observed could be due to epistasis or LD. Epistasis ensues when genetic loci involved have different effects as a function of the alleles present at other genetic loci, whereas LD results from marker loci that are linked to causal polymorphisms, which may vary as a function of background genetic ancestry.
Of the three loci (BM4307, BMS2270, and BMS4017), identified with alleles derived from domestic cattle in the populations studied, only BMS4017 was ancestry informative (Table 3). None of the three loci were significantly associated with weight or height. Therefore, domestic cattle nuclear alleles at these loci in bison do not appear to significantly effect body size. In light of the detection of domestic cattle gene introgression in many public and private bison populations (Ward et al. 1999; Halbert 2003; Halbert et al. 2005), this result is encouraging for bison conservationists.
From the findings of this study, we can conclude that incorporating population stratification into the linear model influences association results through improved model fit as evidenced by the change in R2 between the reduced and full models. This indicates lower type I error rates and, therefore, increased statistical power to detect trait loci due to reduction in residual variance (Li 1969; Purcell and Sham 2004). Furthermore, the results confirm that genotype–phenotype associations may depend on background ancestry.
A more comprehensive and thorough genomewide association analysis similar to the one done by Schnabel et al. (2003) could be considered as a follow-up study to the current one. Regions that are homologous to bovine QTL for weight and height should be considered as a matter of priority. This can be achieved with the use of a more intense marker map (more microsatellite markers or SNPs if available). This would provide a powerful approach, leading to more robust association study.
Acknowledgments
We recognize the park managers and biologists from Badlands National Park, Fort Niobrara National Wildlife Refuge, Theodore Roosevelt National Park, and Wind Cave National Park for providing samples and phenotypic data. This study was supported in part by the National Park Service (00CRAG0036), the U.S. Geological Survey (00CRAG0020), the Turner Foundation (20011326), and the National Institutes of Health (T32HL072757, K25DK062817, and R01ES009912).
References
- Basrur, P. K., and Y. S. Moon, 1967. Chromosomes of cattle, bison, and their hybrid, the Cattalo. Am. J. Vet. Res. 28: 1319–1325. [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 2000. On adaptive control of the false discovery rate in multiple testing with independent statistics. J. R. Stat. Soc. 57: 289–300. [Google Scholar]
- Berger, J., and C. Cunningham, 1994. Bison: Mating and Conservation in Small Populations. Columbia University Press, New York.
- Berger, J., and M. Peacock, 1988. Variability in size-weight relationships of Bison bison. J. Mamm. 67: 618–623. [Google Scholar]
- Casas, E., J. W. Keele, S. D. Shackelford, M. Koohmaraie, T. S. Sonstegard et al., 1998. Association of the muscular hypertrophy locus with carcass traits in beef cattle. J. Anim. Sci. 76: 468–473. [DOI] [PubMed] [Google Scholar]
- Casas, E., S. D. Shackelford, J. W. Keele, R. T. Stone, S. M. Kappes et al., 2000. Quantitative trait loci affecting growth and carcass composition of cattle segregating alternate forms of myostatin. J. Anim. Sci. 78: 560–569. [DOI] [PubMed] [Google Scholar]
- Casas, E., R. T. Stone, J. W. Keele, S. D. Shackelford, S. M. Kappes et al., 2001. A comprehensive search form quantitative trait loci affecting growth and carcass composition of cattle segregating alternative forms of the myostatin gene. J. Anim. Sci. 79: 854–860. [DOI] [PubMed] [Google Scholar]
- Chakraborty, R., and K. M. Weiss, 1988. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA 85: 9119–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coder, G. D., 1975. The national movement to preserve the American buffalo in the United States and Canada between 1880 and 1920. Ph.D. Dissertation, Ohio State University, Columbus, OH.
- Elo, K. T., J. Vilkki, D. Dekoning, R. J. Velmala and A. V. Makki-Tanila, 1999. A quantitative trait locus for live weight maps to bovine chromosome 23. Mamm. Genome 10: 831–835. [DOI] [PubMed] [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Ewens, E. J., and R. S. Spielman, 1995. The transmission/disequilibrium test: history, subdivision, and admixture. Am. J. Hum. Genet. 57: 455–464. [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multi-locus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garn, S. M, and S. D. Pesick, 1982. Comparison of the Benn index and other body mass indices in nutritional assessment. Am. J. Clin. Nutr. 36(4): 573–575. [DOI] [PubMed] [Google Scholar]
- Gauderman, W. J., 2003. Candidate gene association for a quantitative trait, using parent-offspring trios. Genet. Epidemiol. 25: 327–338. [DOI] [PubMed] [Google Scholar]
- Grosz, M. D., and M. D. MacNeil, 2001. Putative quantitative trait locus affecting birth weight on bovine chromosome 2. J. Anim. Sci. 79: 68–72. [DOI] [PubMed] [Google Scholar]
- Halbert, N. D., 2003. The utilization of genetic markers to resolve modern management issues in historic bison populations: implications for species conservation. Ph.D. Dissertation, Texas A&M University, College Station, TX.
- Halbert, N. D., T. Raudsepp, B. P. Chowdhary and J. N. Derr, 2004. Conservation genetic analysis of the Texas State Bison Herd. J. Mamm. 85: 924–931. [Google Scholar]
- Halbert, N. D., T. J. Ward, R. D. Schnabel, J. F. Taylor and J. N. Derr, 2005. Conservation genomics: disequilibrium mapping of domestic cattle chromosomal segments in North American bison populations. Mol. Ecol. 14: 2343–2362. [DOI] [PubMed] [Google Scholar]
- Hartl, G. B., R. Goltenboth, M. Grillitsch and R. Willing, 1988. On the biochemical systematics of the Bovini. Biochem. Syst. Ecol. 16: 575–579. [Google Scholar]
- Hoggart, C. J., E. J. Parra, M. D. Shriver, C. Bonilla, R. A. Kittles et al., 2004. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 74: 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberty, C. J., and J. D. Morris, 1989. Multivariate analysis versus multiple univariate analyses. Psychol. Bull. 105: 302–308. [Google Scholar]
- Ihara, N., A. Takasuga, K. Mizoshita, H. Takeda, M. Sugimoto et al., 2004. A comprehensive genetic map of the cattle genome based on 3802 microsatellites. Genome Res. 14: 1987–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J.-J., F. Farnir, J. Savell and J. F. Taylor, 2003. Detection of QTL for growth and beef carcass fatness traits in a cross between Bos taurus (Angus) and Bos indicus (Brahman) cattle. J. Anim. Sci. 81: 1933–1942. [DOI] [PubMed] [Google Scholar]
- Lautenberger, J. A., J. C. Stephens, S. J. O'Brien and M. W. Smith, 2000. Significant admixture linkage disequilibrium across 30 cM around the FY locus in African Americans. Am. J. Hum. Genet. 66: 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levene, H., 1960. Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling, pp. 278–292. Stanford University Press, Stanford, CA.
- Li, C., 1969. Population subdivision with respect to multiple alleles. Ann. Hum. Genet. 33: 2329. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Manel, S., P. Brthier and G. Luikart, 2002. Detecting wildlife poaching: identifying the origin of individuals with Bayesian assignment tests and multilocus genotypes. Conserv. Biol. 16: 650–659. [Google Scholar]
- McKeigue, P. M., 1997. Mapping genes that underlie ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am. J. Hum. Genet. 60: 188–196. [PMC free article] [PubMed] [Google Scholar]
- McKeigue, P. M., 1998. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am. J. Hum. Genet. 63: 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue, P. M., 2005. Prospects for admixture mapping of complex traits. Am. J. Hum. Genet. 76: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momens, G., A. Van Zeveren and L. J. Peelman, 1998. Effectiveness of bovine microsatellites in resolving paternity cases in American bison, Bison bison L. Anim. Genet. 29: 12–18. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., and S. Tavaré, 2002. Linkage disequilibrium: what history has to tell. Trends Genet. 18: 83–90. [DOI] [PubMed] [Google Scholar]
- Parra, E. J., A. Marcini, J. Akey, J. Martinson, M. A. Batzer et al., 1998. Estimating African American admixture proportions by use of population-specific alleles. Am. J. Hum. Genet. 63: 1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff, C. L., E. J. Parra, C. Bonilla, K. Heister, P. M. McKeigue et al., 2001. Population structure in admixed populations: effect of admixture dynamics on the pattern of linkage disequilibrium. Am. J. Hum. Genet. 68: 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polziehn, R. O., C. Strobeck, J. Sheraton and R. Beech, 1995. Bovine mtDNA discovered in North American bison populations. Conserv. Biol. 9: 1638–1643. [Google Scholar]
- Pritchard, J. K., and P. Donnelly, 2001. Case-control studies of association in structured or admixed populations. Theor. Popul. Biol. 60: 227–237. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and W. Wen, 2003. Documentation for STRUCTURE Software, Version 2 (http://pritch.bsd.Uchicago/edu).
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell, S., and P. Sham, 2004. Properties of structured association approaches to detecting population stratification. Hum. Hered. 58: 93–105. [DOI] [PubMed] [Google Scholar]
- Risch, N., and K. Merikangas, 1996. The future of genetic studies of complex human diseases. Science 273: 1516–1517. [DOI] [PubMed] [Google Scholar]
- Ritz, L. R., M.-L. Glowatzi-Mullis, D. E. Machugh and C. Gaillard, 2000. Phylogenetic analysis of the tribe bovine using microsatellites. Anim. Genet. 31: 178–185. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., T. Burke, K. Elo, M. W. Feldman, P. J. Freidlin et al., 2001. Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics 159: 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel, R. D., J. F. Taylor and J. N. Derr, 2003. Development of a linkage map and QTL scan for growth traits in North American bison. Cytogenet. Genome Res. 102: 59–64. [DOI] [PubMed] [Google Scholar]
- Shriver, M. D., E. J. Parra, D. C. Bonilla, H. Norton, C. Jovel et al., 2003. Skin pigmentation, biological ancestry and admixture mapping. Hum. Genet. 112: 387–399. [DOI] [PubMed] [Google Scholar]
- Stone, R. T., J. W. Keele, S. D. Shackelford, S. M., Kappes and M. Koohamiriae, 1999. A primary screen of the bovine genome for quantitative trait loci affecting carcass and growth traits. J. Anim. Sci. 77: 1379–1384. [DOI] [PubMed] [Google Scholar]
- Turakulov, R., and S. Easteal, 2003. Number of SNP loci needed to detect population structure. Hum. Hered. 55: 37–45. [DOI] [PubMed] [Google Scholar]
- Wall, D. A., S. K. Davis and B. M. Read, 1992. Phylogenetic relationships in the subfamily Bovinae (Mammalia: Artiodactyla) based on ribosomal DNA. J. Mamm. 73: 262–275. [Google Scholar]
- Ward, T. J., 2000. An evaluation of the outcome of inter-specific hybridization events coincident with a dramatic demographic decline in North America bison. Ph.D. Dissertation, Texas A&M University, College Station, TX.
- Ward, T. J., J. P. Bielawski, S. K. Davis, J. W. Templeton and J. N. Derr, 1999. Identification of domestic cattle hybrids in wild cattle and bison species: a general approach using mtDNA markers and the parametric bootstrap. Anim. Conserv. 2: 51–57. [Google Scholar]
- Wilson, G. A., and C. M. Strobeck, 1999. Genetic variation within and relatedness among wood and plains bison populations. Genome 42: 483–496. [PubMed] [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15: 322–354. [DOI] [PubMed] [Google Scholar]
- Ying, K. L., and D. G. Peden, 1977. Chromosomal homology of wood bison and plains bison. Can. J. Zool. 55: 1759–1762. [Google Scholar]