

Abstract
Objective:
This review describes the pathologic and epidemiologic features of gastrointestinal stromal tumor (GIST) as well as the contemporary management of this tumor. The integration of surgery and treatment with targeted molecular agents in the treatment of GIST is highlighted.
Summary Background Data:
GIST is the most common mesenchymal tumor of the gastrointestinal tract. Its cellular origin from the interstitial cell of Cajal and distinctness from smooth muscles tumors were only recently appreciated. The discovery of the centrality of KIT proto-oncogene mutations in the pathogenesis of this tumor, and the development of imatinib mesylate, a specific inhibitor of KIT tyrosine kinase function have revolutionized the treatment of GIST.
Methods:
We conducted a review of the English literature on GIST. The pathology, epidemiology, diagnosis, and treatment of this tumor are summarized with particular emphasis on recent developments in the field.
Results:
GIST is a rare tumor that usually arises from the stomach or small intestine. It is characterized by immunohistochemical staining for KIT. Treatment of primary localized tumors is surgical. The benefit of adjuvant treatment with the KIT tyrosine kinase inhibitor imatinib is the subject of investigation. The treatment of unresectable, recurrent, or metastatic GIST is primarily imatinib treatment. The integration of surgery or ablative modalities is often employed, particularly when all disease is amenable to gross resection or destruction, or when GIST becomes resistant to imatinib. Newer tyrosine kinase inhibitors, such as sunitinib are the subject of ongoing investigation.
Conclusions:
The treatment paradigm for GIST has required the integration of surgery and molecular therapy and this will likely serve as a paradigm for the treatment of other solid tumors as targeted agents are developed.
This review summarizes the pathology, epidemiology, diagnosis, and treatment of gastrointestinal stromal tumor. The integration of molecularly targeted agents and surgery is highlighted.
Although gastrointestinal stromal tumor (GIST) is an uncommon tumor, it has recently received considerable attention because it serves as a model for the molecular therapy for cancer. Imatinib mesylate (STI 571, Gleevec; Novartis Pharmaceuticals, Basel Switzerland), a selective inhibitor of KIT protein, achieves dramatic responses in metastatic GIST since the KIT gene is usually mutated. While the treatment of primary, localized GIST remains surgical, the integration of surgery and molecular therapy for primary or metastatic GIST is under intense investigation and will serve as the basis for combined therapy in other malignancies.
Background
Despite its rarity, GIST is the most common mesenchymal tumor (ie, sarcoma) of the gastrointestinal tract and most often arises in the stomach or small intestine. GISTs are thought to originate from the interstitial cell of Cajal (ICC). ICCs are located in and around the myenteric plexus and are thought to function as intestinal pacemaker cells that regulate intestinal motility. On the basis of their histologic appearance, GISTs were previously thought to share smooth muscle differentiation. Historically, GISTs were thus often misclassified as leiomyomas or leiomyosarcomas. Subsequently, it has been determined that GISTs have distinct ultrastructural features and immunophenotypical markers compared with smooth muscle and smooth muscle tumors. The most notable immunohistological marker for GISTs is KIT (CD 117), with at least 95% of cases of staining positive. KIT is expressed only on differentiated ICCs, mast cells, and melanocytes corroborating a shared lineage between GISTs and ICCs. KIT is also expressed in development by hematopoietic stem cells, pro-B cell and pro-T cell lymphocytes, and progenitor cells of dendritic, erythroid, megakaryotic, and myeloid lineages. In recent studies comparing gene expression between GIST and other sarcomas, GISTs were shown to have a distinct and fairly homogenous profile once again confirming that GIST is a distinct entity.1–3
Epidemiology
GIST has a slight male predominance. Although GIST has been reported in people of all ages, most patients are between 40 and 80 years old at the time of diagnosis, with a median age of approximately 60 years.4–8 Nearly all GISTs are sporadic. About a dozen families with germline mutations have been reported, most of which have had a KIT mutation,9 but one carried a platelet-derived growth factor-α (PDGFRα) mutation.10 GIST can also occur in association with the hereditary syndromes Von Recklinghausen disease and Carney's triad (gastric GIST, paraganglioma, and pulmonary chondroma).
The former discrepancies in the classification of GIST have made its incidence difficult to determine. Data from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute (NCI) reported in 1995 indicate that 500 to 600 new cases are diagnosed per year in the United States.11 The rapid accrual of patients into recent clinical trials has made it clear that the previous estimates understated the true incidence. A recent population-based study from Sweden suggests an incidence of about 13 cases per million persons per year.12 There is no information on the true incidence in the United States; but extrapolating from these newer data, it may be as high as a 3000 to 5000 cases per year.
GIST can occur anywhere along the GI tract but most commonly arises in the stomach (65%) or small intestine (25%). About 5% to 10% of GISTs are located in the colon and rectum and another 5% in the esophagus.4,12,13 While GIST is the most common mesenchymal tumor in most portions of the GI tract, leiomyomas predominate in the esophagus. Rarely, GISTs develop in the omentum, mesentery, or retroperitoneum.
Diagnosis
Clinical Diagnosis
The diagnosis of GIST is often made only after laparotomy and formal pathologic examination. Because of the rarity of this tumor, it is often not suspected prior to surgery. The preoperative diagnosis of GIST requires a high degree of suspicion and familiarity with its radiologic appearance.
The clinical presentation of GIST varies widely. In a population-based study, approximately 70% of patients with GIST were symptomatic, 20% were asymptomatic, and 10% were detected at autopsy. GISTs that caused symptoms tended to be larger with an average size of 6 cm versus 2 cm for asymptomatic GISTs and 1.5 cm for GISTs detected at autopsy.12 Asymptomatic GISTs are often found incidentally on physical examination, radiologic imaging, endoscopy, or laparotomy. Symptoms are most commonly related to mass effect or bleeding. GISTs can grow very large before producing symptoms as they tend to displace adjacent structures without invasion. Large tumors can cause vague abdominal discomfort, pain, bloating, early satiety, or increased abdominal girth. When GISTs erode into the lumen of the GI tract, they can induce significant hemorrhage or anemia from occult bleeding. GISTs can also rupture into the peritoneum causing life-threatening hemorrhage. In addition to symptoms from mass effect or bleeding, GISTs can cause dysphagia when located in the esophagus, biliary obstruction when located around the ampulla of Vater, and intussusception or rarely intestinal obstruction in the small bowel.14
As computed tomography (CT) and magnetic resonance imaging (MRI) are often used to evaluate abdominal symptoms, GIST is commonly detected on these studies. Contrast-enhanced CT and MRI are useful to evaluate the primary tumor as well as the liver and peritoneum, which are the most common sites of metastasis. A primary tumor is typically a well-circumscribed and often highly vascular mass closely associated with the stomach or intestine. GISTs often appear heterogeneous due to necrosis or intratumoral hemorrhage. On CT, GIST usually appears as a hyperdense-enhancing mass. Hypermetabolic uptake on 18Fluorodeoxyglucose (FDG) positron emission tomography (PET) is highly sensitive but not specific for the diagnosis of GIST. 18FDG PET can provide functional information about the response of a tumor to treatment.
Endoscopy may be useful in the diagnosis of gastric or colorectal GIST. On endoscopy, GIST appears as a submucosal mass. Endoscopic ultrasound can be useful to confirm that a tumor originates from the bowel wall and not the mucosa. As GIST seldom initially metastasizes to the chest, a chest radiograph is satisfactory to assess the thorax. Bone scans are necessary only to evaluate specific symptoms.
Percutaneous biopsy should rarely be used for resectable primary GIST. Theoretically, biopsies can precipitate tumor rupture and lead to tumor dissemination or hemorrhage. Furthermore, it can often be difficult to reliably diagnose GIST from a percutaneous biopsy, especially if only a fine-needle aspiration is performed. Even core biopsies may be inadequate if necrotic or hemorrhagic tissue is sampled. Percutaneous biopsy is more appropriate in cases where it will change the clinical management. For example, biopsy may be useful when another diagnosis, such as lymphoma, that would not benefit from surgical resection is entertained. Biopsy is also indicated in cases where the mass is marginally resectable and neoadjuvant imatinib treatment is desirable.
Pathologic Diagnosis
Histologically, GISTs can be characterized as spindle-cell type (70%), epithelioid type (20%), or rarely mixed type where both features are present.15 Spindle-cell GIST appears as uniform fusiform cells in intersecting fascicles or whorls. Epithelioid GISTs typically appear as rounded cells in a nested pattern. GISTs usually have scant stroma and uniform cytology with fibrillary eosinophilic cytoplasm and nuclei containing fine chromatin and inconspicuous nucleoli.
The characteristic immunohistochemical profile of GIST has proven very helpful in diagnosis. Most importantly, GISTs are almost always immunopositive for KIT. KIT overexpression is usually related to mutations in the KIT gene, although PDGFRα mutations and other unknown mechanism also appear to result in KIT overexpression without KIT gene mutations. There is variability in the level of KIT expression, although typically staining is very intense. Furthermore, 60% to 70% are positive for CD34, 30% to 40% are positive for smooth-muscle actin (SMA), and approximately 5% are positive for the S-100 protein. In contrast to smooth muscle tumors, GIST rarely expresses desmin and, when it does, it is invariably focal with only a small number of immunopositive cells.15 The diagnosis of a KIT-negative GIST requires an expert pathologist and depends on tissue morphology as well as genotyping the tumor for a KIT or PDGFRα mutation as some tumors negative for KIT byimmunohistochemistry have been shown to have either mutation.16,17
Treatment of Primary Disease
For patients with primary, localized GIST, surgery represents the only chance for cure. Surgical resection typically carries little morbidity for most tumors less than 10 cm that are confined to the stomach or intestine and extensive surgery is not usually required. Resection of GIST can usually be accomplished with a wedge resection of the stomach or a segmental resection of the small intestine. Every effort should be taken to ensure negative margins, and wide margins have not been shown to be beneficial. As GIST rarely metastasizes to lymph nodes, lymphadenectomy is not routinely indicated. GISTs usually displace adjacent tissues without infiltrating them, and thus they can usually be lifted away from surrounding structures. When GISTs are densely adherent to adjacent organs, en bloc resection should be performed. GISTs are soft and friable so special care needs to be taken to avoid intraoperative rupture, which increases the risk of recurrence. While laparoscopic resection of small GISTs may be technically possible, it should only be undertaken when it will not increase the chance of tumor rupture. When surgical margins are microscopically positive on final pathologic review, further management depends on whether the surgeon thinks the margin is truly positive or is an artifact of the tissue processing, whether the area of positive margin could be identified on repeat exploration, and whether further resection is technically feasible.
Complete gross resection is possible in approximately 85% of patients with primary, localized tumors.4,6,8,18 Negative microscopic margins are achieved in 70% to 95% of these completely resected cases.4–6,8,18 At least 50% of patients develop tumor recurrence after complete resection of localized GIST and 5-year survival is usually about 50%.4,5,18 Results of published series reporting outcome for completely resected primary, localized GIST are shown in Table 1.
TABLE 1. Outcome of Complete Surgical Resection of Primary, Localized GIST

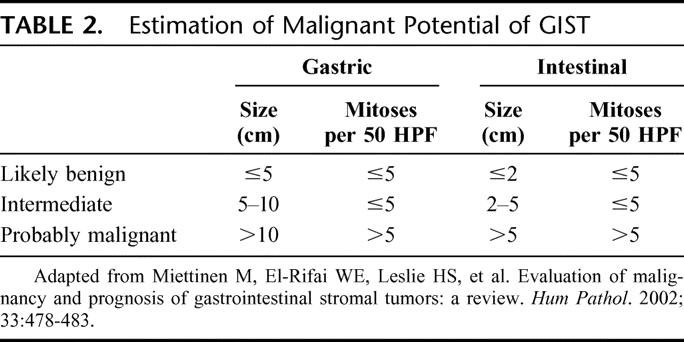
It is difficult to determine whether any particular GIST will recur after complete resection. It is now accepted that all GISTs can exhibit malignant behavior and none, except perhaps <1 cm tumors, can be labeled as definitely benign based on clinicopathologic features. Tumor size and mitotic index are the 2 most important prognostic variables for GIST.19 Site of origin within the GI tract has also been identified as a prognostic factor.13 Gastric GISTs tend to have a more favorable clinical course than those from the small intestine. Colonic GISTs recur at least as often as those from the small bowel. Esophageal GISTs are often more advanced on diagnosis and have a poor outcome. The particular kinase mutation may have prognostic importance as well, although current data are conflicting as to whether KIT mutations, and in particular KIT exon 11 mutations, correlate with a better or worse outcome.6,20–23 A commonly accepted system for grouping GISTs based on the risk of recurrence is shown in Table 2.13
TABLE 2. Estimation of Malignant Potential of GIST
Before the specific tyrosine kinase inhibitor imatinib was applied to GIST in 2000, GIST proved refractory to any treatment other than surgery, including conventional chemotherapy and radiation therapy. Thus, historically the standard of care after surgical resection was observation alone. The success of imatinib in the treatment of metastatic GIST and the significant risk of recurrence of GIST with surgery alone have prompted investigation into the benefit of imatinib when combined with complete surgical resection. The American College of Surgeons Oncology Group (ACOSOG) is conducting 2 prospective trials for completely resected primary, localized GIST. The Z9000 trial, which met accrual in September 2003, is evaluating recurrence and survival in 106 patients who underwent complete resection of a high-risk GIST (≥10 cm, intraperitoneal rupture or bleeding, or multifocal tumors) and then were treated for 1 year with imatinib 400 mg/day. The Z9001 trial is a randomized, double-blind trial in which patients who have undergone complete resection of a GIST ≥3 cm are either given imatinib 400 mg/day or placebo for 1 year. More than 400 of the 672 patients planned have been accrued as of August 2005. In Europe, there are 2 open trials prospectively assessing adjuvant imatinib in the treatment of GIST. The European Organization for Research and Treatment of Cancer (EORTC) trial 62024 is randomizing patients with intermediate- or high-risk GIST to no treatment or open-label imatinib 400 mg/day for 2 years. The Scandinavian Sarcoma Group XVIII trial is testing the duration of adjuvant imatinib treatment. In this trial, patients with complete gross resection of high-risk primary GIST with or without metastasis are randomized to imatinib 400 mg/day for either 1 or 3 years.
The success of imatinib in the treatment of GIST has also changed the paradigm for the approach to marginally resectable primary tumors. When, due to the size and location of the tumor, resection would require the risk of severe organ dysfunction or where negative margins would be difficult to achieve, it may be advisable to treat with imatinib first to downsize the tumor so as to make complete resection easier and safer to achieve. (See algorithm in Fig. 1.) This approach is currently being studied by the Radiation Therapy Oncology Group Phase II trial 0132, which is evaluating the role of neoadjuvant imatinib in a prospective nonrandomized fashion.
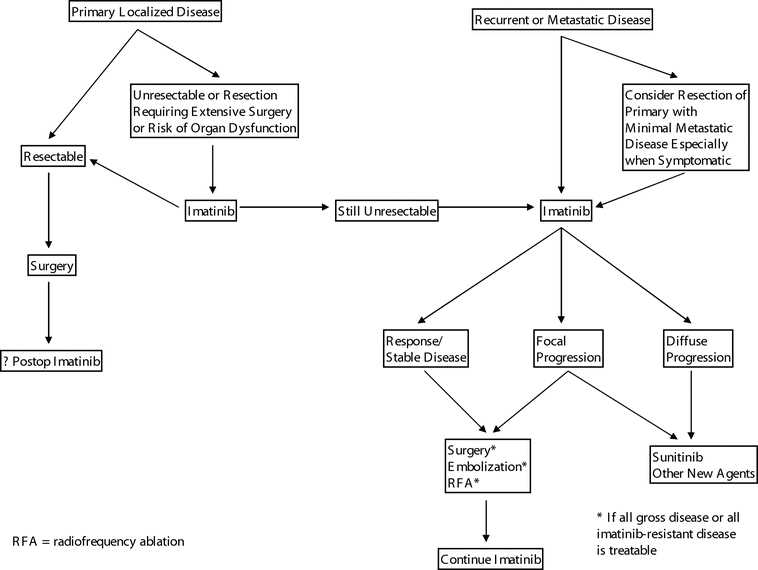
FIGURE 1. Algorithm for the treatment of GIST.
There is no evidence to determine the proper postoperative follow-up of patients with completely resected GIST. Although there is no proof that earlier detection of recurrent GIST improves survival, there is now therapy that can halt the progression of disease in most patients. It thus appears reasonable to perform routine postoperative surveillance. Most recurrences occur within the first 3 to 5 years, so the 2 consensus statements advocate the most intense surveillance during this interval. The National Comprehensive Cancer Network guidelines recommend CT scans of the abdomen and pelvis with intravenous contrast every 3 to 6 months during this interval and yearly afterward.24 The European Society of Medical Oncology guidelines stratify intensity of surveillance with risk of recurrence.25 For tumors >5 cm in size or with >5 mitoses/HPF, CT scans are recommended every 3 to 4 months for 3 years followed by every 6 months for 2 additional years and yearly thereafter. For smaller and less mitotically active tumors, CT scans are recommended every 6 months for 5 years.
Treatment of Recurrent and Metastatic Disease
Unfortunately, approximately two thirds of patients with GIST will experience recurrence or metastasis during the course of their disease. About 20% to 30% of patients have metastases at initial diagnosis and another 50% will develop recurrence. The median time to recurrence after surgery is 19 to 25 months.4,5,8,18 The first recurrence is typically in the abdomen with metastases to lung and bone sometimes developing later. The initial site of recurrence involves the liver in 65%, the peritoneal surfaces in 50%, and both in about 20%.4,18 Because of the multifocal nature of recurrent GIST and its predilection for further recurrence after resection, recurrent and metastatic GIST should be managed similarly.
Surgery alone is usually not effective treatment of recurrent GIST. Complete resection can only be achieved less than half of the time for patients with peritoneal recurrence. Although removal of peritoneal disease is usually technically simple as it tends lie on the peritoneal surface without invading the structures below, most often the disease is multifocal even when cross-sectional imaging suggests it may be limited. Recurrence after resection of peritoneal disease is the rule rather than the exception. When GIST is metastatic to the liver, it also tends to be multifocal and diffuse and thus not amenable to resection. In patients who have resectable liver lesions, recurrence is almost uniform after partial hepatectomy, and the 5-year survival was 30% in the era prior to the advent of imatinib.26
Imatinib Mesylate
The use of imatinib mesylate in the treatment of GIST has revolutionized the management of this disease and has made GIST a paradigm for the treatment of solid tumors with molecularly targeted therapy. The discovery that mutations in the KIT gene causing increased KIT protein function drive the oncogenesis of most GISTs was reported in 1998. Only 2 years later, imatinib, a potent inhibitor of KIT signaling, wasfirst used in the care of a patient with metastatic GIST. The ensuing 5 years have established the safety and efficacy of this drug and have further delineated its clinical impact andits limitations. Ongoing studies will help integrate other treatments with imatinib therapy. The history of the development of imatinib and its application to GIST is instructive of the rapid pace that is possible in the translation of fundamental knowledge of cancer pathogenesis into the implementation of new treatment strategies.
In 1998, Hirota et al found mutations in the KIT gene of 5 of 6 GISTs they examined.27 KIT was originally discovered as an oncogene in a feline sarcoma retrovirus, and its cellular homologue was found to encode for a transmembrane receptor tyrosine kinase signaling molecule. The ligand of KIT (known as KIT ligand, stem-cell factor, steel factor, or mast-cell growth factor) causes dimerization of KIT molecules on the cell surface when it binds to KIT. The resulting activation of KIT's tyrosine kinase moiety causes subsequent autophosphorylation and phosphorylation of downstream signaling molecules. This results in inhibition of apoptosis and in increased cell proliferation. KIT function is important in normal gametogenesis, melanocytogenesis, erythropoiesis, lymphopoiesis, megakaryopoiesis, as well as mast cell development and function. In GIST, KIT mutations cause transcription of a protein that is constitutively active and results in ligand-independent signaling. In large studies, activating KIT mutations have been found in approximately 80% to 90% of GISTs.28,29 KIT mutations appear to play a central role in the oncogenesis of GIST that is somewhat unique in solid tumors. Activating KIT mutations in transgenic mice are sufficient for the development of GIST30 as is seen in human families carrying these mutations. The fact that imatinib has such dramatic activity against GIST confirms the centrality of the KIT pathway in this tumor.
In 2000, a patient with metastatic GIST who had failed other available treatments was started on imatinib. Imatinib had recently been developed as an orally administered inhibitor of the tyrosine kinase function of the bcr-abl fusion protein for the treatment of chronic myeloid leukemia.31 Within weeks of starting imatinib, the patient exhibited an objective clinical response. By MRI there was a dramatic reduction in tumor volume, by 18FDG-PET there was elimination of hypermetabolic uptake, and histopathologic examination of serial biopsies showed decreased tumor density.32 These results led to the initiation of clinical trials formally testing the efficacy of imatinib in metastatic GIST. Remarkably, prospective trials have shown that approximately 50% of patients will have a response to imatinib and about 75% to 85% will have at least stable disease (Table 3).33–38 Approximately 70% of patients with metastatic disease will be alive 2 years after starting imatinib mesylate, and about 50% will be free of progression.37 By comparison, patients with metastatic GIST treated with doxorubicin had a 2-year survival of about 20%,37 and only 50% of patients undergoing surgical resection of metastatic GIST were alive at 14 months.39
TABLE 3. Trials of Imatinib Mesylate in Metastatic GIST

The ideal dose of imatinib has not been determined. Two trials showed no added benefit with doses greater than 400 mg/day.36,38 While the toxicity of 800 mg/day is definitely greater than that of 400 g/day,36,37 one trial showed a small increase in progression-free survival with the higher dose (Table 3).37 The standard starting dose is currently 400 mg/day, and dose escalation can be considered for patients who are refractory or who become resistant to that dose. Major toxicities of imatinib therapy are usually manageable and include mild fatigue, periorbital edema, diarrhea, muscle cramps, nausea, abdominal pain, and rash.
Imatinib is now considered first-line treatment of metastatic GIST. In rare circumstances, such as primary GIST with synchronous, low-volume metastatic disease, especially when symptomatic, surgical resection followed by imatinib may be used. Although little is known about the use of imatinib in the treatment of the small subset of GIST without KIT mutations, there may be clinical benefit.40 Although imatinib halts progression of disease in the majority of patients with metastatic GIST, complete responses are rare. Thus, imatinib should be though of as an oncostatic agent, rather than as a cytotoxic agent. Indeed, a recent study confirmed that stopping imatinib is associated with an increased risk of disease progression; however, it is not known whether discontinuation of imatinib followed by reintroduction when disease progresses is associated with a reduction in survival.41 At this time, in the setting of stable or responsive disease, life-long treatment is recommended and withdrawal is generally not advocated unless toxicities are unmanageable.
Response to imatinib treatment can be followed by CT or 18FDG-PET. CT is the preferred modality for serial monitoring as it provides more anatomic information. On CT, changes associated with imatinib treatment include regression in size or change in appearance from hypervascular to hypo-attenuating and homogenous. On 18FDG-PET, GIST lesions usually become “cold” (Fig. 2). Indeed, changes on 18FDG-PET are seen within a few days after starting therapy. While response to treatment of solid tumors has traditionally been determined by size measurements such as the World Health Organization criteria42 or the Response Evaluation Criteria in Solid Tumors (RECIST) criteria,43 treatment effects of imatinib on GIST may correlate poorly with these guidelines. Response may be better assessed by changes in tumor density on CT or changes in metabolism by 18FDG-PET. In one study, although approximately 86% of patients had stable or progressive disease based on RECIST criteria 2 months after starting imatinib, 55% of patients had >10% decrease in tumor density measured in Hounsfield Units and 70% of patients had >60% reduction in 18FDG uptake, with good correlation between these 2 measurements.44 Another study showed that overall survival and time to progression after starting imatinib correlated with response on 18FDG-PET on the first follow-up study but not response on the first follow-up CT as measured by RECIST criteria.45 The measurement of functional changes of tumors by CT density or metabolic activity on 18FDG-PET appears to be a lesson learned from GIST with relevance to the treatment of other solid tumors with targeted agents.
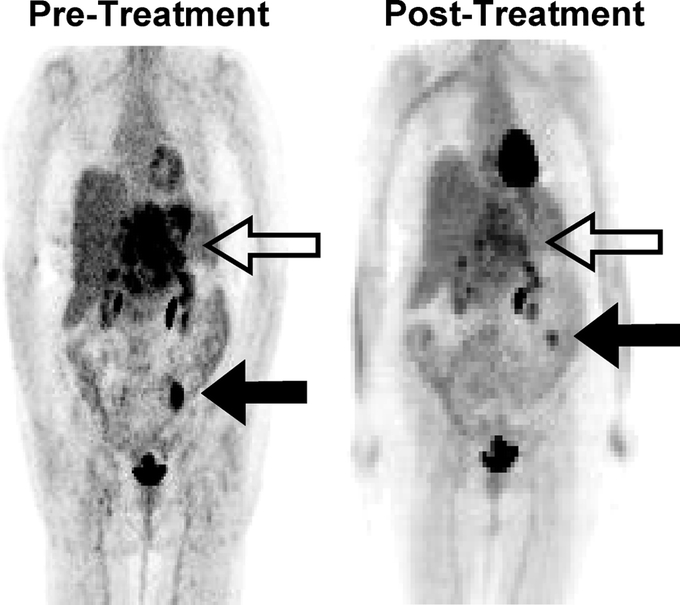
After maximal response to imatinib (generally within 2–6 months after initiating treatment), surgery should be considered if all gross disease can be resected, as resistance may be more likely to develop otherwise. Unfortunately, it is unlikely that the benefit of surgery for imatinib-stable disease can be tested in a randomized trial. In patients who develop focal resistance with some tumors progressing on imatinib and others remaining stable, surgery can be considered for the progressive disease, although the benefit of this approach is unknown. (Fig. 3 shows a CT scan of a patient who underwent surgery for imatinib-refractory metastatic disease.) For the control of liver metastases, radiofrequency ablation and hepatic artery embolization may be used in lieu of formal hepatic resection (Fig. 1). It is unknown whether cytoreduction (ie, resection when it is known all gross disease cannot be removed) for those with stable disease on imatinib is beneficial in reducing the chance of developing resistant clones. Newer targeted agents are now available, and there is general consensus that a second agent should be used in the setting of multifocal resistance to imatinib.
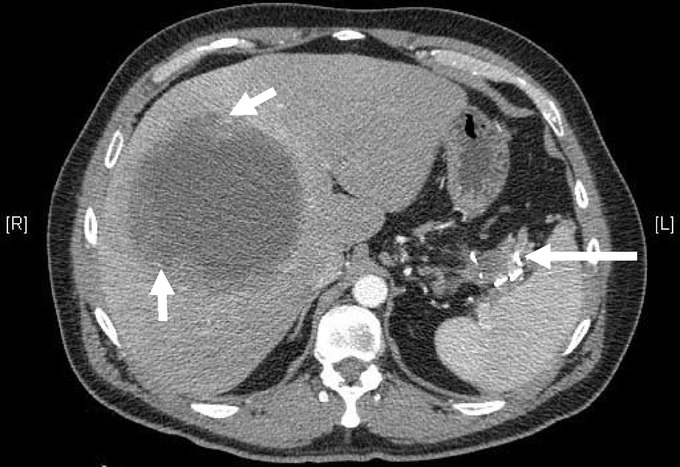
FIGURE 3. CT showing gastric GIST metastatic to the liver in a patient who was subsequently treated with surgical resection. The short white arrows show the large liver metastasis, and the large white arrows show the residual primary tumor. The patient presented with a left upper quadrant tumor that was partially resected. He was then treated with imatinib mesylate with good response, but over a 3-year course developed a liver lesion that increased in size despite dose escalation. The patient was treated with an extended right hepatectomy with diaphragm resection as well as a partial gastrectomy, splenectomy, and distal pancreatectomy. The left upper quadrant mass showed necrosis but no viable tumor on pathologic review.
The molecular and genetic basis for the variable responsiveness to imatinib as well as the development of resistance is beginning to be understood. Metastatic GISTs with mutations in exon 11 of KIT (66% of cases) have a 67% to 83% response rate to imatinib compared with an approximately 40% response rate for cases with KIT exon 9 mutations (16% of cases).29,46,47 A subgroup of GISTs without KIT mutations have been shown to have mutations in the homologous tyrosine kinase PDGFRα.48 PDGFRα mutations, which occur in about 2% of cases, can also have a favorable response to imatinib except those with D842V mutations.29 Approximately 11% of GISTs do not have a known tyrosine kinase mutation, and these cases have a 32% response rate to imatinib.29,46,47 In a multivariate analysis, a KIT exon 11 mutation was the single best predictor of an objective response to imatinib.47
While only a minority (<15%) of patients have primary resistance to imatinib and develop worsening disease despite therapy, half the patients will become resistant by 2 years after imatinib initiation. The most common mechanism of acquired resistance is secondary KIT mutation.49–52 This is concordant with the finding that in chronic myeloid leukemia treated with imatinib, second site mutation in BCR-ABL is the primary mechanism of resistance.53,54 Often, novel mutations arise that have not been found in imatinib-naive GIST.50–52,55 Some tumors, indeed, may acquire multiple new tyrosine kinase mutations during the course of treatment.52,56 The other identified mechanism of imatinib resistance is KIT or PDGFα amplification, but this occurs infrequently.
Newer Agents
Several new agents are under evaluation in metastatic GIST, particularly imatinib-resistant GIST. SU11248 (Sunitinib malate, Pfizer, New York, NY) is another small molecule inhibitor of tyrosine kinases with activity against KIT and PDGFR as well as the vascular endothelial cell growth factor (VEGF) and fms-related tyrosine kinase 3 (Flt3) receptor. In phase I/II trials, patients with metastatic GIST who are intolerant of or resistant to imatinib and treated with SU11248 have an 8% to 15% response rate, with an additional 39% to 58% having stable disease.57,58 Some of those appear to have benefit lasting greater than 1 year with sunitinib treatment.58,59 Not unexpectedly, a recent phase III trial randomized patients to sunitinib or placebo who were intolerant to or progressing through imatinib. Sunitinib showed a significant benefit in time to progression (approximately 5-month increase) and overall survival despite the fact that patients who progressed through placebo were crossed over to sunitinib.60 Signaling molecules downstream from KIT have been proposed as targets, and clinical trials are currently evaluating the use of the multikinase inhibitor AMG706 (Amgen, Inc., Thousand Oaks, CA), the mTOR inhibitor RAD001 (Novartis) in combination with imatinib, and the protein kinase C inhibitor PKC412 (Novartis) in combination with imatinib. As GISTs are often very vascular, the use of inhibitors of VEGF such as bevacizumab (Avastin, Genentech) has been proposed, and indeed, sunitinib inhibits VEGF in addition to KIT and PDGFR. Furthermore, the design of second-generation KIT inhibitors, particularly with the ability to inhibit KIT despite secondary KIT mutations, is underway.
CONCLUSION
The rapid application of imatinib for the treatment of metastatic GIST heralded the beginning of targeted therapy for solid malignancies. Nevertheless, time has shown the limitations of treating GIST with this single agent alone. At diagnosis, most GISTs are localized and amenable to surgical resection. Surgical resection remains the only known chance for cure. Future and ongoing investigations will delineate the most effective ways to integrate surgery and targeted therapy to reduce recurrence after resection of primary disease and to prolong survival in metastatic disease. The lessons learned in GIST will likely be relevant to the future use of molecular therapy for other cancers.
Footnotes
Supported by Grant No. CA102613.
Reprints: Ronald P. DeMatteo, MD, FACS, Memorial Sloan-Kettering Cancer Center, Box 203, 1275 York Avenue, New York, NY 10021. E-mail: dematter@mskcc.org.
REFERENCES
- 1.Allander SV, Nupponen NN, Ringner M, et al. Gastrointestinal stromal tumors with KIT mutations exhibit a remarkably homogeneous gene expression profile. Cancer Res. 2001;61:8624–8628. [PubMed] [Google Scholar]
- 2.Nielsen TO, West RB, Linn SC, et al. Molecular characterisation of soft tissue tumours: a gene expression study. Lancet. 2002;359:1301–1307. [DOI] [PubMed] [Google Scholar]
- 3.Segal NH, Pavlidis P, Antonescu CR, et al. Classification and subtype prediction of adult soft tissue sarcoma by functional genomics. Am J Pathol. 2003;163:691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeMatteo RP, Lewis JJ, Leung D, et al. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pierie J-PEN, Choudry U, Muzikansky A, et al. The effect of surgery and grade on outcome of gastrointestinal stromal tumors. Arch Surg. 2001;136:383–389. [DOI] [PubMed] [Google Scholar]
- 6.Singer S, Rubin BP, Lux ML, et al. Prognostic value of KIT mutation type, mitotic activity, and histologic subtype in gastrointestinal stromal tumors. J Clin Oncol. 2002;20:3898–3905. [DOI] [PubMed] [Google Scholar]
- 7.Fujimoto Y, Nakanishi Y, Yoshimura K, et al. Clinicopathologic study of primary malignant gastrointestinal stromal tumor of the stomach, with special reference to prognostic factors: analysis of results in 140 surgically resected patients. Gastric Cancer. 2003;6:39–48. [DOI] [PubMed] [Google Scholar]
- 8.Langer C, Gunawan B, Schuler P, et al. Prognostic factors influencing surgical management and outcome of gastrointestinal stromal tumours. Br J Surg. 2003;90:332–339. [DOI] [PubMed] [Google Scholar]
- 9.Nishida T, Hirota S, Taniguchi M, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. [DOI] [PubMed] [Google Scholar]
- 10.Chompret A, Kannengiesser C, Barrois M, et al. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology. 2004;126:318–321. [DOI] [PubMed] [Google Scholar]
- 11.Thomas RM, Sobin LH. Gastrointestinal cancer. Cancer. 1995;75(suppl 1):154–70. [DOI] [PubMed] [Google Scholar]
- 12.Kindblom LG. Gastrointestinal Stromal Tumors; Diagnosis, Epidemiology, Prognosis. ASCO Annual Meeting, Chicago, 2003. [Google Scholar]
- 13.Emory TS, Sobin LH, Lukes L, et al. Prognosis of gastrointestinal smooth-muscle (stromal) tumors: dependence on anatomic site. Am J Surg Pathol. 1999;23:82–87. [DOI] [PubMed] [Google Scholar]
- 14.Dematteo RPRP, Maki RGRG, Antonescu CC, et al. Targeted molecular therapy for cancer: the application of STI571 to gastrointestinal stromal tumor. Curr Probl Surg. 2003;40:144–193. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher CDM, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33:459–465. [DOI] [PubMed] [Google Scholar]
- 16.Medeiros F, Corless CL, Duensing A, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28:889–894. [DOI] [PubMed] [Google Scholar]
- 17.Tzen CY, Mau BL. Analysis of CD117-negative gastrointestinal stromal tumors. World J Gastroenterol. 2005;11:1052–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crosby JA, Catton CN, Davis A, et al. Malignant gastrointestinal stromal tumors of the small intestine: a review of 50 cases from a prospective database. Ann Surg Oncol. 2001;8:50–59. [DOI] [PubMed] [Google Scholar]
- 19.Miettinen M, El-Rifai WE, Leslie HS, et al. Evaluation of malignancy and prognosis of gastrointestinal stromal tumors: a review. Hum Pathol. 2002;33:478–483. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian S, West RB, Corless CL, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene. 2004;23:7780–7790. [DOI] [PubMed] [Google Scholar]
- 21.Lasota J, Dansonka-Mieszkowska A, Sobin LH, et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004;84:874–883. [DOI] [PubMed] [Google Scholar]
- 22.Taniguchi M, Nishida T, Hirota S, et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res. 1999;59:4297–4300. [PubMed] [Google Scholar]
- 23.Kim TW, Lee H, Kang Y-K, et al. Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clin Cancer Res. 2004;10:3076–3081. [DOI] [PubMed] [Google Scholar]
- 24.Demetri GD, Benjamin R, Blanke CD, et al. Optimal management of patients with gastrointestinal stromal tumors (GIST): expansion and update of NCCN Clinical Practice Guidelines. J Comprehensive Cancer Network. 2004;2(suppl 1):1–26. [PubMed] [Google Scholar]
- 25.Blay JY, Bonvalot S, Casali P, et al. Consensus meeting for the management of gastrointestinal stromal tumors: Report of the GIST Consensus Conference of 20–21 March 2004, under the auspices of ESMO. Ann Oncol. 2005;16:566–578. [DOI] [PubMed] [Google Scholar]
- 26.DeMatteo RP, Shah A, Fong Y, et al. Results of hepatic resection for sarcoma metastatic to liver. Ann Surg. 2001;234:540–547; discussion 547–548. [DOI] [PMC free article] [PubMed]
- 27.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. [DOI] [PubMed] [Google Scholar]
- 28.Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10:3282–3290. [DOI] [PubMed] [Google Scholar]
- 29.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. [DOI] [PubMed] [Google Scholar]
- 30.Sommer G, Agosti V, Ehlers I, et al. Gastrointestinal stromal tumors in a mouse model by targeted mutation of the Kit receptor tyrosine kinase. Proc Natl Acad Sci USA. 2003;100:6706–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. [DOI] [PubMed] [Google Scholar]
- 32.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. [DOI] [PubMed] [Google Scholar]
- 33.van Oosterom AT, Judson I, Verweij J, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001;358:1421–1423. [DOI] [PubMed] [Google Scholar]
- 34.Heinrich MC, Rubin BP, Longley BJ, et al. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol. 2002;33:484–495. [DOI] [PubMed] [Google Scholar]
- 35.van Oosterom AT, Judson IR, Verweij J, et al. Update of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: a report of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2002;38(suppl 5):83–87. [DOI] [PubMed] [Google Scholar]
- 36.Benjamin RS, Rankin C, Fletcher C, et al. Phase III dose-randomized study of imatinib mesylate (STI571) for GIST: Intergroup S0033 early results [Abstr 3271]. Proc Am Soc Clin Oncol. 2003;22:814. [Google Scholar]
- 37.Verweij PJ, Casali PG, Zalcberg PJ, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–1134. [DOI] [PubMed] [Google Scholar]
- 38.Blanke C, Joensuu H, Demetri G, et al. Long-term follow up of advanced gastrointestinal stromal tumor (GIST) patients treated with imatinib mesylate. 2004 Gastrointestinal Cancers Symposium, San Francisco, 2004. [Google Scholar]
- 39.Mudan SS, Conlon KC, Woodruff JM, et al. Salvage surgery for patients with recurrent gastrointestinal sarcoma: prognostic factors to guide patient selection. Cancer. 2000;88:66–74. [DOI] [PubMed] [Google Scholar]
- 40.Blackstein ME, Rankin C, Fletcher C, et al. Clinical benefit of Imatinib in patients (pts) with metastatic gastrointestinal stromal tumors (GIST) negative for the expression of CD117 in the S0033 trial. J Clin Oncol. 2005;23(16 suppl):9010. [Google Scholar]
- 41.Le Cesne A, Perol D, Ray-Coquard I, et al. Interruption of imatinib (IM) in GIST patients with advanced disease: updated results of the prospective French Sarcoma Group randomized phase III trial on survival and quality of life. J Clin Oncol. 2005;23(16 suppl):9031.16339756 [Google Scholar]
- 42.Miller AB, Hoogstraten B, Staquet M, et al. Reporting results of cancer treatment. Cancer. 1981;47:207–214. [DOI] [PubMed] [Google Scholar]
- 43.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. [DOI] [PubMed] [Google Scholar]
- 44.Choi H, Charnsangavej C, de Castro Faria S, et al. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol. 2004;183:1619–1628. [DOI] [PubMed] [Google Scholar]
- 45.Goerres GW, Stupp R, Barghouth G, et al. The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylate. Eur J Nucl Med Mol Imaging. 2005;32:153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–695. [DOI] [PubMed] [Google Scholar]
- 47.Heinrich MC, Shoemaker JS, Corless CL, et al. Correlation of target kinase genotype with clinical activity of imatinib mesylate (IM) in patients with metastatic GI stromal tumors (GISTs) expressing KIT (KIT+). J Clin Oncol. 2005;23(16 suppl):7. [Google Scholar]
- 48.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. [DOI] [PubMed] [Google Scholar]
- 49.Fletcher JA, Corless CL, Dimitrijevic S, et al. Mechanisms of resistance to imatinib mesylate (IM) in advanced gastrointestinal stromal tumor (GIST) [Abstr 3275]. Proc Am Soc Clin Oncol. 2003;22:815. [Google Scholar]
- 50.Chen LL, Trent JC, Wu EF, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64:5913–5919. [DOI] [PubMed] [Google Scholar]
- 51.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–279. [DOI] [PubMed] [Google Scholar]
- 52.Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to Imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–4190. [DOI] [PubMed] [Google Scholar]
- 53.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. [DOI] [PubMed] [Google Scholar]
- 54.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. [DOI] [PubMed] [Google Scholar]
- 55.Tamborini E, Bonadiman L, Greco A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology. 2004;127:294–299. [DOI] [PubMed] [Google Scholar]
- 56.Wardelmann E, Thomas N, Merkelbach-Bruse S, et al. Acquired resistance to imatinib in gastrointestinal stromal tumours caused by multiple KIT mutations. Lancet Oncol. 2005;6:249–251. [DOI] [PubMed] [Google Scholar]
- 57.Demetri GD, Desai J, Fletcher JA, et al. SU11248, a multi-targeted tyrosine kinase inhibitor, can overcome imatinib (IM) resistance caused by diverse genomic mechanisms in patients with metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol. 2004;22(14 suppl):3001. [Google Scholar]
- 58.Desai J, Maki R, Heinrich MC, et al. Activity and tolerability of the multi-targeted tyrosine kinase inhibitor SU011248 in patients with metastatic gastrointestinal stromal tumor (GIST) refractory to imatinib mesylate. 2004 Gastrointestinal Cancers Symposium, San Francisco, 2004. [Google Scholar]
- 59.Maki RG, Fletcher JA, Heinrich MC, et al. Results from a continuation trial of SU11248 in patients (pts) with imatinib (IM)-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol. 2005;23(16 suppl):9011. [Google Scholar]
- 60.Demetri GD, van Oosterom AT, Blackstein M, et al. Phase 3, multicenter, randomized, double-blind, placebo-controlled trial of SU11248 in patients following failure of imatinib for metastatic GIST. J Clin Oncol. 2005;23(16 suppl):4000. [Google Scholar]
- 61.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. [DOI] [PubMed] [Google Scholar]