

Abstract
BACKGROUND
Neonatal alloimmune thrombocytopenia (NATP) is an important cause of morbidity and mortality in the newborn. Optimal management of subsequent pregnancies requires knowledge of the alloantigen that caused maternal immunization, but this is possible only in a minority of cases. This study investigated whether this can be explained in part by maternal immunization against the “rare” alloantigen HPA-9b (Maxa), implicated previously only in a single NATP case.
STUDY DESIGN AND METHODS
Archived paternal DNA from unresolved cases of NATP and normal individuals was typed for platelet (PLT)-specific antigens with real-time polymerase chain reaction and direct sequencing. PLT-specific alloantibodies were characterized by flow cytometry and solid-phase enzyme-linked immunosorbent assay. Recombinant GPIIb/IIIa was expressed in stably transfected Chinese hamster ovary cells. Clinical information was obtained directly from attending physicians.
RESULTS
Six of 217 fathers were positive for the presence of HPA-9b (Maxa), an incidence about seven times that in the general population. In each of five cases studied, maternal serum samples reacted with intact paternal PLTs and paternal GPIIb/IIIa. Only one of three serum samples tested recognized recombinant GPIIb/IIIa carrying the HPA-9b (Maxa) mutation. These seemingly discrepant reactions may reflect different requirements for oligosaccharides linked to residues close to the mutation in GPIIb that determines HPA-9b (Maxa). NATP in the affected children was severe and was associated with intracranial hemorrhage in three of six infants on whom information was obtained.
CONCLUSIONS
Maternal immunization against HPA-9b (Maxa) is an important cause of NATP and should be considered in cases of apparent NATP not resolved on the basis of maternal-fetal incompatibility for “common” PLT antigens.
ABBREVIATIONS: ACE = antigen capture enzyme-linked immunosorbent assay, CHO = Chinese hamster ovary, ICH(s) = intracranial hemorrhage(s), MACE = modified antigen capture enzyme-linked immunosorbent assay, MAIPA = monoclonal antibody-specific immobilization of platelet antigens, NATP = neonatal alloimmune thrombocytopenia
Neonatal alloimmune thrombocytopenia (NATP) is caused by passively transmitted maternal antibodies specific for fetal platelet (PLT) antigens inherited from the father.1–3 This condition occurs about once in 1000 live births and is a significant cause of morbidity and mortality in the newborn.4,5 Advances in the understanding of PLT-specific alloantigen systems and improvements in diagnostic techniques have made it possible to identify maternal-fetal incompatibility for specific PLT alloantigens in many NATP cases, but others go unresolved despite use of the best available diagnostic techniques.6 A possible reason for failure to identify specific PLT alloantigens as the immunizing agent in cases of apparent NATP is that the sensitizing agent may be one of at least 11 rare (“private”) alloantigens identified in individual cases of NATP7–17 and not ordinarily considered in the laboratory workup. We investigated this possibility by evaluating archived blood samples from fathers of NATP cases in which maternal immunization against one of the “common” PLT-specific antigens was not identified. Six fathers were found to be positive for the private alloantigen HPA-9b (Maxa), implicated previously as an immunogen capable of causing NATP only in a single instance.8 The HPA-9b (Maxa) determinant is created by a guanine to adenine substitution in GPIIb nucleotide 2602 (relative to the A in the ATG start codon), resulting in a valine to methionine substitution. In this report, we describe clinical and serologic characteristics of NATP associated with maternal-fetal incompatibility for HPA-9b (Maxa) in this group of patients.
MATERIALS AND METHODS
Typing of genomic DNA for HPA-9b (Maxa) and HPA-3a/b (Baka/b)
Genomic DNA was isolated from peripheral blood white blood cells (WBCs) or buccal swabs with a DNA blood kit (QIAgen, Valencia, CA) according to the manufacturer’s directions. GPIIb exons 25 to 26 carrying the HPA-9b (Maxa) polymorphism were amplified in a nested polymerase chain reaction (PCR) with the primer pairs and conditions listed in Table 1. The primary reaction, optimized by a PCR optimization kit (Opti-Prime, Stratagene, La Jolla, CA), was performed with 0.2 μg of genomic DNA with 29 pmol of primers 25S and 26A and 0.05 mmol per L dNTPs in Opti-Prime Buffer 2 (10 mmol/L Tris-HCl, pH 8.3, 1.5 mmol/L MgCl2, and 75 mmol/L KCl). A total of 2.5 units of Biolase DNA polymerase were added after denaturation at 95°C for 5 minutes. Amplification was performed in 35 cycles under the conditions listed in Table 1. The primary PCR mixture was then diluted 50-fold and 1 μL was used as the template for the nested reaction with 25 pmol each of n25S and n26A (Table 1). The nested reaction was carried out in the same buffer conditions as the primary reaction except with 40 cycles of amplification. The final amplification products were electrophoresed, and DNA bands were excised and purified with a GFX PCR DNA and gel band purification kit (GE Healthcare Waukesha, WI). Automated DNA sequence analysis was performed in both directions on a genetic analyzer (ABI 3100, Applied Biosystems, Foster City, CA). The HPA-3a/b (Baka/b) locus18 is located 18 nucleotides downstream from the HPA-9 (Max) locus within this amplified region of DNA,8 permitting HPA-9 and HPA-3 genotypes to be determined from the same sequencing reaction.
TABLE 1.
Primers for HPA-9a/b (Maxa/b) amplification
| Name | Sequence | PCR conditions |
|---|---|---|
| 25S | S* 5′-CCTCAGCATCCACCTTCCGGG-3′ | 58°C 30 sec, 72°C 30 sec, 95°C 30 sec |
| 26A | A* 5′-GAGAACTGGATCCTGAAGCC-3′ | |
| n25S | S 5′-GCCCTCCGACCTGCTCTACATC-3′ | 66°C 45 sec, 72°C 30 sec, 95°C 30 sec |
| n25A | A 5′-GCTCGGGCTCTGGCAGGAAG-3′ | |
| Sex25 | S 5′-GCCTCCTGTCAACCCT-3′ | 60°C 1 min, 95°C 25 sec |
| M | A Maxa-specific 5′-GCAGCCCCCAGTCCAT-3′ | |
| W | A wild-type-specific 5′-GCAGCCCCCAGTCCAC-3′ | |
| SMax | S 5′-CTCTCAAGATGGACTGGG-3′† | 50°C 30 sec, 72°C 1.5 min, 95°C 30 sec |
| AMax | A 5′-CCCAGTCCATCTTGAGAG-3′† | 50°C 30 sec, 72°C 30 sec, 95°C 30 sec |
| S1255 | S 5′-CAAGTGCTGGTGTTCCTG-3′ | |
| A2865 | A 5′-CAGCACAAACTGATCCAGAGGC-3′ |
S = sense; A = antisense.
Bold nucleotide is mutagenic.
Typing for HPA-9b (Maxa) in the general population with allele-specific real-time PCR
Blood samples were obtained from 1048 blood donors identified only by racial background. DNA was extracted from WBCs as described above, and HPA-9b (Maxa) typing was performed with real-time PCR analysis on a real-time multicolor detection system (iCycler iQ, Bio-Rad, Hercules, CA) with SYBR Green double-stranded DNA detection. The PCR procedure was performed with an upstream sense primer, Sex25 (Table 1), and either antisense primer M, specific for the HPA-9b (Maxa) sequence (Table 1), or antisense primer W, specific for the common allele sequence (Table 1). The specificity of the reaction was optimized with a PCR system kit (Fail-Safe, Epicentre Technology, Madison, WI), which provided all the required reagents including SYBR Green. Fail-Safe Buffer G was found to be optimal for amplification specificity and maximum product signal. Reactions were performed with 6.25 pmol of each primer (either S25+W or S25+M) (Table 1) with 125 ng of genomic DNA and 1.25 units of Fail-Safe PCR enzyme mix. Real-time amplification was achieved with denaturation at 95°C for 2 minutes followed by 40 cycles of 95°C for 25 seconds and 60°C for 1 minute. A typical real-time PCR procedure demonstrating the presence of the HPA-9b (Maxa) mutation is shown in Fig. 1.
Fig. 1.
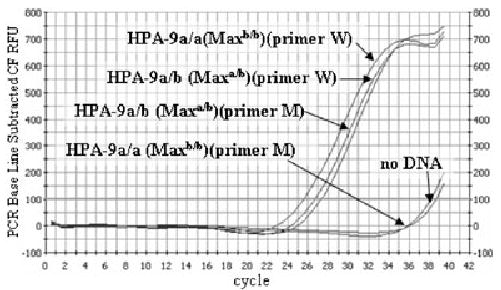
Detection of the HPA-9b (Maxa) mutation with real-time PCR. HPA-9a/b and HPA-9a/a DNA samples were amplified in the presence of SYBR Green with flanking primer Sex25 and the HPA-9b-specific primer (M) or the common allele-specific primer (W) curve fit relative fluorescence units (Table 1). Specific products are apparent after 21 cycles; primer-dimers are not apparent after 34 cycles.
Construction of cDNA encoding GPIIb V837M (HPA-9b (Maxa))
Throughout this report, nucleotide 1 refers to the A of the ATG translation start codon of GPIIb. Site-specific mutagenesis was performed with human GPIIb cDNA in vector pcDNA3 (a mammalian expression vector from Invitrogen [Carlsbad, CA] containing the neomycin-selectable marker) with overlapping polymerase chain reaction (PCR) as described previously.19 Briefly, two overlapping mutagenic primers (Smax and Amax) possessing the G to A base-pair substitution at nucleotide 2602 (corresponding to the HPA-9b (Maxa) polymorphism) were used in conjunction with flanking primers, S1255 or A2865, to generate a PCR product possessing the HPA-9b (Maxa) polymorphism. After gel purification, the PCR product was digested with restriction enzymes ClaI and Sse8387II (New England Biolabs, Beverly, MA). The digested PCR product was then subcloned into human GPIIb cDNA in pcDNA3 digested with the same restriction enzymes, thereby replacing the wild-type ClaI-Sse8387II fragment with the PCR product possessing the HPA-9b (Maxa) mutation.
Generation of stable Chinese hamster ovary cell lines expressing GPIIb carrying the HPA-9b (Maxa) mutation
Chinese hamster ovary (CHO) cells were cotransfected with mutated (HPA-9b (Maxa) GPIIb or nonmutated cDNA encoding GPIIb in mammalian expression vector pcDNA3 (neomycin selection marker, Invitrogen) and nonmutated GPIIIa in pcDNA3.1, a mammalian expression vector containing a Zeocin-selectable marker (Invitrogen) with Lipofectamine (Invitrogen) by use of the manufacturer’s suggested protocol. Stable cells lines were selected with 0.2 mg per mL Zeocin and 0.6 mg per mL neomycin 3 days after infection. Cells expressing GPIIb/IIIa were sorted with the GPIIb/IIIa-specific monoclonal antibody (MoAb) AP2 (anti-GPIIb/IIIa complex) on a cell sorter (FACStar Plus, Becton Dickinson, Franklin Lakes, NJ). Cells stably expressing GPIIb (HPA-9b (Maxa))/IIIa or GPIIb/IIIa were maintained in αMEM (Invitrogen) supplemented with 10 percent fetal bovine serum, glutamine, 0.2 mg per mL Zeocin, and 0.6 mg per mL neomycin.
Serologic studies
Maternal serum samples were initially tested against paternal PLTs and PLTs from group O normal donors having known PLT alloantigen phenotypes utilizing flow cytometry and a flow cytometer (FACSCalibur, Becton Dickinson) as previously described.6,20 Antibodies recognizing glycoprotein complexes GPIIb/IIIa, Ib/IX, Ia/IIa, GPIV, and Class I HLA were detected in solid-phase enzyme-linked immunosorbent assays (ELISAs) with antigen capture ELISA (ACE)21 and modified antigen capture ELISA (MACE).22 The normal PLT panel used in these assays contained at least one member carrying each of the HPA-1a/b (PlA1/A2), HPA-2a/b (Koa/b), HPA-3a/b (Baka/b), HPA-4a (Pena), and HPA-5a/b (Brb/a) alloantigen systems. Maternal serum samples were also screened for antibodies reactive with Class I HLA antigens with a solid phase ELISA test (QuikScreen, GTI, Waukesha, WI).23
PLT alloantigen typing
PLT phenotyping for HPA-1a/b and HPA-3a/b was performed serologically with ACE21 and genotyping for HPA-2a/b, HPA-4a/b, and HPA-5a/b was performed with allele-specific PCR.24
Clinical information
Clinical histories and laboratory findings made in the perinatal period were characterized by direct communication with attending physicians.
RESULTS
Incidence of HPA-9b (Maxa) among fathers of unresolved NATP cases and in the general population
Of 217 archived DNA samples studied, 6 (2.8%) were found to be heterozygous for HPA-9b (Maxa) by direct sequencing of PCR products. A sequencing result demonstrating the G to A substitution at position 2602 typical of the HPA-9b (Maxa) mutation is shown in Fig. 2. In contrast, only 4 of 1048 randomly selected blood donors (0.38%) typed with real-time PCR were positive for this marker, yielding an estimated gene frequency of approximately 0.002, assuming that each positive individual was heterozygous for the variant of GPIIb that carries HPA-9b (Maxa). This figure is slightly less than the value of 0.003 found by Noris and coworkers8 in a study of 500 normal persons of Northern European ancestry. Each of the HPA-9b (Maxa)–positive fathers and the HPA-9b (Maxa)–positive blood donors identified themselves as “Caucasians.” None of 15 African American, 14 Hispanic, 3 Asian, and 3 Native American individuals was HPA-9b (Maxa)–positive. The incidence of HPA-9b (Maxa) among fathers of unresolved NATP cases was about seven times the incidence in the blood donor population, a difference that was highly significant (p < 0.001).
Fig. 2.
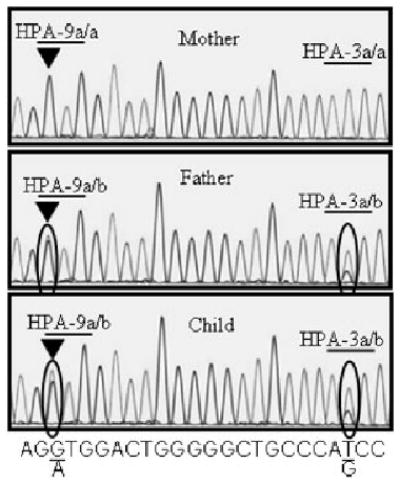
Sequence analysis of the HPA-9 (Max) locus in Family 4. PCR products generated with primers n25S and n25A were subjected to automated sequence analysis with primer n25S (see Table 1). DNA of father and child had both A and G nucleotides at position 2602 (encircled, left) indicating heterozygosity for HPA-9b (Maxa). Mother’s DNA contained only G at this position. Father and son both had both T and G at position 2621 (encircled, right) indicating heterozygosity for HPA-3a/b (Baka/b), whereas the mother had only T at this position, indicating that her genotype was HPA-3a/a (Baka/a).
Clinical findings made in NATP cases in which the father was HPA-9b (Maxa)–positive
Details of the clinical course and laboratory findings made in the index cases and siblings subsequently fathered by the same HPA-9b (Maxa)–positive individuals were obtained for five cases and are summarized in Table 2. It was not possible to obtain follow-up information on the sixth case.
TABLE 2.
Clinical and laboratory findings in infants born to mothers immunized against HPA-9b (Maxa)
| Case | Child | HPA-9b (Maxa) antibody present | Maxa type of infant | PLT nadir (×109/L) | Time (days) to reach PLT count of 100 × 109/L | Bleeding symptoms | ICH | Treatment | Outcome |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | Yes | Positive | 5 | 18 | Extensive petechiae at 12 hr | Small ICH | Random PLT transfusions, Days 1, 6, and 13 | No neurologic sequelae |
| 2 | Yes | Positive | 6 | 10 | Extensive petechiae and purpura at birth | Major ICH | Maternal and random PLT transfusion, Days 1 and 6 | Permanent disability | |
| 2 | 1 | Yes | Positive | 20 | 5 | Moderate petechiae at birth | No | None | Recovery |
| 2 | No | Negative | 225 | None | No | None | Normal infant | ||
| 3 | 1 | Yes | Positive | 7 | 18 | Moderate petechiae at birth | Small ICH | PLT transfusion, Days 1 and 7 | No neurologic |
| 2 | No | Negative | 235 | None | No | None | |||
| 4 | 1 | Yes | Positive | 6 | 30 | Moderate petechiae at birth | No | IV gamma globulin at 3 and 4 weeks of age | Recovery |
| 2 | Yes | Positive | 237 | None | No | Prenatal IV gamma globulin (last 2 months) | Normal infant | ||
| 5 | 1 | Yes | Positive | 30 | 5 | Moderate petechiae at birth | No | Random PLT transfusion, Day 2 | Recovery |
| 2 | Yes | Positive | 231 | None | No | Prenatal IV gamma globulin (last 2 months) | Normal infant |
Firstborn infants
In all five cases, the firstborn infant had severe thrombocytopenia, with a PLT nadir ranging from 5 × 109 to 30 × 109 per L. Each child developed moderate or severe petechial hemorrhages on the limbs and trunk in the first 12 hours after birth. Infants born to Mothers 1 and 3 had small intracranial hemorrhages (ICHs) detected on computed axial tomography scan but exhibited no neurologic abnormalities. PLTs from random donors were transfused to the infants born to Mothers 1, 3, and 5 and produced posttransfusion PLT increments of at least 50 ×109 per L in each instance. Between 5 and 30 days elapsed before PLT levels stabilized at a level of 100 × 109 per L or higher in the five affected infants (Table 2). The infant born to Mother 4 had petechiae at birth but no blood studies were performed. Because of intermittent petechiae observed at home, a blood count was performed at 3 weeks of age and revealed a PLT level of 6 ×109 per L. This infant was given intravenous (IV) gammaglobulin at 3 and 4 weeks of age and achieved a stable PLT count in the normal range at 32 days of age. Typing performed after birth on DNA obtained by buccal swab from the five infants showed that each was positive for HPA-9b (Maxa).
Second-born infants
The second infant born to the mother of Case 1 had extensive petechiae and ecchymoses at birth and a PLT count of 6 × 109 per L. Despite transfusion of washed maternal PLTs, a major intracerebral hemorrhage occurred leading to severe mental and physical disability. The second children born to the mothers of Cases 2 and 3 had normal PLT counts at birth. Subsequent typing of these infants showed that both were HPA-9b (Maxa)–negative. Typing performed on amniotic cells during the second pregnancies of Mothers 4 and 5 showed that both fetuses were HPA-9b (Maxa)–positive. In light of this finding, each mother received infusions of 1.0 g per kg IV gammaglobulin weekly during the last 2 months of gestation. Both infants had normal PLT counts at birth.
Serologic findings
Maternal serum obtained from Mothers 1, 3, 4, and 5 after the birth of their first child reacted with PLTs from the corresponding father in flow cytometry but not with PLTs from normal donors known to be positive for antigens of the HPA-1 to -5 systems. A serum sample from the mother of Case 2 was positive against the father’s PLTs and PLTs from normal donors, but contained broadly reactive HLA antibodies that contributed to these reactions. No HLA antibodies were detected in serum samples from the other four mothers. When tested in the ACE against GPIIb/IIIa from father’s PLTs immobilized with MoAb AP2 specific for the GPIIb/IIIa complex, negative reactions were obtained with serum from each of the five mothers. Positive reactions, however, were obtained in all five cases when the mothers’ serum samples were tested against the fathers’ GPIIb/IIIa in the MACE assay using monoclonal AP2 for antigen capture. Serum from the mother of Case 1 also gave a positive reaction against the father’s GPIIb/IIIa in the MoAb-specific immobilization of PLT antigens (MAIPA) assay (similar in principle to MACE).25 The other maternal serum samples were not tested in MAIPA. The strongest reaction in MACE was given by serum from the mother of Case 4, yielding an optical density (OD) of 0.544, significantly higher than the OD of 0.039 obtained with normal serum. No antibodies reactive with GPIb/IX, GPIa/IIa, or GPIV were detected in the maternal serum samples with the MACE assay. The relatively weak reactions of maternal serum in MACE against GPIIb carrying the HPA-9b antigen contrast with much stronger reactions commonly obtained with maternal antibodies recognizing the HPA-1a (PlA1) determinant, carried on GPIIIa.
The finding that each maternal antibody reacted with paternal GPIIb/IIIa and that each father and each affected infant was positive for HPA-9b (Maxa) provided strong, but circumstantial evidence that the antibodies were HPA-9b (Maxa)–specific. To define specificity further, we studied the reactions of serum from Cases 1, 4, and 5 against GPIIb/IIIa mutated to contain a valine-to-methionine substitution at position 837 of GPIIb8 and expressed in CHO cells. As shown in Fig. 3, serum from the mother of Case 1 recognized mutant but not unmodified GPIIb/IIIa on the transfected cells. Nevertheless, only equivocal reactions were obtained with serum samples from the mothers of Cases 4 and 5 (not shown). The mothers of Cases 2 and 3 were studied during their second pregnancies, at which time antibody was not present (Table 2).
Fig. 3.

Reactions of serum samples from the mother of Case 1 with CHO cells expressing mutated (GPIIb Met837) or nonmutated GPIIb/IIIa. (A) The mother’s serum sample reacted with GPIIb/IIIa containing the HPA-9b (Maxa) mutation (open histograms) but not with nonmutated GPIIb/IIIa (closed histograms). Anti-HPA-1a (PlA1) antibody reacted with both constructs equally well (B), whereas normal serum failed to recognize either (C).
Each HPA-9b (Maxa)–positive individual was also positive for HPA-3b
The (HPA-3a/b) antigens result from an isoleucine-to-serine substitution at position 843 of GPIIb, six amino acid residues away from the HPA-9b (Maxa) site at position 837.18 Noris and colleagues8 subcloned PCR products comprising nucleotides that encode HPA-9b (Maxa) and HPA-3a/b (Baka/b) from HPA-9b-positive individuals, subjected these to restriction fragment analysis, and found that the HPA-9b mutation was present only on GPIIb alleles that encode HPA-3b (Bakb), indicating that HPA-9b (Maxa) is probably linked to HPA-3b (Bakb). Consistent with this, we found that each of 13 HPA-9b (Maxa)–positive individuals (9 from NATP families and 4 from the general population) was HPA-3b (Bakb)–positive. Although HPA-9b (Maxa) may be found naturally only on alleles that also encode HPA-3b (Bakb), our finding that HPA-9b (Maxa)–specific antibody from Mother 1 reacts with the HPA-9b (Maxa) form of GPIIb modified to contain the HPA-9b (Maxa) mutation (Fig. 3) shows that the HPA-3b (Bakb) mutation is not an absolute requirement for the reaction of that particular antibody.
DISCUSSION
The five (and possibly six) cases of NATP described here had severe neonatal thrombocytopenia apparently caused by transplacentally acquired antibodies specific for HPA-9b (Maxa). In each family, a firstborn infant was affected, as was true also of the single case described by Noris and colleagues,8 indicating that maternal immunization against HPA-9b (Maxa), as with other PLT-specific alloantigens,1,5 can occur during the first pregnancy. Each of the affected infants had severe thrombocytopenia lasting from 5 to 30 days. Random PLT transfusions produced satisfactory posttransfusion increments, as would be expected because of the rarity of the HPA-9b (Maxa) antigen in the general population.
Three of the six infants developed ICH in the postnatal period. Two recovered without sequelae, but the third was left with permanent disability. The single previously reported case of NATP caused by anti-HPA-9b (Maxa) also had an ICH.8 It has been estimated that 10 to 15 percent of infants with NATP caused by maternal alloantibodies specific for HPA-1a (PlA1)experience ICH.1,2 Development of this complication in four of seven infants with NATP caused by anti-HPA-9b (Maxa) is consistent with the possibility that HPA-9b-specific antibodies may be more prone to cause intracerebral bleeding in a fetus or newborn infant than other PLT-specific alloantibodies, although Noris and associates8 were unable to show that anti-HPA-9b (Maxa) inhibited the response of HPA-9b–positive PLTs to ADP, collagen, or epinephrine.8 It is of particular interest that the second infants born to Mothers 4 and 5 had normal PLT counts at birth despite maternal-fetal incompatibility for HPA-9b (Maxa) and the presence of anti-HPA-9b in maternal plasma. As noted, each of these mothers was given high dose IV gamma globulin during the last 2 months of pregnancy because of reports indicating that this treatment can reduce the severity of thrombocytopenia in infants born to mothers immunized against a fetal antigen.1,26 Although exceptions have been reported,27 clinical observations indicate that NATP generally is at least as severe in a second antigen-incompatible infant born to an untreated mother than it was in the firstborn.1 This was certainly the case in the second infant born to Mother 1, who experienced a severe ICH leading to permanent disability. It seems likely, but is impossible to prove, that prenatal treatment with IV gamma globulin favorably influenced the PLT levels in the second infants born to Mothers 4 and 5.
In the cases studied, each of the maternal serum samples reacted with paternal PLTs but not with normal donor PLTs carrying antigens of the HPA-1 to -5 systems in a flow cytometric assay. Nevertheless, the serum samples failed to recognize paternal GPIIb/IIIa captured in ELISA plates by immobilized MoAb AP2 specific for the GPIIb/IIIa complex (ACE assay). Positive reactions were obtained in all five cases when the mother’s serum sample was tested against GPIIb/IIIa from the father’s PLTs in MACE, an assay similar to the widely used MAIPA25 in which intact PLTs are incubated with antibody-containing serum, washed, and then lysed with nonionic detergent. Soluble immune complexes consisting of antibody bound to GPIIb/IIIa are then captured with a GPIIb/IIIa-specific MoAb and the associated human antibody is detected with appropriate secondary reagents.22
The inconsistent and relatively weak reactions of the maternal serum samples with GPIIb/IIIa from HPA-9b (Maxa)–positive paternal PLTs in solid-phase assays and with recombinant GPIIb/IIIa bearing the HPA-9b mutation is in distinct contrast to antibodies reactive with HPA-1a (PlA1), which routinely give strong positive reactions in these assays. Failure of the antibodies to react with paternal GPIIb/IIIa in the ACE assay was not due to competition between anti-HPA-9b (Maxa) and AP2, the MoAb used to immobilize the target GPIIb/IIIa complex because AP2 was suitable for detection of anti-HPA-9b in the MACE assay and can be used to detect most HPA-3b (Bakb)-specific antibodies,28 which recognize an epitope at position 843 in the GPIIb heavy chain only six residues away from the HPA-9b (Maxa) site at position 837. We recently described two HPA-3b (Bakb)–specific maternal antibodies from cases of severe NATP that could be detected in assays with intact PLTs but not in MACE, MAIPA, or ACE and suggested that these antibodies recognize a “labile” aspect of the HPA-3b (Bakb) determinant that is lost when the GPIIb/IIIa complex is solubilized with detergent. A recent report described a HPA-3b (Bakb)–specific antibody with similar properties.29 Findings made with maternal serum samples from Cases 1 through 5 are consistent with the possibility that the HPA-9b (Maxa) epitope is lost when GPIIb/IIIa is solubilized and captured with a monoclonal before adding anti-HPA-9b (ACE assay), but is partially preserved when antibody is allowed to react with its target before solubilization (MACE or MAIPA).
Reactions of HPA-9b (Maxa) antibodies could also be influenced by oligosaccharides located on serine residues at positions 845 and 847 in GPIIb that are potential sites for O-glycosylation. HPA-3a (Baka)–specific antibodies are known to differ from one another in their serologic behavior28,30–32 based at least in part on differing requirements for O-linked oligosaccharide residues, presumably located at positions 845 and/or 847.30,33–35 One biochemical study provided direct evidence that Ser847 is, in fact, glycosylated.35 Because the HPA-9b (Maxa) antigen appears to be expressed only on the HPA-3b (Bakb) allele of GPIIb (Ser843), it is possible that residue 843 is also linked to an oligosaccharide that could influence the reactions of anti-HPA-9b. Thus, HPA-9b (Maxa)-specific antibodies, like those specific for HPA-3a (Baka) and for blood group antigens M and N36–38 may differ from one another in their requirements for oligosaccharides linked to adjacent amino acids. Reaction of the antibody from Mother 1 but not those from Mothers 4 or 5 with recombinant GPIIb/IIIa carrying the HPA-9b (Maxa) mutation (Fig. 3) could reflect minor structural differences in O-glycosylation of rGPIIb/IIIa by CHO cells relative to the native human molecule.39 Alternatively, the antibodies from Mothers 4 and 5 may require the HPA-3b (Bakb) mutation at position 843 for their reaction with HPA-9b (Maxa). Further studies with panels of HPA-9b (Maxa)–specific antibodies, HPA-9b–positive PLTs, and rGPIIb/IIIa will be required to resolve these issues.
The finding that HPA-9b (Maxa) was approximately seven times more prevalent among fathers of NATP cases not explained by maternal-fetal incompatibility for more common PLT-specific antigens suggests that HPA-9b may be especially prone to cause maternal immunization during pregnancy. Davoren and coworkers6 recently analyzed more than 1000 cases of NATP caused by maternal immunization against known PLT-specific antigens6 and found that HPA-1a (PlA1), HPA-5b(Bra), HPA-1b (PlA2), and HPA-3a (Baka) were the immunogens in 81, 9, 4, and 2 percent, respectively. We found five and possibly six cases of NATP caused by maternal immunization against HPA-9b (Maxa) among a total of 217 cases studied, some of which may not have had actual NATP. This experience suggests that HPA-9b (Maxa), despite its rarity in the general population, may be the third most important trigger for NATP, after HPA-1a (PlA1) and HPA-5b (Bra) and that paternal typing for HPA-9b (Maxa) should be performed in cases of suspected NATP not resolved in the initial laboratory evaluation. Studies to determine whether maternal immunization against other “rare” PLT alloantigens is more common than was thought appear indicated.
Footnotes
Supported by Grant HL-13629 from the National Heart, Lung, and Blood Institute.
References
- 1.Bussel JB. Alloimmune thrombocytopenia in the fetus and newborn. Semin Thromb Hemost. 2001;27:245–52. doi: 10.1055/s-2001-15254. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan C. Alloimmune thrombocytopenia of the fetus and the newborn. Blood Rev. 2002;16:69–72. doi: 10.1054/blre.2001.0187. [DOI] [PubMed] [Google Scholar]
- 3.Murphy MF, Manley R, Roberts D. Neonatal alloimmune thrombocytopenia. Haematologica. 1999;4(84 Suppl EHA4):110–4. [PubMed] [Google Scholar]
- 4.Blanchette VS, Chen L, de Friedberg ZS, et al. Alloimmunization to the PlA1 platelet antigen: results of a prospective study. Br J Haematol. 1990;74:209–15. doi: 10.1111/j.1365-2141.1990.tb02567.x. [DOI] [PubMed] [Google Scholar]
- 5.Williamson LM, Hackett G, Rennie J, et al. The natural history of fetomaternal alloimmunization to the platelet-specific antigen HPA-1a (PlA1, Zwa) as determined by antenatal screening. Blood. 1998;92:2280–7. [PubMed] [Google Scholar]
- 6.Davoren A, Curtis BR, Aster RH, et al. Human platelet antigen-specific alloantibodies implicated in 1162 cases of neonatal alloimmune thrombocytopenia. Transfusion. 2004;44:1220–5. doi: 10.1111/j.1537-2995.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang R, McFarland JG, Kekomaki R, et al. Amino acid 489 is encoded by a mutational “hot spot” on the beta 3 integrin chain: the CA/TU human platelet alloantigen system. Blood. 1993;82:3386–91. [PubMed] [Google Scholar]
- 8.Noris P, Simsek S, Bruijne-Admiraal LG, et al. Max(a), a new low-frequency platelet-specific antigen localized on glycoprotein IIb, is associated with neonatal alloimmune thrombocytopenia. Blood. 1995;86:1019–26. [PubMed] [Google Scholar]
- 9.Simsek S, Folman C, van der Schoot CE, et al. The Arg633His substitution responsible for the private platelet antigen Gro(a) unravelled by SSCP analysis and direct sequencing. Br J Haematol. 1997;97:330–5. doi: 10.1046/j.1365-2141.1997.502696.x. [DOI] [PubMed] [Google Scholar]
- 10.Santoso S, Kalb R, Kroll H, et al. A point mutation leads to an unpaired cysteine residue and a molecular weight polymorphism of a functional platelet beta 3 integrin subunit: the Sra alloantigen system of GPIIIa. J Biol Chem. 1994;269:8439–44. [PubMed] [Google Scholar]
- 11.Santoso S, Amrhein J, Hofmann HA, et al. A point mutation Thr(799) Met on the alpha(2) integrin leads to the formation of new human platelet alloantigen Sit(a) and affects collagen-induced aggregation. Blood. 1999;94:4103–11. [PubMed] [Google Scholar]
- 12.Santoso S, Kiefel V, Richter IG, et al. A functional platelet fibrinogen receptor with a deletion in the cysteine-rich repeat region of the beta(3) integrin: the Oe(a) alloantigen in neonatal alloimmune thrombocytopenia. Blood. 2002;99:1205–14. doi: 10.1182/blood.v99.4.1205. [DOI] [PubMed] [Google Scholar]
- 13.Jallu V, Meunier M, Brement M, et al. A new platelet polymorphism Duv(a+), localized within the RGD binding domain of glycoprotein IIIa, is associated with neonatal thrombocytopenia. Blood. 2002;99:4449–56. doi: 10.1182/blood.v99.12.4449. [DOI] [PubMed] [Google Scholar]
- 14.Kuijpers RW, Simsek S, Faber NM, et al. Single point mutation in human glycoprotein IIIa is associated with a new platelet-specific alloantigen (Mo) involved in neonatal alloimmune thrombocytopenia. Blood. 1993;81:70–6. [PubMed] [Google Scholar]
- 15.Sachs UJ, Kiefel V, Bohringer M, et al. Single amino acid substitution in human platelet glycoprotein Ibbeta is responsible for the formation of the platelet-specific alloantigen Iy(a) Blood. 2000;95:1849–55. [PubMed] [Google Scholar]
- 16.Peyruchaud O, Bourre F, Morel-Kopp MC, et al. HPA-10w(b) (La(a)): genetic determination of a new platelet-specific alloantigen on glycoprotein IIIa and its expression in COS-7 cells. Blood. 1997;89:2422–8. [PubMed] [Google Scholar]
- 17.Unkelbach K, Kalb R, Breitfeld C, et al. New polymorphism on platelet glycoprotein IIIa gene recognized by endonuclease Msp I: implications for PlA typing by allele-specific restriction analysis. Transfusion. 1994;34:592–5. doi: 10.1046/j.1537-2995.1994.34794330013.x. [DOI] [PubMed] [Google Scholar]
- 18.Lyman S, Aster RH, Visentin GP, et al. Polymorphism of human platelet membrane glycoprotein IIb associated with the Baka/Bakb alloantigen system. Blood. 1990;75:2343–8. [PubMed] [Google Scholar]
- 19.Peterson JA, Nyree CE, Newman PJ, et al. A site involving the “hybrid” and PSI homology domains of GPIIIa (beta 3-integrin subunit) is a common target for antibodies associated with quinine-induced immune thrombocytopenia. Blood. 2003;101:937–42. doi: 10.1182/blood-2002-07-2336. [DOI] [PubMed] [Google Scholar]
- 20.Curtis BR, Edwards JT, Hessner MJ, et al. Blood group A and B antigens are strongly expressed on platelets of some individuals. Blood. 2000;96:1574–81. [PubMed] [Google Scholar]
- 21.Furihata K, Nugent DJ, Bissonette A, et al. On the association of the platelet-specific alloantigen, Pena, with glycoprotein IIIa: evidence for heterogeneity of glycoprotein IIIa. J Clin Invest. 1987;80:1624–30. doi: 10.1172/JCI113250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menitove JE, Pereira J, Hoffman R, et al. Cyclic thrombocytopenia of apparent autoimmune etiology. Blood. 1989;73:1561–9. [PubMed] [Google Scholar]
- 23.Lucas DP, Paparounis ML, Myers L, et al. Detection of HLA class I-specific antibodies by the QuikScreen enzyme-linked immunosorbent assay. Clin Diagn Lab Immunol. 1997;4:252–7. doi: 10.1128/cdli.4.3.252-257.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skogen B, Bellissimo DB, Hessner MJ, et al. Rapid determination of platelet alloantigen genotypes by polymerase chain reaction using allele-specific primers. Transfusion. 1994;34:955–60. doi: 10.1046/j.1537-2995.1994.341195065032.x. [DOI] [PubMed] [Google Scholar]
- 25.Kiefel V, Santoso S, Weisheit M, et al. Monoclonal antibody-specific immobilization of platelet antigens (MAIPA): a new tool for the identification of platelet-reactive antibodies. Blood. 1987;70:1722–6. [PubMed] [Google Scholar]
- 26.Bussel JB, Berkowitz RL, Lynch L, et al. Antenatal management of alloimmune thrombocytopenia with intravenous gamma-globulin: a randomized trial of the addition of low-dose steroid to intravenous gamma-globulin. Am J Obstet Gynecol. 1996;174:1414–23. doi: 10.1016/s0002-9378(96)70582-3. [DOI] [PubMed] [Google Scholar]
- 27.Groves AM, Clough V, Stevens R. Neonatal alloimmune thrombocytopenia may be less severe in a subsequent pregnancy. Pediatr Hematol Oncol. 2003;20:393–8. [PubMed] [Google Scholar]
- 28.Harrison CR, Curtis BR, McFarland JG, et al. Severe neonatal alloimmune thrombocytopenia caused by antibodies to human platelet antigen 3a (Baka) detectable only in whole platelet assays. Transfusion. 2003;43:1398–402. doi: 10.1046/j.1537-2995.2003.00533.x. [DOI] [PubMed] [Google Scholar]
- 29.Kataoka S, Kobayashi H, Chiba K, et al. Neonatal alloimmune thrombocytopenia due to an antibody against a labile component of human platelet antigen-3b (Bakb) Transfus Med. 2004;14:419–23. doi: 10.1111/j.1365-3148.2004.00537.x. [DOI] [PubMed] [Google Scholar]
- 30.Take H, Tomiyama Y, Shibata Y, et al. Demonstration of the heterogeneity of epitopes of the platelet-specific alloantigen, Baka. Br J Haematol. 1990;76:395–400. doi: 10.1111/j.1365-2141.1990.tb06374.x. [DOI] [PubMed] [Google Scholar]
- 31.Teramura G, Slichter SJ. Report on the Sixth International Society of Blood Transfusion Platelet Serology Workshop. Transfusion. 1996;36:75–81. doi: 10.1046/j.1537-2995.1996.36196190520.x. [DOI] [PubMed] [Google Scholar]
- 32.Lin M, Shieh SH, Liang DC, et al. Neonatal alloimmune thrombocytopenia in Taiwan due to an antibody against a labile component of HPA-3a (Baka) Vox Sang. 1995;69:336–40. doi: 10.1111/j.1423-0410.1995.tb00369.x. [DOI] [PubMed] [Google Scholar]
- 33.Djaffar I, Vilette D, Pidard D, et al. Human platelet antigen 3 (HPA-3): localization of the determinant of the alloantibody Lek (a) (HPA-3a) to the C-terminus of platelet glycoprotein IIb heavy chain and contribution of O-linked carbohydrates. Thromb Haemost. 1993;69:485–9. [PubMed] [Google Scholar]
- 34.Goldberger A, Kolodziej M, Poncz M, et al. Effect of single amino acid substitutions on the formation of the PlA and Bak alloantigenic epitopes. Blood. 1991;78:681–7. [PubMed] [Google Scholar]
- 35.Calvete JJ, Muniz-Diaz E. Localization of an O-glycosylation site in the alpha-subunit of the human platelet integrin GPIIb/IIIa involved in Baka (HPA-3a) alloantigen expression. FEBS Lett. 1993;328:30–4. doi: 10.1016/0014-5793(93)80959-x. [DOI] [PubMed] [Google Scholar]
- 36.Judd WJ, Issitt PD, Pavone BG, et al. Antibodies that define NANA-independent MN-system antigens. Transfusion. 1979;19:12–8. doi: 10.1046/j.1537-2995.1979.19179160260.x. [DOI] [PubMed] [Google Scholar]
- 37.Blackall DP, Ugorski M, Pahlsson P, et al. A molecular biologic approach to study the fine specificity of antibodies directed to the MN human blood group antigens. J Immunol. 1994;152:2241–7. [PubMed] [Google Scholar]
- 38.Sadler JE, Paulson JC, Hill RL. The role of sialic acid in the expression of human MN blood group antigens. J Biol Chem. 1979;254:2112–9. [PubMed] [Google Scholar]
- 39.Pahlsson P, Blackall DP, Ugorski M, et al. Biochemical characterization of the O-glycans on recombinant glycophorin A expressed in Chinese hamster ovary cells. Glycoconj J. 1994;11:43–50. doi: 10.1007/BF00732431. [DOI] [PubMed] [Google Scholar]