

Abstract
Cutaneous malignant melanoma remains the leading cause of skin cancer death in industrialized countries. Clinical and histological variables that predict survival, such as Breslow’s index, tumor size, ulceration, or vascular invasion have been identified in malignant melanoma. Nevertheless, the potential relevance of biological variables still awaits an in-depth exploration. Using tissue microarrays (TMAs), we retrospectively analyzed 165 malignant melanoma samples from 88 patients corresponding to distinct histological progression phases, radial, vertical, and metastases. A panel of 39 different antibodies for cell cycle, apoptosis, melanoma antigens, transcription factors, DNA mismatch repair, and other proteins was used. Integrating the information, the study has identified expression profiles distinguishing specific melanoma progression stages. Most of the detected alterations were linked to the control of cell cycle G1/S transition; cyclin D1 was expressed in radial cases 48% (12 of 25) with significant lost of expression in vertical cases 14% (9 of 65), P = 0.002; whereas p16INK4a (89% in vertical versus 71% in metastatic cases, P = 0.009) and p27KIP1 (76% in radial versus 45% in vertical cases, P = 0.010) were diminished in advanced stages. The study also defines a combination of biological markers associated with shorter overall survival in patients with vertical growth phase melanoma, that provided a predictor model with four antibodies (Ki67, p16INK4a, p21CIP1, and Bcl-6). This predictor model was validated using an independent series of 72 vertical growth phase melanoma patients.
Cutaneous malignant melanoma, the most aggressive common skin tumor, is characterized by a multi-factorial etiology. Sun exposure and genetic susceptibility have been proposed as major etiological and predisposing factors that somehow explain the reported increased incidence.1,2 Metastatic melanoma tumors have a poor response to any therapy, with a 5-year survival probability ranging from 0 to 30%.3–5
Cutaneous malignant melanoma develops in three different stages, from radial to vertical growth phases and metastatic disease. The underlying molecular events that explain malignant melanoma genesis have been only partially characterized, and only a small number of genes have been identified as playing key roles in melanoma initiation and progression. Among these, some cell cycle regulators, apoptotic, signal transduction, cell adhesion, and matrix digestion genes have been demonstrated to be deregulated in this neoplasm.6–11
The variability of clinical behavior in patients with malignant melanoma is only partially explained by clinical and histological data, and we need to identify biological variables of use in assigning patients to specific risk groups. Although this aim has been achieved for single markers, malignant melanoma has benefited little from the use of massive molecular analytical techniques. The use of tissue microarrays (TMAs) is a technique suitable for malignant melanoma progression study, since it is a powerful tool that allows hundreds of specimens to be interrogated simultaneously with a panel of antibodies or probes directed against selected targets. Information obtained with TMAs not only contributes to the identification of key molecular events, but also facilitates the development of outcome predictor models of unprecedented accuracy.12–14
In this study, we have collected a large series of human malignant melanomas of different stages, and factor, signal-transduction, transcription-factor, differentiation, and melanoma-specific antigens. The study identifies a set of stage-specific markers, and allows a predictor model to be developed that identifies groups of patients with increased risk of death.
Materials and Methods
Malignant Melanoma Samples
A total of 175 tissue samples from common nevi (n = 10), primary radial growth phase malignant melanoma (n = 28), primary vertical growth phase malignant melanoma (n = 66), skin (n = 34), and nodal (n = 37) melanoma metastases were examined. Primary (radial and vertical phases) and metastatic tumors used to construct the TMAs were all taken from a series of 88 patients consecutively diagnosed at the 12 Octubre University Hospital, Madrid, between 1995 and 2000. In 2 of 88 cases, biopsies from both vertical and radial phases were included. In 30 of 88 patients, samples corresponding to primary (vertical phase in all cases) and metastatic disease were included. In 11 of 88 patients it was possible to include biopsies from both cutaneous and lymph node metastases. Additional metastases (8 of 88 cases) from patients from whom primary tumors could not be obtained were also included in the study. There was no history of familial occurrence in the selected patients.
All patients were treated according to standard criteria: surgical resection in all primary cases and surgical resection (when possible) plus chemotherapy, if indicated, in cases with demonstrated metastases. For survival analysis, all included samples corresponded to the initial diagnostic specimens before any further treatment.
Paraffin blocks were selected on the basis of the availability of suitable formalin-fixed, paraffin-embedded tissue (at least 1-mm thick). Hematoxylin and eosin-stained slides were available for review in all cases measured as Breslow’s index, and histological progression phase were defined according to standard criteria15,16 (also see additional information at http://ajp.amjpathol.org). Approval for this study was obtained from the 12 Octubre Hospital Institutional Review Board. Informed consent, when necessary, was obtained following the guidelines of the Helsinki Declaration.
Generation of Tissue Arrays
For characterization of immunohistochemical protein expression, we constructed six different tissue arrays containing the distinctive histological stages of the tumor. Each tissue array was assembled as previously described.12 Briefly, two 1.5-mm-diameter cylinders of tissue were taken from representative areas of each archival paraffin block and arrayed into a new recipient paraffin block with a custom-built precision instrument (Beecher Instruments, Silver Spring, MD) according to the previously described method.12
These tissues had been fixed in 4% formalin (Panreac, Barcelona, Spain) and paraffin-embedded according to routine procedures. Areas chosen for the cylinder core had high tumor cellularity. To evaluate the most adequate part of each primary tumor we selected the invasive border of the tumor in large lesions and all tumor cells in smaller samples. In addition, normal tissues (skin, tonsil, and reactive lymphoid tissue) and three different cell lines with known cell cycle alterations were placed adjacent to tumoral tissues to serve as internal controls and to ensure the quality of staining slides. Initial sections were stained for hematoxylin and eosin to verify the histopathological findings.
Immunohistochemistry
Three-micrometer tissue sections from the TMA blocks were sectioned and applied to special immunohistochemistry coated slides (DAKO, Glostrup, Denmark). These slides were baked overnight in a 56°C oven. Sections were deparaffinized in xylene for 20 minutes, rehydrated through a graded ethanol series and washed with phosphate-buffered saline. Antigen retrieval was achieved by a 2-minute heat treatment in a pressure cooker, containing 1 liter of 10 mmol/L sodium citrate buffer, (pH 6.5, DAKO) that had been previously brought to the boil. Before staining, endogenous peroxidase activity was quenched with 1.5% hydrogen peroxide (DAKO) in methanol for 10 minutes.
Immunohistochemical staining was performed on these sections using 39 different antibodies, (source and concentration are listed in Table 1). Antibodies used in this study were selected for reporting on key aspects of cell cycle, apoptosis, signal transduction, and melanoma differentiation. We have also considered the status of the markers described in previous studies in this tumor type. After incubation, in the case of nuclear markers, immunodetection was performed with the LSAB Visualization System (DAKO) using 3,3′-diaminobenzidine chromogen as substrate, according to the manufacturer’s instructions. In addition, whenever possible, cytoplasmic markers were visualized with the APAAP System (alkaline phosphatase anti-alkaline phosphatase, DAKO), using neo-fuchsine chromogen as substrate to rule out the possibility of a role of endogenous melanin in the observed reactivity. All sections were counter-stained with hematoxylin (DAKO). Negative controls were obtained by omitting the primary antibody.
Table 1.
Parameters Measured and Antibodies Tested in the Study, Indicating Source, Dilution, Visualization Method, Pattern of Reactivity, Threshold and Positive Control (Evaluated in Skin, Tonsil, and Reactive Lymph Node Tissue)
| Protein | Clone | Source | Dilution | Visualization system | Reactivity | Threshold | Internal control |
|---|---|---|---|---|---|---|---|
| Bcl2 | 124 | DAKO | 1:25 | LSAB/DAB | pos/neg | >10% positive cells | Small lymphocytes |
| Bcl6 | PG-B6p | DAKO | 1:10 | LSAB/DAB | pos/neg | Any nuclear expressi | CG B-cells in lymph node |
| Bclx-L | 2H12 | ZYMED | 1:10 | APAAP/NF | pos/neg | >10% positive cells | Cell lines |
| Bmi 1 | POLY | Santa Cruz | 1:100 | ENVISI/DAB | pos/neg | >10% positive cells | Mantle cells, normal epithelium |
| Caveolin | 2297 | Transduction | 1:25 | APAAP/NF | high/low | ≥50% positive cells | Endothelial cells |
| Cyclin A | 6E6 | Novocastra | 1:100 | LSAB/DAB | pos/neg | ≥5% positive cells | Proliferating cells (G2/M) |
| Cyclin B1 | 7A9 | Novocastra | 1:25 | LSAB/DAB | pos/neg | ≥5% positive cells | Proliferating cells (G2/M) |
| Cyclin D1 | DCS-6 | DAKO | 1:100 | LSAB/DAB | high/low | ≥30% positive cells | Endothelial cells and macrophages |
| Cyclin D3 | DCS-22 | Novocastra | 1:10 | LSAB/DAB | pos/neg | ≥5% positive cells | Proliferating cells |
| Cyclin E | 13A3 | Novocastra | 1:10 | LSAB/DAB | pos/neg | ≥10% positive cells | Proliferating cells |
| CDK1 | 1 | Transduction | 1:1500 | LSAB/DAB | pos/neg | ≥5% positive cells | Proliferating cells, basal cells in skin |
| CDK2 | 8D4 | NeoMarkers | 1:500 | LSAB/DAB | pos/neg | ≥5% positive cells | Proliferating cells |
| CDK6 | K6.83 | Chemicon | 1:10 | LSAB/DAB | pos/neg | ≥5% positive cells | TMA controls |
| C-kit | POLYC | DAKO | 1:50 | APAAP/NF | high/low | ≥50% positive cells membranous express | Melanocytes in normal skin |
| Hdm2 | IF2 | Oncogen | 1:10 | LSAB/DAB | pos/neg | Any nuclear expres. | Macrophages, endothelial cells |
| HMB 45 | HMB 45 | BIOGENEX | 1:10 | APAAP/NF | pos/neg | ≥50% positive cells | Melanocytes in normal skin |
| HLA* DP, DQ, DR | J576 | CNIO | 1:150 | LSAB/DAB | pos/neg | Any nuclear expres. | GC |
| Ki67 | MIB1 | DAKO | 1:100 | LSAB/DAB | high/low | ≥20% positive cells | Proliferating cells |
| MELAN A | A103 | DAKO | 1:10 | APAAP/NF | pos/neg | ≥50% positive cells | Melanocytes in normal skin |
| MEL 18 | POLY | Santa Cruz | 1:100 | LSAB/DAB | high/low | ≥50% positive cells | Mantle cells, plasma cells |
| MLH 1 | G168-15 | Pharmingenn | 1:100 | APAAP/NF | high/low | ≥50% positive cells | Normal skin |
| MSH 2 | FE11 | ONCOGEN | 1:100 | APAAP/NF | high/low | ≥70% positive cells | Normal skin |
| MUM-1 | POLYCL | Santa Cruz | 1:200 | LSAB/DAB | high/low | ≥30% positive cells | Plasma cells, GC |
| NFKappaB | F-6 | Santa Cruz | 1:200 | LSAB/DAB | pos/neg | Any nuclear expres | Cell lines |
| P16 | F12 | Santa Cruz | 1:50 | LSAB/DAB | high/low | ≥50% positive cells | Epithelial cells in normal skin |
| P21 | EA10 | Oncogene | 1:50 | LSAB/DAB | pos/neg | ≥10% positive cells | Scattered GC cells |
| P27 | 57 | Transduction Lab | 1:1000 | LSAB/DAB | pos/neg | ≥10% positive cells | Resting lymphoid cells, upper epidermis in normal skin |
| P53 | DO-7 | Novocastra | 1:50 | LSAB/DAB | pos/neg | ≥10% positive cells | Scattered GC cells |
| PH1** | CIB | 1:2 | LSAB/DAB | pos/neg | Any nuclear expres | ||
| PKCβ | 28 | Serotec | 1:500 | APAAP/NF | pos/neg | ≥10% positive cells | Endothelial cells, adipocytes, macrophages |
| RING 1B** | 3RING1 | CIB | 1 | LSAB/DAB | pos/neg | Any nuclear expres | Mantle cells |
| Rb | G3-245 | BD | 1:250 | LS | pos/neg | ≥10% positive cells | Proliferating cells |
| Rb P | POLYCL | PharMingen Santa Cruz | 1:500 | AB/DAB LSAB/DAB | pos/neg | ≥10% positive cells | Proliferating cells |
| RYBP** | POLYCL | CIB | 1:25 | LSAB/DAB | pos/neg | Any nuclear express | Endothelial cells, plasma cells |
| S-100 | POLYCL | DAKO | 1:500 | APAAP/NF | pos/neg | ≥30% positive cells | Normal melanocytes, nerves |
| Survivin | POLYCL ONAL | RD Systems | 1:1500 | APAAP/NF | pos/neg | ≥2% positive cells (nuclear expression) | Scattered GC cells |
| SKP2 | 1G12E9 | ZYMED | 1:10 | LSAB/DAB | pos/neg | >5% positive cells | Proliferating cells |
| STAT1 | C-136 | Santa Cruz | 1:50 | LSAB/DAB | pos/neg | Any nuclear expression | Reactive lymphocytes macrophages |
| Topo-isomerase | Ki-S1 | DAKO | 1:400 | LSAB/DAB | pos/neg | ≥10% positive cells | Proliferating cells |
APAAP, alkaline phosphatase anti-alkaline phosphatase; NF, neo-fuchsine; pos/neg, positive/negative.
BD: BD Biosciences/Transduction Labs (San Diego, California), DAKO Corp (Carpenteria, California), Zymed (South San Francisco, California).
Antibodies provided by the Monoclonal Antibodies Unit* (CNIO) and by Dr. Miguel Angel Vidal** (CIB-CSIC, Madrid, Spain).
Scoring Systems
Immunostaining intensity was evaluated by two different pathologists (S.R.A.G. and J.L.R.P.) and scored using uniform and clear cut-off criteria, to maintain the reproducibility of the method. Discrepancies were resolved by simultaneous re-evaluation. Briefly, the result of immunostaining was recorded as negative or positive, and low versus high expression, taking into account the expression in tumoral cells and the specific cut-off for each marker (see Table 1 for definition of positive phenotype). As a general criterion, the cut-offs were selected to facilitate reproducibility, and when possible, to translate biological events. Selected sample replicates were highly reproducible in our series (96%), as other authors have found.9,14 STAT1-positive expression was considered only with respect to the nuclear signal that translates the active form of this protein.
Statistics
Clinical data and immunohistochemistry scoring were performed blind, and data were compiled only after all analyses were completed. Fisher’s exact test was used to test whether a specific protein-expression profile was associated with a particular histological stage of melanoma progression. Significance was concluded for values of P < 0.05. All statistical tests were two-sided. To avoid the influence of multiple testing, adjusted P values were also computed using the method proposed by Hochberg and Benjamini17 to minimize the false discovery rate.
Complete follow-up survival data for 6 years or more were available in 60 vertical malignant melanoma cases. The overall survival (OS) predictive value of the studied markers these cases was explored using Kaplan-Meier, log-rank test, and Cox regression analysis. A multivariate model was developed by backwards elimination, starting with all markers with a P value lower than 0.10 in the univariate analysis. This statistical significance level was selected to allow for optimal discovery in the initial phases of this analysis. From the final model for each patient a score was computed taking the linear predictor from the multivariate model.
Validation Set
Given the exploratory nature of the study, an independent series of 72 vertical growth phase melanoma cases was included to validate the prognostic value of the set of prognostic markers. This validation set was similar to the training set and included patients with vertical growth phase melanoma diagnosed from 1986 through 1998.
Validation was carried out in two ways: first, the previously mentioned score was computed using the linear predictor from the multivariate model in the original sample and the statistical significance of this score was tested. Differences in survival according to this score are presented graphically for both the original and the validation set of patients. For this representation, score values were collapsed into three risk groups, dividing the whole range into three equally spaced categories. Second, the same multivariate model was fitted in the validation sample to test whether the hazard ratios (HRs) obtained in this set of patients were different from those in the original model. All statistical analyses were carried out using the Stata Statistical Program (StataCorp 2001, Release 7.0).
Results
Clinical Data
The series comprises 88 patients diagnosed with cutaneous malignant melanoma (41 males and 47 females). Their ages at the time of diagnosis ranged from 21 to 91 years (mean, 58 years). The average ages of patients with radial and vertical growth phases were 28 and 61 years and the percentage metastases were 0% and 55%, respectively. The survival analysis was restricted to vertical growth melanomas patients with follow-up information (n = 60) as, in this series, only these cases developed metastases and some of them died. In the patients with vertical growth phase melanomas the median follow-up was 61 months (range, 2 to 186 months). During the 6 years considered in the survival analysis, 4 were lost to follow-up, 19 patients died of melanoma, 4 patients died of other causes, and 33 patients remained alive.
Expression Profiling at Each Stage of Malignant Melanoma Progression
Immunohistochemical analysis of protein expression was carried out on 28 radial and 66 vertical growth phase malignant melanomas, 71 metastatic malignant melanoma, and 10 nevus specimens. The results of expression profiling at each stage are detailed in Table 2 and illustrated in Figure 1 and Figure 2, where each phase in the growth of malignant melanoma is distinguished by a specific constellation of markers. Nevertheless, some of the most significant features are worth comment: S-100 was positive in all but one case (the exception being a metastasis). HMB45 and melan A (specific markers of melanocytic origin) were constitutively expressed in the majority of primary malignant melanomas [95.6% (88 of 92) for HMB45 and 92.4% (85 of 92) for melan A] and in most metastatic cases, [81% (55 of 68) and 75% (50 of 67), for HMB45 and melan A, respectively; see Table 2]. Ki-67, a proliferation marker, was significantly higher in vertical growth phase melanoma and metastases than in nevi and radial growth phase tumors.
Table 2.
Contingency Analysis (Fisher’s Exact Test) Comparing the Level of Protein Expression in Each Progression Step of Cutaneous Malignant Melanoma
| Nevus
|
Radial/Nevus
|
Radial
|
Vertical/Radial
|
Vertical
|
Met/Vertical
|
Metastasis
|
Met/Nevus
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c/to | %(+)N | p | p* | c/to | %(+)R | p | p* | c/to | %(+)V | p | p* | c/to | %(+)M | p | p* | |
| Melanoma specific proteins | ||||||||||||||||
| HMB45 | 10/10 | 100% | 1.000 | 1.000 | 26/26 | 100% | 0.574 | 0.831 | 62/66 | 94% | 0.036 | 0.108 | 55/68 | 81% | 0.199 | 0.310 |
| Melan A | 2/10 | 20% | 0.000 | 0.000 | 25/26 | 96% | 0.668 | 0.905 | 60/66 | 91% | 0.021 | 0.084 | 50/67 | 75% | 0.001 | 0.007 |
| S-100 | 10/10 | 100% | 1.000 | 1.000 | 26/26 | 100% | 1.000 | 1.000 | 66/66 | 100% | 1.000 | 1.000 | 68/69 | 99% | 1.000 | 1.000 |
| Cell Cycle | ||||||||||||||||
| Cyclin A | 0/10 | 0% | 0.003 | 0.016 | 14/26 | 54% | 0.347 | 0.607 | 43/66 | 65% | 0.344 | 0.602 | 49/66 | 74% | 0.000 | 0.000 |
| Cyclin B1 | 0/10 | 0% | 0.303 | 0.489 | 4/25 | 16% | 0.410 | 0.662 | 16/63 | 25% | 1.000 | 1.000 | 15/58 | 26% | 0.102 | 0.209 |
| Cyclin D1 | 0/10 | 0% | 0.007 | 0.028 | 12/25 | 48% | 0.002 | 0.015 | 9/65 | 14% | 0.021 | 0.084 | 21/65 | 32% | 0.053 | 0.131 |
| Cyclin D3 | 0/9 | 0% | 0.303 | 0.489 | 6/27 | 22% | 0.073 | 0.279 | 5/66 | 8% | 0.009 | 0.050 | 17/66 | 26% | 0.109 | 0.199 |
| Cyclin E | 0/10 | 0% | 1.000 | 1.000 | 2/25 | 8% | 0.335 | 0.704 | 12/66 | 18% | 0.467 | 0.676 | 8/65 | 12% | 0.587 | 0.770 |
| CDK1 | 0/10 | 0% | 0.002 | 0.012 | 15/27 | 56% | 0.364 | 0.612 | 29/66 | 44% | 0.154 | 0.323 | 21/67 | 31% | 0.054 | 0.126 |
| CDK2 | 1/10 | 10% | 0.000 | 0.000 | 27/28 | 86% | 0.001 | 0.012 | 46/66 | 48% | 0.114 | 0.266 | 34/68 | 34% | 0.162 | 0.284 |
| CDK6 | 0/9 | 0% | 0.303 | 0.489 | 6/27 | 22% | 0.227 | 0.561 | 24/66 | 36% | 0.005 | 0.035 | 10/67 | 15% | 0.597 | 0.737 |
| p16 | 9/9 | 100% | 0.553 | 0.774 | 23/26 | 88% | 1.000 | 1.000 | 59/66 | 89% | 0.009 | 0.050 | 48/68 | 71% | 0.102 | 0.209 |
| p21 | 0/10 | 0% | 0.073 | 0.192 | 13/25 | 32% | 0.795 | 1.000 | 28/66 | 27% | 0.100 | 0.263 | 42/66 | 42% | 0.011 | 0.040 |
| p27 | 7/10 | 70% | 0.694 | 0.911 | 19/25 | 76% | 0.010 | 0.053 | 30/66 | 45% | 0.380 | 0.570 | 25/68 | 37% | 0.082 | 0.181 |
| SKP2 | 1/9 | 11% | 0.391 | 0.587 | 8/26 | 31% | 0.346 | 0.632 | 28/65 | 43% | 0.491 | 0.687 | 33/67 | 49% | 0.037 | 0.104 |
| p53 | 7/9 | 78% | 0.620 | 0.840 | 24/28 | 86% | 0.770 | 1.000 | 53/66 | 80% | 0.074 | 0.207 | 61/66 | 92% | 0.196 | 0.317 |
| Rb | 2/10 | 20% | 0.005 | 0.023 | 20/26 | 77% | 1.000 | 1.000 | 49/66 | 74% | 0.353 | 0.593 | 46/69 | 67% | 0.011 | 0.040 |
| pRb | 0/10 | 0% | 0.007 | 0.028 | 12/25 | 48% | 0.345 | 0.659 | 40/66 | 61% | 0.602 | 0.766 | 38/69 | 55% | 0.001 | 0.007 |
| HDM 2 | 1/10 | 10% | 0.012 | 0.042 | 16/28 | 57% | 0.232 | 0.541 | 47/66 | 71% | 0.551 | 0.723 | 51/66 | 77% | 0.000 | 0.000 |
| BCL-6 | 0/10 | 0% | 1.000 | 1.000 | 1/24 | 4% | 1.000 | 1.000 | 5/64 | 8% | 0.115 | 0.254 | 1/65 | 2% | 1.000 | 1.000 |
| Apoptosis | ||||||||||||||||
| BCL-2 | 10/10 | 100% | 0.157 | 0.300 | 20/26 | 77% | 0.336 | 0.672 | 55/63 | 87% | 0.000 | 0.000 | 27/60 | 45% | 0.001 | 0.007 |
| BCLX-L | 0/10 | 0% | 0.145 | 0.305 | 6/23 | 26% | 0.070 | 0.294 | 6/66 | 9% | 0.204 | 0.389 | 12/66 | 18% | 0.348 | 0.487 |
| SURVIVIN | 0/10 | 0% | 0.038 | 0.106 | 10/28 | 36% | 0.002 | 0.015 | 46/65 | 71% | 0.356 | 0.575 | 41/66 | 62% | 0.000 | 0.000 |
| Transcription factors | ||||||||||||||||
| MUM-1 | 5/10 | 50% | 0.079 | 0.175 | 22/26 | 85% | 0.008 | 0.048 | 36/66 | 55% | 0.000 | 0.000 | 5/70 | 7% | 0.002 | 0.011 |
| PKCB | 0/10 | 0% | 0.078 | 0.182 | 7/24 | 29% | 0.032 | 0.149 | 37/66 | 56% | 0.000 | 0.000 | 12/68 | 18% | 0.346 | 0.501 |
| STAT-1 | 0/10 | 0% | 0.034 | 0.102 | 10/25 | 40% | 0.815 | 0.978 | 29/66 | 44% | 0.029 | 0.094 | 17/68 | 25% | 0.107 | 0.204 |
| DNA repair | ||||||||||||||||
| MLH-1 | 9/9 | 100% | 0.077 | 0.190 | 15/22 | 68% | 0.001 | 0.012 | 64/66 | 97% | 0.000 | 0.000 | 40/67 | 60% | 0.023 | 0.074 |
| MSH-2 | 9/10 | 90% | 1.000 | 1.000 | 24/26 | 92% | 0.079 | 0.277 | 65/65 | 100% | 0.028 | 0.098 | 60/66 | 91% | 1.000 | 1.000 |
| Membrane receptor | ||||||||||||||||
| CAVEOLIN | 8/10 | 80% | 0.001 | 0.007 | 4/25 | 16% | 0.283 | 0.626 | 19/66 | 29% | 0.104 | 0.257 | 29/67 | 43% | 0.042 | 0.110 |
| C-KIT | 1/10 | 10% | 0.000 | 0.000 | 22/25 | 88% | 0.000 | 0.000 | 17/66 | 26% | 0.542 | 0.734 | 14/68 | 21% | 0.677 | 0.812 |
| Transcription regulators: Polycomb | ||||||||||||||||
| BMI 1 | 10/10 | 100% | 0.016 | 0.052 | 15/26 | 58% | 0.170 | 0.476 | 26/63 | 41% | 0.860 | 1.000 | 27/69 | 39% | 0.000 | 0.000 |
| PH 1 | 0/10 | 0% | 0.552 | 0.799 | 3/28 | 11% | 0.543 | 0.815 | 11/66 | 17% | 0.181 | 0.362 | 5/66 | 8% | 1.000 | 1.000 |
| RING 1B | 8/10 | 80% | 1.000 | 1.000 | 22/26 | 85% | 0.575 | 0.805 | 61/65 | 78% | 0.002 | 0.017 | 34/66 | 52% | 0.170 | 0.286 |
| RYBP 2 | 0/10 | 0% | 0.152 | 0.304 | 6/25 | 24% | 0.092 | 0.297 | 6/63 | 10% | 1.000 | 1.000 | 6/69 | 9% | 1.000 | 1.000 |
| MEL 18 | 7/9 | 78% | 0.164 | 0.299 | 24/26 | 96% | 0.435 | 0.677 | 58/65 | 89% | 1.000 | 1.000 | 61/68 | 90% | 0.282 | 0.423 |
| Proliferation | ||||||||||||||||
| KI 67 | 0/10 | 0% | 1.000 | 1.000 | 1/28 | 4% | 0.000 | 0.000 | 26/66 | 39% | 0.294 | 0.537 | 32/65 | 49% | 0.004 | 0.019 |
| Others | ||||||||||||||||
| TOPOISO† | 1/10 | 10% | 0.000 | 0.000 | 20/26 | 77% | 1.000 | 1.077 | 52/66 | 79% | 0.015 | 0.070 | 38/66 | 58% | 0.006 | 0.025 |
| HLA‡ | 0/10 | 0% | 1.000 | 1.000 | 0/24 | 0% | 0.183 | 0.480 | 7/66 | 11% | 0.792 | 0.978 | 9/69 | 13% | 0.594 | 0.756 |
Each level is compared with the less aggressive stage (radial growth phase versus nevus, vertical versus radial and metastatic versus vertical). P values comparing nevus versus melanoma are also included (right-hand columns). Data reflect the number of positive cases of the total (c/to), the corresponding percentage and significance (p-value (p) and adjusted p-values (p*)) of the compared phases.
Significant results are indicated in bold. N, nevus; R, radial growth phase; V, vertical growth phase; M, metastases). †, Topoisomerase II. ‡, HLA DP, DQ, DR.
Figure 1.

TMA design for melanoma progression. Cylinders corresponding to nevus (A), radial growth phase (B), vertical growth phase (C), and metastases (D). Immunohistochemical patterns. Core samples with positive and negative (or high and low) expression for selected antibodies are shown. All of the cases are representative of nevus-column A, and of each malignant melanoma progression step-columns B, C, and D. Note the high expression of cyclin D1 in radial (B) and metastases (D) when compared with vertical cases (C) and the loss of p16INK4a expression in metastases. Note also the loss of p27KIP1 and caveolin with tumor progression and the nuclear survivin expression in melanomas when compared with nevi. Original magnification for A–D, ×40; insets, ×600 and ×800, respectively. HE, hematoxylin and eosin.
Figure 2.

Signature in melanoma progression. Representative proteins differentially expressed in cutaneous malignant melanoma identified by tissue array analysis. Subheadings indicate protein function and arrows represent up- or down-regulation in radial growth phase melanoma relative to nevus; vertical growth phase relative to radial growth phase and metastasis versus vertical growth phase. If there was no difference in protein expression between one phase and the comparative phase, the name of the marker is not included. See also Table 2 for differences in protein expression. A total of 175 specimens were evaluated, including 10 nevus, 28 radial malignant melanoma, 66 vertical malignant melanoma, and 71 metastases (34 skin metastases and 37 nodal metastases).
High levels of expression of specific cyclins (cyclin A, cyclin D1, and cyclin D3) and CDKs (CDK1 and CDK 2) were found when comparing nevus to malignant melanoma. Particularly striking was cyclin D1, which was negative in all nevi and was markedly expressed in radial growth phase malignant melanoma (12 of 25, 48%) and metastases (21 of 65, 32%). Surprisingly, the expression decreased significantly in the deepest component of the primary vertical growth phase tumor (see Figure 1). In the case of cyclin D3, high levels of expression were found only in metastatic stages. We found no significant change in cyclin E protein expression in any melanocytic lesions; however, CDK 2, the normal cyclin E partner, was more strongly expressed in radial growth phase melanoma (27 of 28, 86%) than in nevus (0%) and its expression progressively diminished in the subsequent steps (46 of 66, 48% in vertical cases and 34 of 68, 34% in metastases, respectively).
In the group of cell cycle suppressors, loss of the p16INK4a protein was observed to be associated with advanced stages, with significantly lower expression levels in metastatic cases (48 of 68, 71%) compared with primary tumors (82 of 92, 89.13%) and benign lesions (9 of 9, 100%). The loss of p27KIP1 protein expression with melanoma progression was significant in vertical growth phase (30 of 66, 45%) compared with radial growth phase (19 of 25, 76%, P = 0.010).
Nuclear expression of the apoptotic inhibitor survivin was negative in all nevi (10 cases), and commonly expressed in malignant melanoma (97 of 159 cases, 61%), showing notably increased expression with melanoma progression. Regarding other proteins, caveolin, a membrane-scaffolding protein, experienced significant losses in primary malignant melanomas compared with nevi. MUM-1/IRF4, a known transcription factor detected in most primary melanocytic lesions (5 of 10, 50% benign nevi and 58 of 92, 70% of primary melanomas in our series, respectively), was progressively lost with tumoral advance and had a very low level of expression in metastatic cases (only 5 of 70, constituting 7% of positive cases).
Simultaneous Analysis of Vertical versus Metastatic Melanoma in the Same Patients
The existence of changes in the expression of a set of relevant molecules, when comparing vertical growth phase with metastatic melanoma, was confirmed in a restricted series of 30 patients, in which simultaneous analysis of samples representative from both stages was performed (Table 3).
Table 3.
Comparative Expression of Primary versus Metastatic Tumors in the Same Patient (30 Patients)
| Marker | Vertical (+) vs. metastases (−) | Vertical (−) vs. metastases (+) | Vertical (+) vs. metastases (+) | Vertical (−) vs. metastases (−) | Missing values* |
|---|---|---|---|---|---|
| BCL-2 | 9/30 (30%) | 1/30 (3.3%) | 9/30 (30%) | 4/30 (13.3%) | 7/30 (23.3%) |
| Cyclin D1 | 1/30 (3.3%) | 4/30 (13.3%) | 3/30 (10%) | 17/30 (56.6%) | 5/30 (16.6%) |
| Cyclin D3 | 2/30 (6.6%) | 3/30 (10%) | 1/30 (3.3%) | 21/30 (70%) | 3/30 (10%) |
| CDK 6 | 10/30 (33.3%) | 4/30 (13.3%) | 3/30 (10%) | 10/30 (33.3%) | 3/30 (10%) |
| HMB 45 | 3/30 (10%) | 1/30 (3.3%) | 23/30 (76.6%) | 0/30 (0%) | 3/30 (10%) |
| Melan A | 7/30 (23.3%) | 1/30 (3.3%) | 17/30 (56.6%) | 1/30 (3.3%) | 4/30 (13.3%) |
| MLH 1 | 13/30 (43.3%) | 0/30 (0%) | 13/30 (43.3%) | 0/30 (0%) | 3/30 (10%) |
| MSH 2 | 2/30 (6.6%) | 0/30 (0%) | 24/30 (80%) | 0/30 (0%) | 4/30 (13.3%) |
| MUM 1 | 11/30 (36.6%) | 0/30 (0%) | 1/30 (3.3%) | 17/30 (56.6%) | 1/30 (3.3%) |
| P16 | 8/30 (26.6%) | 2/30 (6.6%) | 17/30 (56.6%) | 0/30 (0%) | 3/30 (10%) |
| P21 | 7/30 (23.3%) | 6/30 (20%) | 5/30 (16.6%) | 9/30 (30%) | 3/30 (10%) |
| PKCB | 12/30 (40%) | 2/30 (6.6%) | 2/30 (6.6%) | 11/30 (36.6%) | 3/30 (10%) |
| RING 1B | 11/30 (36.6%) | 0/30 (0%) | 12/30 (40%) | 3/30 (10%) | 4/30 (13.3%) |
| STAT 1 | 7/30 (23.30%) | 1/30 (3.3%) | 3/30 (10%) | 17/30 (56.6%) | 2/30 (6.6%) |
| Topoisomerase | 4/30 (13.3%) | 3/30 (10%) | 4/30 (13.3%) | 15/30 (50%) | 4/30 (13.3%) |
Vertical (+) versus metastases (−); number of cases positive in vertical phase, lost in metastasis; vertical (−) versus metastases (+), number of cases acquiring the expression of some marker; vertical (+) versus metastases (+), number of cases positive in vertical phase and in metastasis; vertical (−) versus metastases (−), cases without marker expression;
missing values, non-valuable data.
Protein Expression and Survival Analysis
None of the radial growth phase malignant melanoma developed metastases or cause-related death, thus we have only considered vertical growth cases with follow-up information (n = 60) in the survival analysis. The results of this analysis using Kaplan-Meier and Cox regression models are shown in Table 4 (more data concerning the univariate analysis are available in Supplemental Table 1 at http://ajp.amjpathol.org). Shorter overall survival (OS) was significantly related to loss of p16INK4a, high Ki-67, and positive expression of cyclin E, p21CIP1, Bcl-6, Rb, and HLA DP.
Table 4.
Biological Markers Associated with Patient’s Outcome
| 6-year overall survival (from primary vertical growth phase melanoma to death)
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Training set
|
Validation set
|
|||||||||||||
| Univariate analysis
|
Multivariate analysis
|
Multivariate including Breslow
|
Multivariate analysis
|
|||||||||||
| Marker | Total cases | Deaths <72m | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value | Hazard ratio | 95% CI | p value |
| BCL-6 neg | 54 | 14 | 1.00 | 0.001 | 1.00 | <0.001 | 1.00 | 0.002 | 1.00 | 0.002 | ||||
| BCL-6 pos | 5 | 5 | 5.97 | 2.07–17.18 | 8.10 | 2.56–25.6 | 6.59 | 1.92–20.5 | 6.59 | 2.03–21.38 | ||||
| Cyclin E neg | 50 | 12 | 1.00 | 0.011 | ||||||||||
| Cyclin E pos | 10 | 7 | 3.39 | 1.32–8.70 | ||||||||||
| HLA neg | 55 | 15 | 1.00 | 0.029 | ||||||||||
| HLA pos | 5 | 4 | 3.46 | 1.13–10.59 | ||||||||||
| Ki 67 low | 37 | 9 | 1.00 | 0.047 | 0.068 | 1.00 | 0.248 | 1.00 | 0.591 | |||||
| Ki 67 high | 23 | 10 | 2.50 | 1.01–6.17 | 2.41 | 0.94–6.17 | 1.76 | 0.67–4.62 | 1.40 | 0.43–4.44 | ||||
| p16 neg | 6 | 5 | 1.00 | 0.002 | 1.00 | 0.001 | 1.00 | 0.001 | 1.00 | 0.006 | ||||
| p16 pos | 54 | 14 | 0.20 | 0.07–0.57 | 0.13 | 0.04–0.41 | 0.14 | 0.04–0.45 | 0.19 | 0.06–0.61 | ||||
| p21 neg | 44 | 9 | 1.00 | 0.010 | 0.065 | 1.00 | 0.106 | 1.00 | 0.395 | |||||
| p21 pos | 16 | 10 | 3.25 | 1.32–8.02 | 2.36 | 0.95–5.86 | 2.16 | 0.85–5.52 | 1.57 | 0.55–4.46 | ||||
| Rb neg | 16 | 1 | 1.00 | 0.045 | ||||||||||
| Rb pos | 44 | 18 | 7.86 | 1.05–58.91 | ||||||||||
| Breslow | 1.00 | |||||||||||||
| 1.68 | 0.94–2.99 | 0.079 | ||||||||||||
Kaplan-Meier, log rank test and Cox regression multivariate analysis of overall survival (OS), measured at time from primary vertical growth phase to death. The follow-up was 6 years. For the training set the significant p-values for the univariate analysis and the multivariate analysis before and after introducing Breslow’s index are shown. Data from the multivariate analysis in the validation set are also included (right column). (All values from the univariate analysis in the training set are available in Supplemental Table 1 at http://ajp.amjpathol.org).
The Cox multivariate regression model included four biological markers: p16INK4a, Bcl-6, Ki 67, and p21CIP1. The strongest predictors were p16INK4a and Bcl-6. The probability of death was around eight times higher when either p16INK4a was lost or Bcl-6 was positive.
The predictive model was not substantially altered by the inclusion of Breslow’s index as a variable, although the effect of Ki-67 was somewhat attenuated, probably reflecting its close relationship with Breslow’s index. This fact corroborates the view that the markers included in the prognostic model and Breslow’s index each contain independent information. To validate the predictor model, TMAs from an independent series of patients with follow-up information (n = 72) were stained and evaluated for the predictive set of markers.
A risk score was computed for each patient taking the linear predictor from the multivariate model. The formula of the score is depicted below, when each marker is considered 0 if negative and 1 if positive:
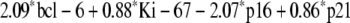 |
When this score was calculated for patients in the validation sample, it was significantly related to the probability of death (P = 0.001).
Figure 3 presents the survival curves for the original and the validation groups collapsing the score into three categories, as described in the Materials and Methods, and specifically demonstrates the existence of a subset of tumors with an unfavorable profile, corresponding to the third risk group. Finally, when the same multivariate model was fitted using the validation sample (see right-hand columns in Table 4) the loss of p16INK4a and the expression of Bcl-6 were again identified as the most significant predictors.
Figure 3.

Kaplan-Meier estimation of overall survival according to the assigned probability in the multivariate analysis. A score was built-up according with the multivariate model. The survival curves for the original and the validation groups are presented. Patients are grouped collapsing the score values into three categories (low, intermediate, and high risk patients) obtained by dividing the whole hazard ratio range into three equally spaced categories (groups).
Discussion
The analysis using TMAs allows the integration not only of the capability for detecting multiple proteins in large series of patients, but also the recognition of discrete cell subpopulations, such as intraepidermal melanocytes, which constitute the bulk of the malignant melanoma in the initial phases of melanoma genesis. The current study demonstrates the suitability of this technique in the study of a classical model in human tumor progression: malignant melanoma. To our knowledge, only a few studies based solely on the expression of single proteins9,18–20 have previously provided evidence of the feasibility of this powerful tool in malignant melanoma samples.
The present paper reports a comparative analysis of the different stages of the malignant melanoma sequence. Thus, each step in malignant melanoma progression is characterized by the expression of a specific profile: including cyclin D1, cyclin A, CDK1, CDK2, STAT1 in radial growth phase; Ki 67, survivin, and PKC-B in vertical growth phase; and cyclin D1, cyclin D3, and loss of p16INK4a, BclII, and MUM1 in metastatic melanoma (Figure 2). The results of this study illustrate how melanoma cells progress, as do other aggressive tumors, through the deregulation of the mechanisms controlling proliferation and escape from programmed cell death.21 Strikingly, progression is meticulously choreographed, and each step is distinguished by the expression of a characteristic set of proteins.
Many of the changes observed in this series involve a key cell cycle step: the progression beyond the G1/S stage. Indeed, most of the cell cycle regulatory proteins found in this study to be deregulated are involved in the control of the G1/S transition, including cyclin D1, D3, cyclin E, CDK2, p16INK4a, p27KIP1, and p21CIP1. Although many of these changes are stage-specific, in general it seems that an increasing degree of expression of cyclins and CDKs, in conjunction with a loss of CDK inhibitors, facilitated the progression to advanced clinical and histological stages. These findings confirm previous observations in single G1/S markers in human malignant melanoma specimens6,7,9,22–24 and is supported by experimental observations in cell lines25,26 and animal models.27–30 Additionally, studies of familiar malignant melanoma have demonstrated mutations in genes regulating G1/S transition, including p16INK4a and CDK4.8,31,32
One of the most striking changes observed in this series is related to cyclin D1 protein. Cyclin D1 is negative in nevi (0 of 10) and expressed in 12 of 25 (48%) radial growth phase malignant melanomas. These findings strongly suggest a role for cyclin D1 in malignant melanoma development. Moreover, this study confirms the previous observation that cyclin D1 is increasingly expressed in malignant melanoma,6,9,33 and also indicates that the precise stage where cyclin D1 is increased is the radial growth phase, showing a trend (P = 0.08) in the metastatic stage. The diminishing expression toward the deep reticular dermis observed in vertical growth phase melanoma cases is consistent with the findings of Sauter and co-workers,9 in which the high level of cyclin D1 expression seemed to be confined to the superficial component of the tumor.
Vertical growth phase malignant melanoma shows a simultaneous increase in proliferation (Ki67) and a decrease in p27KIP1 protein expression (p 0.001) comparing with radial growth phase or benign lesions. Loss of p27KIP1 is required for the G1/S transition, as has been demonstrated in different experimental models.34 The study of SKP2, a protein involved in the ubiquitination and degradation of p27KIP1, shows a constant increase during the different stages of melanoma, which could explain the parallel increase in proliferation and decrease in p27KIP1 expression. A potential role of the extracellular matrix has also been proposed for inducing p27KIP1 degradation in melanoma.35
The significant loss of p16INK4a expression in the present series is correlated with advanced stages of malignant melanoma (metastatic disease). The p16INK4a protein exerts an inhibitory function in the cell cycle by acting at the level of the cyclin D/CDK4-Rb pathway. Inactivation of p16INK4a by homozygous deletions, point mutations, and promoter methylation has been involved in the genesis of malignant melanoma in familial and sporadic cases.10,23,26
Malignant melanomas also feature defective regulation of apoptosis in parallel with progression, as shown by the increasing overexpression of survivin from nevus (0 of 10) to radial (10 of 28, 36%), vertical growth phases (46 of 65, 71%), and metastases (41 of 66, 62%). The frequent expression of survivin in our series is consistent with the results of previous studies,11,36 suggesting that its activation is critical in resistance to apoptosis in malignant melanoma. This observation must be added to those already reported concerning APAF 1 in malignant melanoma.37
The transcription factor MUM-1/IRF4 also seems to play a role in melanoma progression, as previously suggested.18 Interestingly, our results reflect how this expression decreases according to tumor progression, from 85% (22 of 26) in radial and 55% (36 of 66) in vertical growth phase to 7% (5 of 70) in metastases.
Caveolin, a structural membrane-scaffolding protein, is involved in cell-signal transduction and in angiogenesis inhibition.38 It has been proposed that caveolin down-regulation may be associated with loss of apoptotic stimulus induced by caspase-339 and also with a significant role in neovascularization and, consequently, in development of metastatic disease.40–42 In this study, caveolin shows a loss of expression in malignant melanoma compared with nevus, which suggests that down-regulation of caveolin expression may contribute to malignant melanoma development.
This study also develops a predictor model for survival. The proteins included are p16INK4a, Ki67, p21CIP1, and Bcl-6. The data obtained from the overall survival model identify a group of patients with relatively short-term survival (<6 years) that seems to depend on an association between the absence of p16INK4a or the presence of Bcl-6 protein expression in conjunction with either high proliferation (Ki 67 >20) or positive p21CIP1. The absence of p16INK4a expression as a prognostic marker in cutaneous malignant melanoma has previously been noted.10 On the other hand, no data about the expression and significance of Bcl-6 protein in malignant melanomas have been reported to date. In this study, only a small number of invasive melanoma cases (5 of 64, 8%) expressed this protein, but it is noteworthy that all positive cases were strongly associated with poor prognosis in the univariate and the multivariate models. The inclusion of Breslow’s index in this predictive model does not produce any substantial change, thus showing that this index and the biomarkers included represent independent information. The independent series used to validate the model confirmed the adverse prognostic value of both the loss of p16INK4a and the acquisition of retinoblastoma markers.
In summary, a combination of tissue microarrays and clinical and pathological data can facilitate rapid characterization of candidate biomarkers of the development and regulation of progression in malignant melanoma tumors. The present analysis has highlighted the G1/S checkpoint abnormalities as being a critical step in the genesis and progression of malignant melanoma and has successfully identified new candidate proteins involved in melanoma genesis and progression as possible therapy targets.
Supplementary Material
Acknowledgments
We thank the Spanish National Tumor Bank Network, CNIO, and are particularly grateful to Alicia Maroto (12 de Octubre Hospital, Madrid), Dr. Pilar Fernández Segoviano (Getafe Hospital), Dr. José L. Sarasa (Fundación Jiménez Díaz, Madrid), Dr. Anastasio Serrano Egea and Dr. Jesús Razquin Murillo (Virgen de la Luz Hospital, Cuenca), and Dr. Teresa Rivera (Móstoles Hospital) for their help in providing specific tissue samples for this study. Dr. Miguel Angel Vidal (Centro de Investigaciones Biologicas-Consejo Superior de Investigaciones Cientificos) and Dr. Giovanna Roncador (CNIO) have kindly provided some of the antibodies used in this study. We also thank Gonzalo Herrero Tambo for his expertise and excellent assistance with schematic pictures, Dr. David Rimm and Ana Diez for their valuable guidance with TMA construction, and Dr. Phil Mason for his help with the final version of the manuscript.
Footnotes
Address reprint requests to Dr. Miguel A. Piris, Programa de Patología Molecular, Centro Nacional de Investigaciones Oncológicas, C/Melchor Fernández Almagro, 3, Madrid 28029, Spain. E-mail: mapiris@cnio.es.
Supported by grants from the Ministerio de Sanidad y Consumo Fondo de Investigaciones Sanitaries (FIS 01/ 0037–03) and the Ministerio de Ciencia y Tecnologia (MCYT) (Bio2000 CO275/01 and 02), Spain. S. A. is supported by a grant from the Ministerio de Sanidad y Consumo (FIS 01/9269) and the Centro Nacional de Investigaciones Oncologies (CNIO).
References
- Urist MM, Karnell LH. The National Cancer Data Base: report on melanoma. Cancer. 1994;74:782–788. doi: 10.1002/1097-0142(19940715)74:2<782::aid-cncr2820740236>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Sober AJ, Lew RA, Koh HK, Barnhill RL. Epidemiology of cutaneous melanoma: an update. Dermatol Clin. 1991;9:617–629. [PubMed] [Google Scholar]
- Koh HK. Cutaneous melanoma. N Engl J Med. 1991;325:171–182. doi: 10.1056/NEJM199107183250306. [DOI] [PubMed] [Google Scholar]
- Geller AC, Miller DR, Annas GD, Demierre MF, Gilchrest BA, Koh HK. Melanoma incidence and mortality among US whites, 1969–1999. JAMA. 2002;288:1719–1720. doi: 10.1001/jama.288.14.1719. [DOI] [PubMed] [Google Scholar]
- Crowson AN, Margo CM, Mihn MC. The melanocytic proliferations. New York: Wiley-Liss; A Comprehensive Textbook of Pigmented Lesions. 2001:pp 315–332, 501–513. [Google Scholar]
- Maelandsmo GM, Florenes VA, Hovig E, Oyjord T, Engebraaten O, Holm R, Borresen AL, Fodstad O. Involvement of the pRb/p16/cdk4/cyclin D1 pathway in the tumorigenesis of sporadic malignant melanomas. Br J Cancer. 1996;73:909–916. doi: 10.1038/bjc.1996.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florenes VA, Faye RS, Maelandsmo GM, Nesland JM, Holm R. Levels of cyclin D1 and D3 in malignant melanoma: deregulated cyclin D3 expression is associated with poor clinical outcome in superficial melanoma. Clin Cancer Res. 2000;6:3614–3620. [PubMed] [Google Scholar]
- Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, Stoppa-Lyonnet D, Benard J, Bressac-de Paillerets B. Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France: The French Familial Melanoma Study Group. Hum Mol Genet. 1998;7:209–216. doi: 10.1093/hmg/7.2.209. [DOI] [PubMed] [Google Scholar]
- Sauter ER, Yeo UC, von Stemm A, Zhu W, Litwin S, Tichansky DS, Pistritto G, Nesbit M, Pinkel D, Herlyn M, Bastian BC. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62:3200–3206. [PubMed] [Google Scholar]
- Straume O, Sviland L, Akslen LA. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin Cancer Res. 2000;6:1845–1853. [PubMed] [Google Scholar]
- Grossman D, McNiff JM, Li F, Altieri DC. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol. 1999;113:1076–1081. doi: 10.1046/j.1523-1747.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Nocito A, Kononen J, Kallioniemi OP, Sauter G. Tissue microarrays (TMAs) for high-throughput molecular pathology research. Int J Cancer. 2001;94:1–5. doi: 10.1002/ijc.1385. [DOI] [PubMed] [Google Scholar]
- Garcia JF, Camacho FI, Morente M, Fraga M, Montalban C, Bellas TA, Castano A, Diez A, Flores T, Martin C, Martinez MA, Mazorra F, Menarguez J, Mestre MJ, Mollejo M, Saez AI, Sanchez L, Piris MA. Hodgkin and Reed-Sternberg cells harbor alterations in the major tumor suppressor pathways and cell-cycle checkpoints: analyses using tissue microarrays. Blood. 2003;101:681–689. doi: 10.1182/blood-2002-04-1128. [DOI] [PubMed] [Google Scholar]
- Breslow A. Thickness, cross-sectional areas, and depth of invasion in the prognosis of cutaneous melanoma. Ann Surg. 1970;172:902–908. doi: 10.1097/00000658-197011000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WH, Jr, Elder DE, Guerry Dt, Braitman LE, Trock BJ, Schultz D, Synnestvedt M, Halpern AC. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst. 1989;81:1893–1904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate and practical and powerful approach to multiple testing. J R Stat Soc. 1995;B57:289–300. [Google Scholar]
- Natkunam Y, Warnke RA, Montgomery K, Falini B, van De Rijn M. Analysis of MUM1/IRF4 protein expression using tissue microarrays and immunohistochemistry. Mod Pathol. 2001;14:686–694. doi: 10.1038/modpathol.3880373. [DOI] [PubMed] [Google Scholar]
- Kielhorn E, Provost E, Olsen D, D’Aquila TG, Smith BL, Camp RL, Rimm DL. Tissue microarray-based analysis shows phospho-β-catenin expression in malignant melanoma is associated with poor outcome. Int J Cancer. 2003;103:652–656. doi: 10.1002/ijc.10893. [DOI] [PubMed] [Google Scholar]
- Polsky D, Bastian BC, Hazan C, Melzer K, Pack J, Houghton A, Busam K, Cordon-Cardo C, Osman I. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Res. 2001;61:7642–7646. [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- Sauroja I, Smeds J, Vlaykova T, Kumar R, Talve L, Hahka-Kemppinen M, Punnonen K, Jansen CT, Hemminki K, Pyrhonen S. Analysis of G(1)/S checkpoint regulators in metastatic melanoma. Genes Chromosomes Cancer. 2000;28:404–414. doi: 10.1002/1098-2264(200008)28:4<404::aid-gcc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Sparrow LE, Eldon MJ, English DR, Heenan PJ. p16 and p21WAF1 protein expression in melanocytic tumors by immunohistochemistry. Am J Dermatopathol. 1998;20:255–261. doi: 10.1097/00000372-199806000-00006. [DOI] [PubMed] [Google Scholar]
- Florenes VA, Maelandsmo GM, Kerbel RS, Slingerland JM, Nesland JM, Holm R. Protein expression of the cell-cycle inhibitor p27Kip1 in malignant melanoma: inverse correlation with disease-free survival. Am J Pathol. 1998;153:305–312. doi: 10.1016/S0002-9440(10)65572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Li G, Tron VA, Trotter MJ, Ho VC. Expression of cell cycle regulators in human cutaneous malignant melanoma. Melanoma Res. 1999;9:148–154. doi: 10.1097/00008390-199904000-00006. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Hill SP, Evans-Whipp TJ, Chin L, Orlow SJ, Easty DJ, Cheong SC, Beach D, DePinho RA, Bennett DC. p16(Ink4a) in melanocyte senescence and differentiation. J Natl Cancer Inst. 2002;94:446–454. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- Sotillo R, Garcia JF, Ortega S, Martin J, Dubus P, Barbacid M, Malumbres M. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci USA. 2001;98:13312–13317. doi: 10.1073/pnas.241338598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang FC, Merlino G, Chin L. Genetic dissection of melanoma pathways in the mouse. Semin Cancer Biol. 2001;11:261–268. doi: 10.1006/scbi.2000.0376. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol. 2001;21:2144–2153. doi: 10.1128/MCB.21.6.2144-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietze MK, Chin L. Murine models of malignant melanoma. Mol Med Today. 2000;6:408–410. doi: 10.1016/s1357-4310(00)01781-0. [DOI] [PubMed] [Google Scholar]
- Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, Hayward N, Dracopoli NC. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- Walker GJ, Flores JF, Glendening JM, Lin AH, Markl ID, Fountain JW. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer. 1998;22:157–163. doi: 10.1002/(sici)1098-2264(199806)22:2<157::aid-gcc11>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Sanchez-Beato M, Camacho FI, Martinez-Montero JC, Saez AI, Villuendas R, Sanchez-Verde L, Garcia JF, Piris MA. Anomalous high p27/KIP1 expression in a subset of aggressive B-cell lymphomas is associated with cyclin D3 overexpression: p27/KIP1-cyclin D3 co-localization in tumor cells. Blood. 1999;94:765–772. [PubMed] [Google Scholar]
- Henriet P, Zhong ZD, Brooks PC, Weinberg KI, DeClerck YA. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc Natl Acad Sci USA. 2000;97:10026–10031. doi: 10.1073/pnas.170290997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodino C, Cesinaro AM, Ottani D, Fantini F, Giannetti A, Trentini GP, Pincelli C. Communication: expression of the novel inhibitor of apoptosis survivin in normal and neoplastic skin. J Invest Dermatol. 1999;113:415–418. doi: 10.1046/j.1523-1747.1999.00711.x. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273:5419–5422. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- Liu J, Lee P, Galbiati F, Kitsis RN, Lisanti MP. Caveolin-1 expression sensitizes fibroblastic and epithelial cells to apoptotic stimulation. Am J Physiol. 2001;280:C823–C835. doi: 10.1152/ajpcell.2001.280.4.C823. [DOI] [PubMed] [Google Scholar]
- Liu J, Razani B, Tang S, Terman BI, Ware JA, Lisanti MP. Angiogenesis activators and inhibitors differentially regulate caveolin-1 expression and caveolae formation in vascular endothelial cells: angiogenesis inhibitors block vascular endothelial growth factor-induced down-regulation of caveolin-1. J Biol Chem. 1999;274:15781–15785. doi: 10.1074/jbc.274.22.15781. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem. 1997;272:16374–16381. doi: 10.1074/jbc.272.26.16374. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA. 1995;92:1381–1385. doi: 10.1073/pnas.92.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.