

Abstract
Human prion diseases are rare fatal neurodegenerative conditions that occur as acquired, familial, or idiopathic disorders. A key event in their pathogenesis is the accumulation of an altered form of the prion protein, termed PrPSc, in the central nervous system. A novel acquired human prion disease, variant Creutzfeldt-Jakob disease, is thought to result from oral exposure to the bovine spongiform encephalopathy agent. This disease differs from other human prion diseases in its neurological, neuropathological, and biochemical phenotype. We have used immunohistochemistry and Western blot techniques to analyze the tissue distribution and biochemical properties of PrPSc in peripheral tissues in a unique series of nine cases of variant Creutzfeldt-Jakob disease. We have compared this with the distribution and biochemical forms found in all of the major subtypes of sporadic Creutzfeldt-Jakob disease and in a case of iatrogenic Creutzfeldt-Jakob disease associated with growth hormone therapy. The results show that involvement of the lymphoreticular system is a defining feature of variant Creutzfeldt-Jakob disease, but that the biochemical isoform of PrPSc found is influenced by the cell type in which it accumulates.
The human prion diseases or transmissible spongiform encephalopathies are unique in a number of important respects. Firstly, they occur as familial, acquired, or sporadic forms, each of which appears to be transmissible under certain circumstances. Secondly, the responsible agent is notoriously resistant to standard forms of decontamination. Thirdly, they are closely associated with accumulation in the central nervous system of an abnormally folded isoform of a host-encoded glycoprotein, termed PrPSc, which the prion hypothesis equates with the infectious agent.1 PrPSc can be operationally defined by its partial resistance to proteases and as such, it is referred to as PrPres. The normal cellular form of the protein (PrPC) is susceptible to protease degradation and termed PrPsens.
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common human prion disease occurring world wide with a frequency of approximately one per million of the population per annum. Although the cause is unknown, a random stochastic event resulting in the chance conversion of normal cellular PrP (PrPC) to the pathogenic isoform (PrPSc) and its subsequent self-propagation is one possibility. Familial forms of human prion disease are all tightly associated with mutations in the coding sequence of the gene encoding PrP (PRNP), resulting in the formation of a mutant protein that is hypothesized to make conversion to the abnormal isoform considerably more likely. Human prion disease can also be acquired, orally through endocannibalism (kuru), or by medical exposure to contaminated human tissues or derived products (iatrogenic Creutzfeldt-Jakob disease or iCJD) by intracranial, intraocular, or subcutaneous and intramuscular routes.2 Susceptibility, and in some instances disease phenotype, in acquired, familial, and sporadic human prion diseases are substantially influenced by the methionine/valine polymorphism at codon 129 of the PRNP gene.3–6
In 1996 a new variant of Creutzfeldt-Jakob disease (vCJD) was described in the United Kingdom7 that affected young individuals who presented with distinct clinical symptoms8 and a highly stereotyped neuropathology.9 All available evidence points to this disease being the first documented zoonotic human prion disease resulting from exposure (probably oral) to the bovine spongiform encephalopathy (BSE) agent.7,10–13 To date 143 cases of vCJD have been diagnosed in the United Kingdom, but given the potentially lengthy incubation periods involved in acquired prion diseases, the eventual number of cases of vCJD remain hard to predict with any great certainty.14–16
Peripheral tissue involvement in prion disease, as judged by PrPSc accumulation in elements of the lymphoreticular system, is common in sheep scrapie17 and in rodent models;18 however, this has not been a consistently reported feature of sporadic or familial human prion diseases.19,20 In contrast PrPSc accumulates in the tonsil, spleen, lymph node, and appendix in vCJD21,22 and this accumulation, in the case of appendix may precede the clinical onset by several years.23 The presence of PrPSc in peripheral tissues in vCJD may offer diagnostic possibilities.21 Furthermore, because the tonsil and spleen from these cases contain measurable infectivity in mouse bioassay,24 peripheral tissues from those incubating vCJD may also present a risk of iatrogenic transmission. Understanding peripheral pathogenesis is also important in identifying potential therapeutic targets such as peripheral replication, which could present an attractive route to prophylactic treatments.25
We have examined a wide variety of tissues (n = 14) in a series of sCJD (n = 7) and vCJD (n = 9) cases using a combination of Western blot and immunohistochemistry to detect the presence of the disease-associated form of PrP. Our data shows a consistent pattern of peripheral involvement in vCJD that distinguishes it from both sCJD and, interestingly, peripherally acquired iCJD.
Materials and Methods
Cases and Tissue Specimens
Cases of CJD were selected for this study on the basis of the availability of fixed and frozen tissue specimens from a wide range of organs retained at autopsy and the existence of consent for tissue retention and research. Local ethical approval for the use of the material for research has been obtained. All autopsy cases were of UK origin. The brain from each case had previously been examined histologically and biochemically and a definite diagnosis of variant, sporadic, or iatrogenic CJD reached by established criteria.9,26 The case of iCJD was associated with growth hormone therapy. The protease-resistant prion protein (PrPres) isotype found in brain was classified type 1, 2A, or 2B as previously described9 according to accepted nomenclature of Parchi and colleagues5,27,28 A case of clinically possible CJD that was given a final diagnosis of Lewy body dementia was included as a control for neurodegenerative disease without prion involvement and the absence of PrPres from the brain. The polymorphic status of codon 129 of the prion protein gene PRNP of each case was determined by restriction fragment length polymorphism.8 The biochemical and genetic data for each case is summarized in Table 1. All possible codon 129 genotype and PrPres isotype combinations are represented in the sCJD cases chosen (MM1, MV1, MM2A, MV2A, VV2A) except the very rare VV1 class. All vCJD cases were methionine homozygotes and had a type 2B PrPres isotype in brain tissue. The tonsil biopsy material was from a case of vCJD diagnosed outside the UK but thought likely to have resulted from a British exposure to BSE and has been described previously.29
Table 1.
Key to the Diagnostic Classification of the Cases Analyzed
| Case identifier | Diagnosis | PRNP codon 129 genotype | Brain PrPres isotype |
|---|---|---|---|
| C1 | Lewy body dementia | MM | 0 |
| S1 | Sporadic CJD | MV | 1 |
| S2 | Sporadic CJD | MM | 2A |
| S3 | Sporadic CJD | MM | 1 |
| S4 | Sporadic CJD | MV | 2A |
| S5 | Sporadic CJD | VV | 2A |
| S6 | Sporadic CJD | MM | 1 |
| S7 | Sporadic CJD | MM | 1 |
| V1 | Variant CJD | MM | 2B |
| V2 | Variant CJD | MM | 2B |
| V3 | Variant CJD | MM | 2B |
| V4 | Variant CJD | MM | 2B |
| V5 | Variant CJD | MM | 2B |
| V6 | Variant CJD | MM | 2B |
| V7 | Variant CJD | MM | 2B |
| V8 | Variant CJD | MM | 2B |
| V9 | Variant CJD | MM | 2B |
| I1 | Iatrogenic CJD | MV | 2A |
Immunolocalization
PrP was localized in sections of formalin-fixed or periodate-lysine paraformaldehyde-fixed, formic acid-treated tissue by immunohistochemistry. Five-μm paraffin sections were mounted on Superfrost plus slides. PrP immunohistochemistry was performed using the mouse monoclonal antibodies, KG9 (IAH, Compton, UK) and 3F4 (DAKO, Ely, UK) in a protocol that distinguishes PrPSc from the normal cellular form of PrP. Before immunolabeling, sections were taken to water and formalin pigment removed with saturated picric acid. Endogenous peroxidase activity was blocked with 3% H2O2 in methanol for 30 minutes. Sections were pretreated by autoclaving at 121°C in distilled water for 10 minutes, followed by immersion in 96% formic acid for 5 minutes and digestion with proteinase K, diluted to 10 μg/ml in phosphate-buffered saline for 5 minutes. After blocking with normal rabbit serum, nonspecific binding of avidin and biotin was blocked using an avidin/biotin blocking kit (Vector SP-2001; Vector Laboratories, Peterborough, UK), before incubating with the primary antibody KG9 (KG9: 1/20,000 in normal rabbit serum). Selected cases were also immunostained using the same protocol but substituting the monoclonal antibody 3F4 (1:2000 in normal rabbit serum) for KG9. Immunolabeling was completed using the catalyzed signal amplification (CSA) amplification system (DAKO) that is superior in terms of sensitivity to most other immunohistochemical detection systems.30 Labeling was visualized using diaminobenzidine and lightly counterstained with hematoxylin. CR2 (the receptor for the C3d complement C3 fragment) was visualized in certain of these same tissues using the monoclonal antibody CD21 (DAKO). For this antigen, sections were taken to water, endogenous peroxidase activity blocked with 3% H2O2 in methanol, and the material partially digested with trypsin. Subsequent detection used the Vectastain elite avidin/biotin kit (Vector Laboratories).
Image Analysis
For the quantitative assessment of PrP immunostaining, adjacent sections of positive lymphoid follicles from selected organs were cut and stained by immunohistochemistry for KG9 and CD21. Sections were analyzed on a Leica DMR microscope (Leica Microscopy Systems, Milton Keynes, UK) fitted with a Sony 3CCD color video camera (model XC-003P) having a 6.00 mm × 4.96 mm sensing area CCD and 752(H) × 582(V) effective picture elements. A motorized stage (Prior Scientific Instruments Ltd., Cambridge, UK) was fitted to the microscope to provide software control of position and focus. Software developed using Leica Advanced QUIPS, a macro programming facility, was used for automatic image capture, processing, and analysis. For each section, 25 overlapping images were acquired and aligned to form a composite view of the characteristic neuropathology. From this view, four component images were selected at random and those stained for CD21 used to count the number of lymphoid follicles containing follicular dendritic cells. Component images from the adjacent section stained for PrPres using the KG9 antibody were then examined to count the number of follicles that also stained positively for PrPres. All counting was performed by computer-controlled image analysis. The data obtained for each organ was grouped to give an average of the number of stained follicles per section examined.
Western Blot Analysis
Frozen tissues were analyzed by a variation of the Western blotting protocol described previously.9,31 Briefly, 10% w/v extracts were made in nondenaturing conditions by homogenization. The cleared lysate was digested with 50 μg/ml of proteinase K (VWR, Poole, UK) for 1 hour at 37°C. Brain samples were run as twofold serial dilutions of 10% extracts. Proteinase K-treated peripheral tissue extracts, usually 200 μl in the first instance were then concentrated by centrifugation at 21,000 × g for 1 hour at 4°C.32 The resultant pellet was resuspended in denaturing buffer and analyzed by Western blotting using the monoclonal antibody 3F4 (DAKO), enhanced chemiluminescence, and exposure to X-ray film.33 Where peripheral tissue samples were found to give too strong a signal after concentration, the volume of extract used was scaled down appropriately. In preliminary experiments 10% brain extracts were separated by centrifugation into a supernatant and pellet fraction before proteolytic digestion. The pellet was resuspended in the original starting volume of extraction buffer and equivalent amounts of supernatant and pelleted material digested with proteinase K. In separate experiments the efficiency of recovery of PrPres was also investigated by diluting 5 μl of proteinase K-treated 10% brain extract into 200 μl of extraction buffer and recovery by centrifugation using the centrifugation protocol described above.
Results
Sedimentation Properties of PrP from the CJD Brain
Protease-resistance and aggregation are features of PrPSc, which distinguish it from the normal cellular form, PrPC. To test whether centrifugation separates protease-sensitive PrPC (PrPsen) and protease-resistant PrPSc (PrPres) from human brain we centrifuged extracts of frontal cortex from sCJD and vCJD. The supernatant and pellet fractions were analyzed separately with and without digestion with proteinase K (Figure 1). Western blot analysis shows the presence of PrP in both the supernatant and pellet fractions. Treatment with proteinase K shows an absence of detectable PrP in the supernatant fraction. In contrast the pelleted material contains readily detectable protease-resistant PrP. Because these Western blots were loaded with equivalent amounts of tissue extracts (∼500 μg of brain) the results suggest that a simple centrifugation at 21,000 × g effects a quantitative separation of PrPres from PrPsen. PrPres found in the cases of sCJD and vCJD used here differ both in the fragment size and glycosylation ratio (PrPres isotype 1 and 2B, respectively). The efficiency of centrifugal recovery and the conservation of the PrPres isotype were determined by analysis of sCJD and vCJD brain extracts that had been diluted by a factor of 40 in extraction buffer and then concentrated by centrifugation. The pellets were resuspended in the starting volume of extraction buffer then compared with the original extract. Loading of Western blots with equivalent amounts of starting brain extracts (∼500 μg) demonstrates that centrifugal recovery is efficient (Figure 1). Moreover the procedure does not alter the mobility of the fragments. Neither does there seem to be any selectivity for the recovery of any one of the three glycoforms present. Centrifugal concentration therefore offers a simple, efficient method for the concentrated of PrPres from tissues in which it may be present at low abundance and allows isotype analysis of the concentrated material.
Figure 1.

Western blot analysis of standard frontal cortex extracts from sporadic CJD type 1 (1) and variant CJD type 2B (2B). Samples were separated by centrifugation at 21,000 × g into supernatant (S) or pellet (P) fraction. These two fractions were then analyzed before (−) or after (+) digestion with proteinase K as shown in the top and middle panel. In a separate experiment, the same extracts were digested with proteinase K and an aliquot diluted by a factor of 40, collected by centrifugation, and resuspended in the original volume. Analysis of equivalent amounts of the original (−) and the diluted/concentrated samples (+) are shown at the bottom. The 21-kd nonglycosylated fragment diagnostic of type 1 PrPres and the 19-kd nonglycosylated fragment diagnostic of type 2 PrPres are marked in the bottom panel. The positions of the three glycoforms; diglycosylated (D), monoglycosylated (M), and nonglycosylated (N) are also indicated.
Western Blot Analysis of Tonsil by Centrifugal Concentration
Western blot analysis of representative brain and tonsil samples show that PrP is abundant in all samples of frontal cortex (Figure 2A) but that seen in Lewy body dementia is sensitive to protease digestion (Figure 2B). A significant proportion of the signal remains in frontal cortex samples from each case of CJD indicating the presence of abundant PrPres in the brain (Figure 2B). The signal from untreated tonsil samples is considerably lower than that seen in brain (Figure 2C) and the proportion that appears resistant to protease digestion (Figure 2D) is less abundant than that seen in the corresponding brain samples (Figure 2B). Note that the same amount of brain and tonsil tissue (∼1 mg) was loaded in each lane but that the exposure time for brain samples (Figure 2, A and B) was reduced (by a factor of 36) to the minimum possible (5 seconds) in an attempt to avoid signal saturation. These data suggest a gross discrepancy between the amounts of PrP in brain and tonsil and the proportion of that PrP that is resistant to proteolytic degradation. Positive signals from tonsil are most obvious in the cases of vCJD but faint bands of the same mobility are also present in all other lanes (Figure 2D). To distinguish between genuine aggregated protease-resistant PrPSc and potentially artifactual signals seen in tonsil, protease-treated tonsil extracts were enriched for PrPres by centrifugal concentration of 80 μl of a 10% extract and this was compared with 10 μl of unconcentrated 10% extract (Figure 3). The three PrPres glycoforms with similar mobilities to those found in brain samples were seen in samples of vCJD tonsil but no signals were seen in tonsil samples from sCJD and iCJD (Figure 3B). Centrifugal concentration, usually of 200 μl was then routinely used to analyze all other available peripheral tissue samples. Where possible semiquantitative estimates were made of the amount of PrPres present in peripheral tissue extracts compared to that found in a standard sample of vCJD frontal cortex. The principle is illustrated in Figure 4, in which 200 μl of concentrated tonsil extract from vCJD case V7 was analyzed alongside a twofold serial dilution series (starting with 5 μl) of brain extract from the same case. Visual inspection indicates that the signal intensity from the tonsil lies between the fourth (1:16) and fifth (1:32) serial dilution of brain. Hence, 20 mg tissue equivalents of tonsil gave a signal intermediate between 31 μg and 15 μg tissue equivalents of brain, indicating that the abundance of PrPres in the tonsil sample was between 0.08% and 0.16% of that found in the frontal cortex sample from the same case.
Figure 2.

Western blot analysis of frontal cortex (A and B) and tonsil (C and D) analyzed with (B and D) or without (A and C) previous digestion with proteinase K from cases C1 (Lewy body dementia), S5, S6, S7 (sCJD) V4, V5, V7 (vCJD), and I1 (iCJD). All blots were loaded with an equivalent amount of tissue extract and processed together but the exposure time of A and B (frontal cortex) was 1/36th of that for C and D (tonsil).
Figure 3.

Western blot analysis of PrPres in tonsil samples from cases C1 (Lewy body dementia), S5, S6, S7 (sCJD) V4, V5, V7 (vCJD), and I1 (iCJD) before (A) and after (B) centrifugal enrichment for PrPres from an eightfold larger volume of extract. +, Indicates a lane loaded with a one-tenth volume of the frontal cortex from vCJD case V7. Both blots were processed together and exposed for the same length of time.
Figure 4.

Western blot analysis of a dilution series of cerebral cortex (CC) and concentrated tonsil (To) extract from a case of vCJD (V7). The serial dilution was by a factor of two, starting with a loading of 5 μl of a 10% extract of frontal cortex in the first lane. The lane of tonsil represents 200 μl of a 10% extract enriched (×40) by centrifugal concentration for PrPres.
Tissue Distribution of PrPres
Fourteen different tissues from 18 cases were selected for study. The complete set of tissues was not available in the form of both fixed and frozen tissue for each individual case; however, all available tissues for this data set were analyzed resulting in 158 tissues examined by Western blot and the same number examined by immunohistochemistry. The results of the combined Western blot and immunohistochemical analysis of these tissues are shown in Table 2. The pattern of PrPres accumulation falls into discrete groups:
Table 2.
Summary of the Results of Immunohistochemical (IHC) and Western Blot (WB) Analysis of Prpres in Cerebral Cortex (CC), Trigeminal Ganglion (TG), Dorsal Root Ganglion (DRG), Peripheral Nerve (PN), Tonsil (To), Spleen (Sp), Cervical Lymph Node (LN), Appendix (Ap), Adrenal Gland (Ad), Kidney (Ki), Liver (Li), Lung (Lu), Heart (He), and Muscle (Mu) in Cases of Lewy Body Dementia (LBD), Sporadic CJD (sCJD), Variant CJD (vCJD), and Iatrogenic CJD (iCJD)
| Tissue | LBD (C1)
|
sCJD (S1–7)
|
vCJD (V1–9)
|
iCJD (I1)
|
||||
|---|---|---|---|---|---|---|---|---|
| IHC | WB | IHC | WB | IHC | WB | IHC | WB | |
| CC | 0/1 | 0/1 | 7/7 | 7/7 | 9/9 | 9/9 | 1/1 | 1/1 |
| TG | 0/1 | 0/1 | 5/7 | 2/3 | 8/8 | 6/7 | 0/0 | 1/1 |
| DRG | 0/1 | 0/1 | 3/6 | 0/7 | 6/6 | 2/9 | 0/0 | 0/1 |
| PN | 0/1 | 0/1 | 0/7 | 0/7 | 0/8 | 0/8 | 0/1 | 0/1 |
| To | 0/1 | 0/1 | 0/5 | 0/7 | 9/9 | 7/8 | 0/1 | 0/1 |
| Sp | 0/1 | 0/1 | 0/6 | 0/6 | 8/9 | 8/9 | 0/1 | 0/1 |
| LN | 0/0 | 0/1 | 0/0 | 0/7 | 1/1 | 7/9 | 0/0 | 0/1 |
| Ap | 0/1 | 0/1 | 0/5 | 0/3 | 6/6* | 0/8† | 0/1 | 0/1 |
| Ad | 0/1 | 0/1 | 0/5 | 0/3 | 0/7 | 0/6 | 0/1 | 0/1 |
| Ki | 0/1 | 0/1 | 0/7 | 0/3 | 0/7 | 0/5 | 0/0 | 0/1 |
| Li | 0/1 | 0/1 | 0/6 | 0/3 | 0/9 | 0/7 | 0/1 | 0/1 |
| Lu | 0/1 | 0/1 | 0/7 | 0/3 | 0/9 | 0/6 | 0/1 | 0/1 |
| He | 0/1 | 0/1 | 0/7 | 0/3 | 0/8 | 0/5 | 0/1 | 0/1 |
| Mu | 0/1 | 0/1 | 0/7 | 0/7 | 0/9 | 0/9 | 0/1 | 0/1 |
The results are expressed as the number of PrPres-positive cases as proportion of the total number of cases tested for each tissue and technique. Categories in which any positive result was obtained are shown in bold.
Three cases with appendix tissue were not assessable, since fibrosis had obliterated the lymphoid follicles.
See Figure 8.
Absence of PrPres Accumulation in Any Tissue
PrPres was not detectable in any peripheral tissue or brain from the case of Lewy body dementia by Western blotting or immunohistochemistry.
PrPres Accumulation Restricted to the Nervous System
PrPres was detectable in the brain in all cases of CJD, whether sporadic or iatrogenic. In the sCJD cases, the patterns of PrPres accumulation in the brain varied according to the PRNP codon 129 genotype and PrPres isotype, as previously described.27 PrPres was also detectable in the trigeminal ganglia in the majority of the sCJD cases (Table 2, Figure 5). PrPres positivity in the dorsal root ganglia was seen in half of the sCJD cases and was absent from the case of iCJD (Table 2, Figure 5). Peripheral nerve accumulation of PrPres was not seen by either immunohistochemistry or by Western blotting.
Figure 5.

Immunohistochemical analysis of PrP showing intense staining of dorsal root ganglion cells in variant CJD, case V2 (A); staining of occasional dorsal root ganglion cells in sporadic CJD, case S1 (B); no reactivity in the dorsal root ganglion in another sporadic CJD case of different genotype, case S3 (C); variable staining of ganglion cells in the trigeminal ganglion in sporadic CJD, case S4 (D); intense immunoreactivity within germinal centers in the tonsil in variant CJD, case V3 (E); no reactivity in the tonsil in sporadic CJD, case S1 (F); staining of germinal centers in variant CJD in the spleen, appendix, and an ileal Peyer’s patch, respectively, case V2 (G–I); patchy staining within a small germinal center in the thymus in variant CJD, case V4 (J). The monoclonal antibody KG9 was used in all images. Original magnifications: ×400 (A, B, J); ×200 (C, D, G, H); ×100 (E, F, I).
PrPres Accumulation in the Nervous System and Lymphoreticular System
In addition to the nervous system (including trigeminal and dorsal root ganglia), PrPres was detected by immunohistochemistry and Western blot in the majority of cases of vCJD in the tonsil, spleen, and cervical lymph node (Table 2). PrPres was also detectable in germinal centers within the submucosa of the appendix in six of six vCJD cases, exclusively by immunohistochemistry. The remaining three cases with appendix tissue were not assessable because fibrosis in the wall had obliterated the lymphoid follicles. The dorsal root ganglia were positive by immunohistochemistry in all cases of vCJD however only a minority of these were positive by Western blotting (Table 2). Peripheral nerve accumulation of PrPres was not identified in vCJD. In common with sCJD and iCJD, no evidence was seen of PrPres accumulation in adrenal, kidney, lung, heart, or skeletal muscle in vCJD.
Heterogeneity of Peripheral Tissue Involvement
The accumulation of PrPres is widespread in the gray matter of the central nervous system. Although notable morphological features such as plaques can occur, the pattern of staining suggests the involvement of many if not most neurons. In contrast, immunohistochemical staining in peripheral tissues is clearly heterogeneous with the majority of cells unlabeled and a variable minority containing accumulations of PrPres (Figure 5). In the lymphoreticular system the positivity occurs within germinal centers in cells with morphological features consistent with follicular dendritic cells (Figure 5). To estimate the extent of PrPres-positive follicle involvement, counts were made of follicles immunostained with either KG9 for PrPres and CD21 (as a marker of follicular dendritic cells) in tonsil, lymph node, appendix, thymus, and in Peyer’s patches in the gastrointestinal tract from vCJD cases V2, V3, V4, V5, and V7 (Table 3). When expressed as the average number of PrPres-positive follicles found per section, the greatest numbers were found in tonsil followed by lymph node and gastrointestinal tract. Fewer were found in the thymus and the lowest number was found in the appendix. When these numbers were corrected by co-localization with CD21-positive follicles, lymph node had the greatest number followed by gastrointestinal tract, tonsil, and then thymus.
Table 3.
Average Numbers of PrP-Positive (KG9) and CD21-Positive Follicles in Lymph Node (LN), Tonsil (To), Gastrointestinal Tract (GI), Thymus (Thy), and Appendix (Ap) Found in Cases of Variant CJD
| Tissue (number of cases tested) | Average number of KG9-positive follicles per section scored (number of sections scored) | Average number of CD21-positive follicles per section (number of sections scored) | Average number of KG9-positive follicles per section/average number of CD21-positive follicles per section |
|---|---|---|---|
| LN (4) | 1.33 (36) | 0.92 (36) | 1.45 |
| GI (5) | 1.21 (52) | 1.40 (52) | 0.86 |
| To (4) | 3.40 (25) | 4.00 (24) | 0.85 |
| Thy (3) | 0.85 (20) | 1.40 (20) | 0.61 |
| Ap (3) | 0.18 (11) | 0 (12) | — |
The Level of PrPres in Peripheral Tissues
The level of PrPres found in vCJD peripheral tissue samples was estimated by comparison of the relative signal strength of concentrated peripheral tissue samples with a dilution series of a standard brain extract from a case of vCJD (V5). The amount of peripheral tissue extract to be concentrated often required readjustment to bring the unknown signal within the range of the dilution series of brain. Examples of these analyses are shown (Figures 4 and 6). The particular blots shown in Figure 6 were selected to illustrate specific points regarding PrPres isotypes and were not necessarily those from which determinations were made. A determination from the complete series of Western blot results could be made in all seven positive tonsils and all seven positive lymph nodes and these ranged from 0.01 to 10% of the level found in the vCJD brain standard. The range in the six positive vCJD trigeminal ganglia was similar (<0.25 to 25%). The two positive trigeminal ganglia from cases of sCJD fell in the middle of this range.
Figure 6.

Western blot analysis of concentrated extracts of trigeminal ganglion (TG), tonsil (To), and cervical lymph node (LN). The cases are identified as S (sporadic CJD), V (variant CJD), and I (iatrogenic CJD) and the case number using the codes shown in Table 1. A twofold dilution series of frontal cortex from case V5 is shown extending left to right (undiluted, 1:2, 1:4, 1:8). The signals from the tonsil samples shown in B are not comparable to any lanes of the corresponding frontal cortex dilution series and the estimation of PrPres concentration for these particular samples was made from concentrates using less starting tonsil extract.
PrPres Isotypes in Peripheral Tissues
The PrPres isotypes found in peripheral tissues were compared with those found in samples from the brain. A systematic effect of tissue type was found. PrPres in the vCJD lymphoreticular system differed in three respects from that seen in vCJD brain. The glycosylation ratio was further accentuated toward the diglycosylated form (top band) in tonsil, spleen, and lymph node. This is most clearly visible in Figure 6B where the tonsil V5 is shown with a dilution series of brain tissue from the same case. It is also clearly evident in the lymph node samples shown in Figure 6, C and D. Secondly the monoglycosylated form (middle band) was more complex in lymph node and tonsil than that found in brain. When optimally resolved it appeared to be composed of three distinct bands each with a greater mobility than the predominant mononglycosylated (middle) band found in the vCJD brain profile. This is most clearly seen in lymph node from vCJD case V7 shown in Figure 6D. Lastly the nonglycosylated (bottom band) in vCJD tonsil and lymph node migrated faster than the type 2 mobility of vCJD brain and trigeminal ganglia samples (Figure 6D). These differences are not related to postmortem degradation because they are also seen in tonsil biopsies (Figure 7). The PrPres isotype in some trigeminal ganglia also deviated from those seen in samples of the corresponding brain. For example, the glycoform ratio of trigeminal ganglion from vCJD case V7 is the type 2B characteristic of vCJD brain (Figure 6, A and D). However the glycoform ratio of trigeminal ganglion from vCJD case V6 is dominated by monoglycosylated and nonglycosylated forms (middle and bottom bands) and resembles a sporadic type 2A rather than a variant CJD brain isoform pattern (Figure 6, A and D). It was also noted that the trigeminal ganglion in the case of iCJD had a type 1 mobility whereas the brain from this case had been widely sampled previously and consistently found to have a type 2A PrPres isotype (Figure 6A and Figure 2B).
Figure 7.
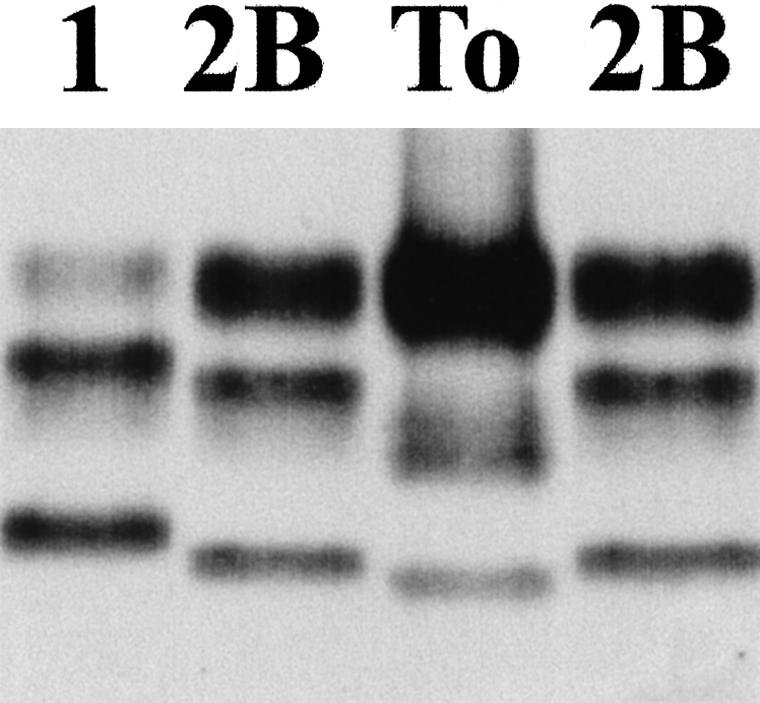
Western blot analysis of PrPres in a concentrated extract made from tonsil biopsy tissue (To). A standard type 1 PrPres isotype from sporadic CJD frontal cortex (1) and a standard type 2B PrPres isotype from variant CJD frontal cortex (2B) are also shown.
Spleen samples from vCJD characteristically gave poorly resolved profiles with only the top (diglycosylated band) clearly defined. A typical example is shown in Figure 8A alongside well-resolved trigeminal ganglion and tonsil samples from the same case (V5). This blot also shows a very faint band with a similar mobility to diglycosylated PrPres in the appendix sample. The weakness of signal coupled with the lack of confirmatory three-band pattern was judged insufficient to score the appendix as positive by Western blot. In comparison the signal from positive sample of vCJD dorsal root ganglion showed all three bands and resembled the pattern seen in other vCJD nervous system samples (Figure 8B).
Figure 8.

Western blot analysis of PrPres in (A) concentrated extracts of trigeminal ganglion (TG), tonsil (To), appendix (Ap), and spleen (Sp) from a case of vCJD (V5) and in (B) concentrated extracts of dorsal root ganglion (DRG) from cases of vCJD and iCJD (V6, V7, I1). Concentrated tonsil from a case vCJD (To V7) and unconcentrated frontal cortex from a case of vCJD (CC V5) are included as positive controls.
Discussion
Studies of the pattern of PrPSc accumulation in different forms of CJD are of importance for four main reasons. Firstly, they aid in the differential diagnosis of this family of human transmissible spongiform encephalopathies or prion diseases. Secondly they provide information pertinent to risk assessment of iatrogenic disease spread. Thirdly, they may help us to understand the sequence of pathogenic events that ultimately result in neuroinvasion and irreversible neurodegeneration and therefore to design rational prophylaxis or therapies. Finally they may help to better define the exact relationship between the protease-resistant prion protein and the transmissible agent in these diseases.
Differences in the physicochemical properties of PrPSc such as the conformation and glycosylation ratio have been advanced as a mechanism whereby prion strain characteristics may be enciphered and faithfully propagated.5,10,34 The protease-resistant prion protein core fragment (PrPres) found in the vCJD brain has an Mr of ∼19 kd and has a highly distinctive glycoform signature that is termed type 410 or type 2B.9,28 Our data indicates that although the type 2B PrPres isoform is consistently found in the vCJD brain, both the protease-resistant core fragment size and glycosylation ratio of can be substantially modified by peripheral tissue-specific influences.
Methodological Considerations
Our study significantly extends the recent report of four cases of vCJD35 by examining a series of peripheral tissues from a larger number of cases of vCJD in greater detail and comparing these with an equal number of relevant controls, namely sCJD, iCJD, and Lewy body dementia. The sensitivity of detection of PrPres in a tissue is a function of the overall tissue load and crucially its distribution. We therefore chose to use a sensitive Western blotting procedure that relies on the aggregated as well as the protease-resistant property of PrPSc for concentration and discrimination.32 This method is rapid and reproducible and avoids precipitation with phosphotungstic acid, which has been shown to change migration of PrPres fragments produced by subsequent proteinase K digestion.35 Western blot analysis of serially diluted standard vCJD brain extracts using the method described here is able to detect the equivalent ∼20 μg of gray matter-enriched cerebral cortex. The analysis of 200 μl of a 10% peripheral tissue extract (20 mg tissue equivalents) should therefore be able to detect PrPres at ∼10−3 of the level found in brain. We combined this Western blot procedure with an optimized immunohistochemical technique for in situ PrPres localization. In principle this combination of complementary techniques is ideally suited to the detection of PrPres whether it is accumulated widely in a tissue but at very low levels or whether it is accumulated at higher levels in a very limited number of cells. It is clear from the results of this study that the likelihood of PrPres detection by Western blotting in lymphoreticular tissues is a function of the number of positive lymphoid follicles (particularly in the appendix) and that a combination of such techniques is essential for a proper evaluation of PrPres accumulation.
Centripetal and Centrifugal Spread
Our data emphasize that the accumulation of PrPres in the lymphoreticular system is a defining feature of vCJD. Not only is PrPres present in lymphoreticular tissues (tonsil, spleen, lymph node, appendix) as judged by Western blot and/or immunohistochemistry in vCJD but it is also demonstrably absent from these same tissues from patients with sCJD and iCJD using the same optimized methodology. This points to fundamental differences in the pathogenesis in these diseases. vCJD is thought to be peripherally acquired, most likely through oral exposure. It is therefore not surprising to find that PrPres accumulation is widely dispersed through the periphery including the immune tissue of the gastrointestinal tract and other major lymphoreticular organs. However, organs such as heart, lung, and liver do not appear to be involved suggesting that the trafficking of the agent is determined by immune function or that other organs either fail to support formation or can efficiently degrade nascent PrPres accumulation. Oral dosing of rodents with the scrapie agent suggests that PrPres accumulation in the lymphoreticular system represents an early lymphoreticular phase, first in gastrointestinal-associated lymphoid system and later in lymph nodes and spleen, both of which precede neuroinvasion.36,37 Subsequent spread appears to use neural pathways of the peripheral nervous system to ultimately invade the brain via both a vagal and a spinal cord route.38,39 A comparison of the final distribution of PrPres at end-stage disease in these rodent models36–39 and that found in the vCJD tissues (this report) suggests that the tissue distribution is determined to a large extent by route of exposure.
Sporadic CJD, on the other hand is thought to arise within the central nervous system itself possibly as a result of a random stochastic event. Hence what limited evidence that has been provided for accumulation outside the brain, in trigeminal ganglion, peripheral nerve, neurosensory retina, and olfactory epithelium in sCJD is best interpreted as evidence of centrifugal spread.20,33,40,41 The finding that a peripherally acquired infection, iCJD can occur without apparent involvement of peripheral tissues is remarkable and indicates the presence of currently unknown agent or host-specified determinants of the route to neuroinvasion.
Risk of Iatrogenic Transmission
The detection of PrPSc provides a simple convenient marker for infectivity. If we accept this then it follows that lymphoreticular tissues in vCJD present a definite risk for iatrogenic transmission of CJD. If the degree of attendant risk correlates directly with PrPres load then vCJD lymph node and tonsil present the greater risk and that presented by gastrointestinal tissue, thymus, and appendix is somewhat lower. PrPres was not detectable in any peripheral nerve samples analyzed here but PrPres was detected in the trigeminal ganglia in variant, sporadic, and iatrogenic CJD, in some instances at levels higher than any tissue other than brain. This is a matter of some concern given that the trigeminal ganglion innervates the oral and nasal cavities and the anterior segment of the eye. It will be important to determine the extent of PrPres deposition along each of the cranial nerves and their targets to obtain a proper risk assessment for surgical and dental procedures. The absence of detectable PrPres in heart, lung, kidney, and liver in this study is reassuring, but it should be remembered that bioassay for infectivity tends to be more sensitive than current PrP-based assays, and that evidence for infectivity in some of these tissues in sCJD has been found in primate transmission studies.42 Moreover all assays are subject to uncertainties arising from sampling, and this is particularly true for large human organs such as brain, lung, and liver and widely distributed tissues such as nerve and muscle. Nevertheless it is noteworthy that neither a previous study35 nor this study can confirm in human disease the observation that PrPres accumulates in muscle tissue of mice infected with scrapie.43 On the basis of the calculations outlined above, if PrPres is present in these muscle samples our data suggest that it is at a level of at least 10−3 below that found in the brain.
PrP Isotype Analysis
PrPres isotype is defined by a combination of the relative mobility (Mr) of the nonglycosylated form and the ratio of the three glycoforms (non-, mono-, and di-glycosylated). The Mr of the nonglycosylated form is dependent on the extent of N-terminal proteinase K-mediated digestion that is itself thought to be a function of differences in protein conformation.5,10,34 The 21-kd type 1 PrPres has an N-terminus at glycine 82 and the 19-kd type 2 PrPres has an N-terminus at serine 97.28 Either the type 1 or 2 mobility has been found in sCJD whereas only type 2 is seen in vCJD brain.28 It is now becoming recognized that the sCJD brain can, in some cases, contain both types.31,44 A situation similar to the one described here where the iCJD case had type 2A in the brain but type 1 in the trigeminal ganglion. This constitutes the first report of a mixed isotype found in an acquired human prion disease.
The biological processes that determine the glycoform ratio of PrPres are poorly understood; nevertheless, there is a clear differentiation between sporadic and variant CJD on the basis of the PrPres glycoform ratio in the brain. vCJD samples are characterized by a predominant diglycosylated form and sCJD samples are characterized by predominance of the monoglycosylated band.9,10 The vCJD brain isoform pattern is shared by BSE brain and by transmissions of BSE to domestic, zoo, and experimental animals,10,12 which has led to the proposal that this represents the glycoform signature of the BSE agent. This glycotype is not however fixed, because the BSE glycoform signature is further accentuated toward diglycosylated forms in vCJD tonsil.21 We confirm this finding here not only in tonsil, but also in lymph node and spleen. In addition, we have identified two hitherto unrecognized aspects of peripheral vCJD isotype variation. First, the nonglycosylated isoform in tonsil and lymph node has a greater mobility than its counterpart in brain. Differences in nonglycosylated PrPres fragment size are usually interpreted as indicating differences in conformation5,10 although the fragment lengths produced are sensitive to the presence or absence of divalent cations45 and the pH at which the proteolytic digestion is performed.46 Secondly the mobility and complexity of the middle (monoglycosylated) band differs between vCJD brain and lymphoreticular tissues. This may depend on the size and complexity of the attached glycans or it may indicate the presence of endogenously truncated diglycosylated products.
These three kinds of difference between brain and lymphoreticular tissue PrPres were systematic and seen in all vCJD samples. They are not artifacts associated with postmortem change because they were also evident in biopsied tonsil. Moreover they can be seen in similar analyses of natural scrapie.47 It would seem therefore that they represent tissue or cell type-specific influences on the conformation and glycosylation of PrP. A fourth type of variation was seen to affect trigeminal ganglia in a case of vCJD analyzed here. In this case the glycoform ratio of the trigeminal ganglion PrPres did not show the BSE glycoform signature found in vCJD brain but rather was typical of sporadic CJD. We have encountered a similar phenomenon previously in the vCJD retina,33 and it may be that here again glycoform ratio can be affected by tissue-specific factors in some instances. Alternatively, it has recently been reported that the retinal PrPres glycoform ratio of animals experimentally infected with the scrapie agent changes during the course of disease48 suggesting an influence of duration of infection. In any event with these findings of glycosylation and presumptive conformational difference in PrPres in different tissues from single individuals infected with the BSE agent it becomes increasingly difficult to maintain a simple direct relationship between agent strain and PrPres isotype. Instead it seems more likely that the prion protein isotype reflects a more subtle interplay between the agent strain, cell types, genotype, and perhaps the pathological process itself, as has recently been suggested on the basis of results using cultured cells to propagate different strains of scrapie.49
Acknowledgments
We thank all of the neuropathologists and their technical staff in the UK for their support of the National CJD Surveillance Unit; Professor Ho-keung Ng of the Prince of Wales Hospital, Hong Kong for the tonsil biopsy specimen; and the laboratory staff in the National CJD Surveillance Unit for invaluable technical support.
Footnotes
Address reprint requests to Mark W. Head, Ph.D., National CJD Surveillance Unit, Bryan Matthews Building, Western General Hospital, Crewe Rd., Edinburgh, EH4 2XU, UK. E-mail: m.w.head@ed.ac.uk.
Supported by the United Kingdom Department of Health, the Scottish Executive, and EC funded project TSELAB (QLK2-CT-2002-81532).
N. S.’s contribution was in fulfillment of an MSc by Research in Life Sciences at the University of Edinburgh; S. S. and S. M. were Special Study Module undergraduate students in the Medical Faculty of the University of Edinburgh.
References
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown P, Preece M, Brandel J-P, Sato T, McShane L, Zerr I, Fletcher A, Will RG, Pocchiari M, Cashman NR, d‘Aignaux JH, Cervenakova L, Fradkin J, Schonberger LB, Collins SJ. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology. 2000;55:1075–1081. doi: 10.1212/wnl.55.8.1075. [DOI] [PubMed] [Google Scholar]
- Deslys JP, Marce D, Dormont D. Similar genetic susceptibility in iatrogenic and sporadic Creutzfeldt-Jakob disease. J Gen Virol. 1994;1:23–27. doi: 10.1099/0022-1317-75-1-23. [DOI] [PubMed] [Google Scholar]
- Monari L, Chen SG, Brown P, Parchi P, Petersen RB, Mikol J, Gray F, Cortelli P, Montagna P, Ghetti B, Goldfarb LG, Gajdusek DC, Lugaresi E, Gambetti P, Autilio-Gambetti L. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proc Natl Acad Sci USA. 1994;91:2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AAF, Trojanowski JQ, Petersen RB, Gambetti P. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–78. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- Lee HS, Brown P, Cervenakova L, Garruto RM, Alpers MP, Gajdusek DC, Goldfarb LG. Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis. 2001;183:192–196. doi: 10.1086/317935. [DOI] [PubMed] [Google Scholar]
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman M, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, Mackenzie J, Estibeiro K, Green AJE, Knight RSG. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000;47:575–582. [PubMed] [Google Scholar]
- Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology. 2000;37:1–9. doi: 10.1046/j.1365-2559.2000.00946.x. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- Scott MR, Will R, Ironside J, Nguyen H-OB, Trembley P, DeArmond SJ, Prusiner SB. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valleron A-J, Boelle P-Y, Will R, Cesbron J-E. Estimation of epidemic size and incubation time based on age and characteristics of vCJD in the United Kingdom. Science. 2001;294:1726–1728. doi: 10.1126/science.1066838. [DOI] [PubMed] [Google Scholar]
- D’Aignaux JNH, Cousens SN, Smith PG. Predictability of UK variant Creutzfeldt-Jakob disease epidemic. Science. 2001;294:1729–1731. doi: 10.1126/science.1064748. [DOI] [PubMed] [Google Scholar]
- Andrews NJ, Farrington CP, Ward H, Cousins SN, Smith PG, Molesworth AM, Knight R, Ironside JW, Will RG. Deaths from variant Creutzfeldt-Jakob disease in the UK. Lancet. 2003;361:751–752. doi: 10.1016/s0140-6736(03)12632-3. [DOI] [PubMed] [Google Scholar]
- Van Keulen LJM, Schreuder BEC, Meloen RH, Mooij-Harkes G, Vromans MEW, Langeveld JPM. Immunohistochemical detection of prion protein in lymphoid tissues of sheep with natural scrapie. J Clin Microbiol. 1996;34:1228–1231. doi: 10.1128/jcm.34.5.1228-1231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride PA, Eikelenbloom P, Kraal G, Fraser H, Bruce ME. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol. 1992;168:413–418. doi: 10.1002/path.1711680412. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Mohri S, Tateishi J. Organ distribution of proteinase-resistant prion protein in human and mice Creutzfeldt-Jakob diseases. J Gen Virol. 1989;70:3371–3397. doi: 10.1099/0022-1317-70-12-3371. [DOI] [PubMed] [Google Scholar]
- Hainfellner JA, Budka H. Disease associated prion protein may deposit in the peripheral nervous system in human transmissible spongiform encephalopathies. Acta Neuropathol. 1999;98:458–460. doi: 10.1007/s004010051109. [DOI] [PubMed] [Google Scholar]
- Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, Frosh A, Tolley N, Bell JE, Spencer M, King A, Al-Sarraj S, Ironside JW, Lantos PL, Collinge J. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet. 1999;353:183–189. doi: 10.1016/s0140-6736(98)12075-5. [DOI] [PubMed] [Google Scholar]
- Joiner S, Linehan J, Brandner S, Wadsworth JDF, Collinge J. Irregular presence of abnormal prion protein in appendix in variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2002;73:597–598. doi: 10.1136/jnnp.73.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton DA, Fathers E, Edwards P, Ironside JW, Zajicek J. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet. 1998;352:703–704. doi: 10.1016/S0140-6736(98)24035-9. [DOI] [PubMed] [Google Scholar]
- Bruce ME, McConnell I, Will RG, Ironside JW. Detection of variant Creutzfeldt-Jakob disease infectivity in extraneural tissues. Lancet. 2001;358:208–209. doi: 10.1016/s0140-6736(01)05411-3. [DOI] [PubMed] [Google Scholar]
- Farquhar C, Dickinson A, Bruce M. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet. 1999;353:117. doi: 10.1016/S0140-6736(98)05395-1. [DOI] [PubMed] [Google Scholar]
- Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Haw JJ, Ironside JW, Jellinger K, Kretzschmar HA, Lantos PL, Masullo C, Schlote W, Tateishi J, Weller RO. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol. 1995;5:459–466. doi: 10.1111/j.1750-3639.1995.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B, Kopp N, Schulz-Schaeffer WJ, Kretzschmar HA, Head MW, Ironside JW, Gambetti P, Chen SG. Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci USA. 2000;97:10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay R, Lau WY, Ng HK, Chan YL, Lyon DJ, van Hasselt CA. Variant Creutzfeldt-Jakob disease in Hong Kong. Hong Kong Med J. 2001;7:296–298. [PubMed] [Google Scholar]
- Sabattini E, Bisgaard K, Ascani S, Poggi S, Piccioli M, Ceccarelli C, Pieri F, Fraternali-Orcioni G, Pileri SA. The En Vision++ system: a new immunohistochemical method for diagnostics and research. Critical comparison with the APAAP,ChemMate CSA, LABC and SABC techniques. J Clin Pathol. 1998;51:506–511. doi: 10.1136/jcp.51.7.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head MW, Tissingh G, Uitdehaag BMJ, Barkhof F, Bunn TJR, Ironside JW, Kamphorst W, Scheltens P. Sporadic Creutzfeldt-Jakob disease in a young Dutch valine homozygote: atypical molecular phenotype. Ann Neurol. 2001;50:258–261. doi: 10.1002/ana.1100. [DOI] [PubMed] [Google Scholar]
- Lee DC, Stenland CJ, Hartwell RC, Ford EK, Cai K, Miller JLC, Gilligan KJ, Rubenstein R, Fournel M, Petteway SR. Monitoring plasma processing steps with a sensitive Western blot assay for the detection of the prion protein. J Virol Methods. 2000;84:77–89. doi: 10.1016/s0166-0934(99)00135-4. [DOI] [PubMed] [Google Scholar]
- Head MW, Northcott V, Rennison K, Ritchie D, McCardle L, Bunn TJR, McLennan N, Ironside JW, Tullo A, Bonshek RE. Prion protein accumulation in eyes of patients with sporadic and variant Creutzfeldt-Jakob disease. Invest Ophthalmol Vis Sci. 2003;44:342–346. doi: 10.1167/iovs.01-1273. [DOI] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet. 2000;358:171–180. doi: 10.1016/s0140-6736(01)05403-4. [DOI] [PubMed] [Google Scholar]
- Maignien T, Lasmezas CI, Beringue Dormont D, Deslys J-P. Pathogenesis of the oral route of infection of mice with scrapie and bovine spongiform encephalopathy agent. J Gen Virol. 1999;80:30335–30342. doi: 10.1099/0022-1317-80-11-3035. [DOI] [PubMed] [Google Scholar]
- Beekes M, McBride PA. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci Lett. 2000;278:181–184. doi: 10.1016/s0304-3940(99)00934-9. [DOI] [PubMed] [Google Scholar]
- Beekes M, McBride PA, Baldauf E. Cerebral targeting indicates vagal spread of infection in hamsters fed scrapie. J Gen Virol. 1999;79:601–607. doi: 10.1099/0022-1317-79-3-601. [DOI] [PubMed] [Google Scholar]
- McBride PA, Beekes M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamster fed with scrapie. Neurosci Lett. 1992;265:135–138. doi: 10.1016/s0304-3940(99)00223-2. [DOI] [PubMed] [Google Scholar]
- Guiroy DC, Shankar SK, Gibbs CJ, Messenheimer JA, Das S, Gajdusek DC. Neuronal degeneration and neurofilament accumulation in the trigeminal ganglion in Creutzfeldt-Jakob diseases. Ann Neurol. 1989;25:102–106. doi: 10.1002/ana.410250119. [DOI] [PubMed] [Google Scholar]
- Zanusso G, Ferrari S, Cardone F, Zampieri P, Pgelati M, Fiorini M, Farinazzo A, Gardiman M, Cavallaro T, Bentivoglio M, Righetti PG, Pochiari M, Rizzuto N, Monaco S. Detection of pathologic prion protein in the olfactory epithelium in sporadic Creutzfeldt-Jakob disease. N Engl J Med. 2003;348:711–719. doi: 10.1056/NEJMoa022043. [DOI] [PubMed] [Google Scholar]
- Brown P, Gibbs CJ, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- Bosque PJ, Ryou C, Telling G, Peretz D, Legname G, DeArmond SJ, Prusiner SB. Prions in skeletal muscle. Proc Natl Acad Sci USA. 2002;99:3812–3817. doi: 10.1073/pnas.052707499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology. 1999;53:2173–2176. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- Wadsworth JDF, Hill AF, Joiner S, Jackson GS, Clarke AR, Collinge J. Strain-specific prion-protein conformation determined by metal ions. Nature Cell Biol. 1999;1:55–60. doi: 10.1038/9030. [DOI] [PubMed] [Google Scholar]
- Zanusso G, Farinazzo A, Fiorini M, Gelati M, Castagna A, Righetti PG, Rizzuto N, Monaco S. pH-dependent prion protein conformation in classical Creutzfeldt-Jakob disease. J Biol Chem. 2001;276:40377–40380. doi: 10.1074/jbc.C100458200. [DOI] [PubMed] [Google Scholar]
- Madec J-Y, Groschup MH, Calavas D, Junghans F, Baron T. Protease-resistant prion protein in brain and lymphoid organs of sheep within a naturally scrapie infected flock. Microbial Pathogenesis. 2000;28:353–362. doi: 10.1006/mpat.2000.0357. [DOI] [PubMed] [Google Scholar]
- Russelakis-Carneiro M, Saborio GP, Anderes L, Soto C. Changes in the glycosylation pattern of prion protein in murine scrapie: implications for the mechanism of neurodegeneration in prion disease. J Biol Chem. 2002;277:36872–36877. doi: 10.1074/jbc.M202229200. [DOI] [PubMed] [Google Scholar]
- Vorberg I, Priola SA. Molecular basis of scrapie glycoform variation. J Biol Chem. 2002;277:36775–36781. doi: 10.1074/jbc.M206865200. [DOI] [PubMed] [Google Scholar]