

Abstract
Caspase-3 is an effector of apoptosis in experimental models of Parkinson's disease (PD). However, its potential role in the human pathology remains to be demonstrated. Using caspase-3 immunohistochemistry on the postmortem human brain, we observed a positive correlation between the degree of neuronal loss in dopaminergic (DA) cell groups affected in the mesencephalon of PD patients and the percentage of caspase-3-positive neurons in these cell groups in control subjects and a significant decrease of caspase-3-positive pigmented neurons in the substantia nigra pars compacta of PD patients compared with controls that also could be observed in an animal model of PD. This suggests that neurons expressing caspase-3 are more sensitive to the pathological process than those that do not express the protein. In addition, using an antibody raised against activated caspase-3, the percentage of active caspase-3-positive neurons among DA neurons was significantly higher in PD patients than in controls. Finally, electron microscopy analysis in the human brain and in vitro data suggest that caspase-3 activation precedes and is not a consequence of apoptotic cell death in PD.
The pathological hallmarks of Parkinson's disease (PD) are a loss of dopaminergic (DA) neurons in the mesencephalon and the presence of Lewy bodies in altered neurons. The exact cause of this neuronal loss is still unknown, but recent human postmortem studies have suggested that, in PD, nigral DA neurons die by apoptosis (1–3) as do DA neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice (4, 5), an in vivo model of PD. However, the significance of purely morphological human postmortem features suggestive of apoptosis remained controversial, and the results of investigations into molecular apoptotic markers in PD brains are awaited to confirm the morphologic studies (6).
Extensive in vitro studies in nonneuronal and neuronal cell systems indicate that aspartate-specific cysteine proteases (caspases) are effectors of apoptosis (7). In neurons, several lines of evidence indicate that caspase-3 (CPP32/Yama/Apopain), a 32-kDa cytosolic protein, plays a major role in the executive phase of apoptosis (8, 9). First, cerebral hyperplasia and cellular disorganization are observed in caspase-3-deficient mice (10). Second, neuronal death in experimental models of several acute and chronic neurodegenerative disorders has been associated with activation of caspase-3 (11–13). Third, with special reference to PD, neurotoxins commonly used to induce experimental parkinsonian syndromes, e.g., 1-methyl-4-phenylpyridinium (MPP+) and 6-hydroxydopamine (6-OHDA), have been shown to exert their proapoptotic actions via activation of caspase-3-like proteases in neuronal in vitro models (14–16). To date, however, cellular expression of caspase-3 has not been studied in postmortem brain from patients with PD or any other neurologic disorders. In the present study, we thus analyzed caspase-3 distribution and activation in PD and experimental models of the disease.
Materials and Methods
Patients and Human Brain Tissue.
Mesencephalons were obtained at autopsy from five individuals with no known history of psychiatric or neurologic disorders (control group) and from five patients with histologically confirmed PD (PD group). Age at death and time interval from death to tissue fixation did not differ significantly between the control group [79.6 ± 9.6 years and 25.4 ± 6.4 hr, respectively (mean ± SEM)] and the PD group (70.4 ± 5.3 years and 24.2 ± 5.8 hr, respectively). Within 2 hr of autopsy, tissue was dissected and processed as described previously (17).
For the quantitative caspase-3 analysis, free-floating, 40-μm-thick sections taken at the level of the oculomotor nerve fibers were used. For the quantitative analysis using the CM1 antibody (18), four to five sections covering the whole extent of the substantia nigra pars compacta (SNpc) from its rostral to its caudal pole of four control and four parkinsonian patients were used. Finally, for the ultrastructural analysis using the CM1 antibody, SN tissue fixed according to a different protocol for electron microscopy (19) from one PD patient not included in the previous analysis was analyzed.
MPTP-Intoxicated Mice.
Mice were intoxicated subchronically with MPTP as described elsewhere (5). In brief, 8-week-old male C57BL/6 mice were injected at a 30-mg/kg per day i.p. dosage over a period of 5 days and sacrificed after 21 days (n = 6). A control group was injected with equivalent volumes of 0.9% NaCl (n = 6). The animals were perfused with 4% paraformaldehyde, the brains were removed, and the mesencephalon was cut into 20-μm-thick sections.
Primary Cultures of Rat Mesencephalon.
Primary cultures of rat mesencephalon were prepared as described previously (20). Apoptosis was induced after 6 days of differentiation, with cell-permeant MPP+ (Sigma) at a concentration of 1 μM. Cultures were fixed 12, 24, and 72 hr after initiation of treatment. All experiments were repeated six times.
Immunohistochemistry.
Free-floating, 40-μm-thick sections were pretreated as described previously (17) and incubated with a polyclonal rabbit antibody (1:250, 48 hr at 4°C) raised against amino acid residues 51NNKNFHKSTGMTSRSGT67 located within the caspase-3 p20 subunit for quantitative analysis. In addition, trial experiments were conducted by using a rabbit polyclonal antibody (65906E; PharMingen; 1:250, 48 hr at 4°C) raised against recombinant human caspase-3-His6 as immunogen (21) (1:250, 48 hr at 4°C) or the CM1 antibody raised against the C terminus of the human p17 caspase-3 fragment, 163CRGTELDCGIETD175, specifically recognizing activated (cleaved) caspase-3 (18) (1:2,500, 48 hr at 4°C). The sections were revealed as described previously (19). Sequential double-labeling experiments were performed by using a rabbit polyclonal anti-ubiquitin antibody (Dako; 1:250, 48 hr at 4°C) and the anti-caspase-3 antiserum used for the quantitative analysis or the CM1 antibody as described (22). Because the two antigens were detected with a different localization within the cell, some sections were coincubated with the two antibodies (anti-ubiquitin and caspase-3 or CM1) at the same time for the illustrations. Similarly, mesencephalon sections from MPTP-treated and control mice first were incubated for 48 hr at 4°C with an anti-tyrosine hydroxylase (anti-TH) mouse mAb (1:500; Incstar, Stillwater, MN), TH-positive neurons were localized and counted, and then sections were unstained and reincubated with a rabbit polyclonal antibody (67341A; PharMingen; 1:500, 48 hr at 4°C) recognizing murine procaspase-3, previously tested by Western immunoblotting (data not shown). Control experiments were performed in which one or the other of the primary antisera was omitted. No staining was observed under these conditions.
Fluorescent double-staining experiments were performed on mesencephalic sections mounted on gelatin-double-coated slides, as described previously (23), to analyze simultaneously caspase-3 and TH.
TH immunocytochemistry in primary cultures of rat mesencephalon was performed as described previously by using FITC as fluorochrome (20). Cultures then were incubated with the CM1 antibody at 1:2,500 for 24 hr at 4°C and revealed by using TRITC as fluorochrome. The cell-permeant fluorescent marker Hoechst 33258 (1 μM; Boehringer Mannheim) was added to the cultures for 15 min at room temperature to assess the morphology of normal and apoptotic (i.e., condensed, fragmented) cells.
Western Immunoblotting.
Specificity of the antibodies was analyzed on Western blots of human SN homogenates from control and parkinsonian patients. Homogenates of SNpc were fractionated, and the proteins (50 μg) were separated by PAGE and transferred onto nitrocellulose membranes. Jurkat cell lysates (15 μg) were used as positive control. To positively detect the cleaved p17 fragment of caspase-3 using the CM1 antibody, 15 μg of Jurkat cell lysates was preincubated with 500 ng of human recombinant caspase-3 (PharMingen) for 60 min at 37°C. The membranes were incubated for 48 hr at 4°C with the caspase-3 polyclonal antibody (1:1,000) or the CM1 antibody (1:5,000) and revealed as described (23).
Electron Microscopy.
Ultrastructural analysis of CM1-immunopositive DA neurons was performed as described previously with minor modifications (24).
Regional Quantification and Image Analysis.
For each stained section, caspase-3-positive and -negative pigmented DA neurons were counted in the central gray substance (CGS), ventral tegmental area (VTA), and SNpc by using a computer-based image analysis system (visioscan; Biocom, Les Ulis) (19). The SNpc was subdivided into lateroventral, medioventral, and dorsal tiers according to the regional vulnerability of nigral DA neurons established by Fearnley and Lees (25). Because the degree of neuronal loss and, thus, the absolute number of pigmented neurons varies between PD patients, the results were expressed as the percentage of pigmented neurons that were stained for caspase-3. For activated caspase-3 staining, immunoreactive DA neurons were counted exclusively in the SNpc without regional subdivisions because of the low numbers of immunoreactive neurons detected, which made a statistical analysis of SNpc subdivisions irrelevant. The total number of DA neurons in the mesencephalon was estimated as described previously (19), and the mean percentage of neuronal loss within the CGS, VTA, and SNpc in our sample was calculated. The total number of CM1-positive neurons in the SNpc was estimated by using the same method.
For the mice, the SNpc was delineated based on the distribution of TH-positive cells at identical levels for all mice and analyzed by using the same methodology as for the human postmortem sections.
Cultures were analyzed by phase-contrast and standard epiillumination fluorescence microscopy and by computer-assisted image analysis (IMSTAR, Paris). Counting was performed at ×20 in eight fields per horizontal axis and eight fields per vertical axis per well. Six wells were analyzed per experimental condition.
Statistical Analysis.
Intergroup differences (control vs. PD) in the percentage of caspase-3 immunoreactive neurons were compared by Student's t test or, in the event of failure in normality test, by Mann–Whitney rank sum test. Correlations were determined by linear regression analysis.
Results
Specificity of Antibodies Directed Against the p20 Subunit of Human Caspase-3 and the Cleaved Caspase-3 p17 Subunit.
On Western blots of proteins extracted from the SNpc of three control subjects and three PD patients, two bands were observed by using the antibody directed against the caspase-3 p20 subunit: a 32-kDa band corresponding to the caspase-3 precursor protein and a 30-kDa band representing the processed form of caspase-3 without its prodomain, as described by Kuida et al. (10) (Fig. 1A). No cleaved p10 or p20 caspase-3 fragments were detected either in control or in parkinsonian SNpc protein extracts. The same result was obtained by using the CM1 antibody with regard to pooled SNpc extracts from four parkinsonian patients, as well as untreated Jurkat cell lysates. However, when Jurkat cell lysates were preincubated with human recombinant caspase-3, a cleaved caspase-3 p17 subunit could readily be detected (Fig. 3A). On tissue sections, staining intensity decreased with lower antibody dilutions. No staining was observed when the primary antibody was omitted. Using a commercial antibody directed against caspase-3, an identical pattern of staining was observed on transverse mesencephalon sections from two control subjects and two PD patients (data not shown).
Figure 1.

Characterization of caspase-3 staining. (A) Specificity of the anti-caspase-3 p20 polyclonal rabbit antibody. Western immunoblot of caspase-3 from 50 μg of SNpc protein extracted from three control and three parkinsonian mesencephalons after SDS/PAGE. Molecular mass markers in the first lane are given in kDa and allow identification of caspase-3 protein at molecular masses of 32 kDa (including prodomain) and 30 kDa (without prodomain). (B) High-power photomicrograph showing cytosolic caspase-3 immunostaining of SNpc neuromelanin-containing neurons in transverse sections of control SNpc. (C and D) Immunofluorescent dopaminergic neurons in transverse SNpc sections of a control subject stained with a caspase-3 polyclonal rabbit antibody revealed by fluorescein (C) and a TH monoclonal mouse antibody revealed by rhodamine600 (D). Whereas caspase-3 immunoreactivity is confined to the cytosol, TH staining can be observed in the perikarya and dendrites. The middle portion of the neuron is occupied by neuromelanin. (Bars = 30 μm.)
Figure 3.

Characterization of CM1 staining. (A) Specificity of the anti-CM1 polyclonal rabbit antibody. Western immunoblot of activated caspase-3 from 50 μg of SNpc protein extracted from four parkinsonian mesencephalons (lane a), 15 μg of untreated Jurkat cell lysate (lane b), and 15 μg of Jurkat cell lysate preincubated with 500 ng of human recombinant caspase-3 for 60 min at 37°C (lane c) after SDS/PAGE. Molecular mass markers in the first lane are given in kDa and allow identification of the cleaved p17 subunit at molecular masses of 17 kDa in lane c (A). Note that in the homogenate of the parkinsonian SNpc, activated caspase-3 is not detectable because of its presence in only a few cells. (B–D) High-power photomicrograph showing cytosolic-activated caspase-3 immunostaining (CM1 antibody) of SNpc neuromelanin-containing neurons in transverse sections of control SNpc. (E) High-power photomicrograph showing Lewy bodies stained with antiubiquitin (arrow) and co-CM1 immunostaining of SNpc neuromelanin-containing neurons in transverse sections of PD SNpc. Note the presence of both Lewy bodies and CM1 staining in a single neuron. (Bar = 30 μm.)
Immunohistochemical Detection of Caspase-3 in Control and Parkinsonian Mesencephalon.
At the macroscopic level, caspase-3 immunostaining was observed in all mesencephalic subregions. Among DA cell groups, staining intensity was high in the SNpc (Fig. 1B), moderate in the VTA, and undetectable in the CGS. At the cellular level, both pigmented and nonpigmented neurons were stained. Immunoreactive glial cells were also observed in all mesencephalic subregions. Fluorescent double-staining experiments with caspase-3 and TH in control human mesencephalon sections evidenced the expression of caspase-3 in TH-immunoreactive neurons in the SNpc and VTA but not in the CGS. Whereas TH immunoreactivity was observed in cell perikarya and dendrites, caspase-3 immunoreactivity was confined to the cytosol of the neuronal perikarya (Fig. 1 C and D).
Quantitative Analysis of Caspase-3-Positive Pigmented Neurons in the SNpc and VTA of Control and PD Patients.
The mean total numbers (±SEM) of caspase-3-positive and -negative melanized neurons in the SNpc and VTA of controls and PD patients are given in Table 1. A variable percentage of caspase-3-positive neurons was observed within the different mesencephalic catecholaminergic cell groups in the mesencephalon of control subjects. These percentages showed a positive linear regression with the degree of neuronal loss within the CGS, VTA, and SNpc (df: 1; r = 0.997; P < 0.001), suggesting that expression of caspase-3 in certain populations of DA neurons may contribute to their vulnerability in this neurodegenerative disorder. This finding was in line with the reduced percentage of pigmented neurons that were caspase-3-immunoreactive in the SNpc of patients with PD (10.8 ± 2.4%) as compared with control subjects (45.2 ± 3.8%) (P < 0.001). In contrast, there was no statistically significant difference between these percentages in the VTA (controls: 15.8 ± 3.9%; PD: 17.0 ± 7.0%), which is less affected in PD (17). The percentage of caspase-3-positive neurons among pigmented neurons was also analyzed in subsectors of the SNpc. It was significantly lower in PD patients than in controls in the lateroventral part (P < 0.001) (controls: 68.4 ± 5.5%; PD: 3.8 ± 3.8%), in the medioventral part (P < 0.006) (controls: 30.4 ± 4.9%; PD: 7.4 ± 3.8%), and in the dorsal part (P < 0.001) (controls: 47.2 ± 4.2%; PD: 12.4 ± 3.6%).
Table 1.
Analysis of caspase-3-positive and caspase-3-negative neurons in PD
| Group | No. (mean ± SEM) of melanized
caspase-3-positive neurons
|
No. (mean ± SEM) of
melanized caspase-3-negative neurons
|
||
|---|---|---|---|---|
| SNpc | VTA | SNpc | VTA | |
| Control | 388 ± 77 | 20 ± 7 | 442 ± 43 | 120 ± 47 |
| PD | 7 ± 2 | 3 ± 1 | 68 ± 15 | 16 ± 4 |
An analysis of Lewy body-containing DA neurons detected by anti-ubiquitin staining within the SNpc of the five PD patients revealed that 52.5 ± 4.0% of the neurons containing Lewy bodies were also immunoreactive for caspase-3 (Fig. 2). This percentage was significantly higher than the overall percentage of caspase-3-postive neurons among all DA neurons (10.8 ± 2.4%) in the same group of PD patients (P < 0.001).
Figure 2.
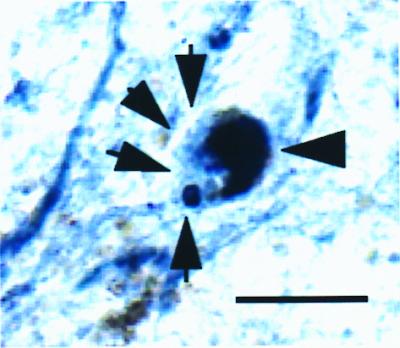
High-power photomicrograph showing Lewy bodies stained for ubiquitin (single arrow) and caspase-3 (triple arrows) in a SNpc neuromelanin-containing (thick arrow) neuron in a transverse section of PD SNpc. Note that ubiquitin-containing fibers and extraneuronal melanin can be seen. (Bar = 30 μm.)
Quantitative Analysis of Caspase-3-Positive DA Neurons in the SNpc of MPTP-Treated and Control Mice.
To test whether similar decreases in caspase-3 immunoreactivity in DA neurons could be observed in an animal model of PD, C57BL/6 mice were treated with a subchronic regimen of MPTP reported to induce apoptotic degeneration of DA SNpc neurons (5) and sacrificed after 21 days when the degeneration process of DA neurons is completed (data not shown). The MPTP group had a significantly lower percentage of caspase-3-positive neurons among TH-positive neurons (53.0 ± 3.7%) than control mice (67.7 ± 2.4%) (P = 0.008).
Quantitative Analysis of Activated, Caspase-3-Positive Pigmented Neurons in the SNpc of Control and PD Patients.
Using an antibody directed against the C terminus of the p17 fragment of caspase-3 (CM1) and, thus, specific for the activated form of caspase-3 (Fig. 3 B–E), we observed 13.6 ± 1.7 CM1-positive, melanized neurons per SNpc section in control subjects and 7.2 ± 1.1 in PD patients. In the SNpc, based on the estimated total numbers, the proportion of DA neurons that were CM1-positive was significantly higher in PD patients than in control subjects (PD: 6.5 ± 3.9%; controls: 1.2 ± 0.2%; Mann–Whitney rank sum test: P = 0.03). Occasionally, it was possible to detect activated caspase-3 in Lewy body-containing neurons (Fig. 3F).
Electron Microscopy Analysis of Nigral DA Neurons Expressing Activated Caspase-3 in PD.
Cytosolic, activated caspase-3 staining was observed in DA SNpc neurons at the ultrastructural level. These neurons typically exhibited a condensed perinuclear endoplasmic reticulum (ER) that was suggestive of increased protein synthesis (Fig. 4A). DA neurons displaying classical morphological features of apoptosis, i.e., chromatin condensation, were CM1-negative (Fig. 4B). Finally, dopaminergic neurons negative for CM1 and without apoptotic features did not display dense perinuclear ER aggregates (Fig. 4C).
Figure 4.

Electron microscopy photomicrograph of PD SNpc showing melanized dopaminergic neurons recognizable by their neuromelanin-containing vesicles: with cytosolic precipitates (thin arrows) corresponding to activated caspase-3 staining (CM1 antibody) [A; note the dense perinuclear endoplasmic reticulum (thick arrows)]; with apoptotic morphology including chromatin condensation (arrows) (B; note that no CM1 staining can be detected); and displaying neither morphological features of apoptosis nor CM1 staining (C; note that the endoplasmic reticulum is less dense than in Fig. 5A). (Bars = 1 μm.)
Does Caspase-3 Activation Precede Cell Death in Primary DA Cultures Treated with MPP+?
To test whether caspase-3 activation precedes morphological features of apoptosis as suggested by our electron microscopy study, low concentrations (1 μM) of MPP+, reported to induce apoptosis, were applied in primary DA neurons (14). Cultures fixed 12 hr after treatment showed a significantly higher percentage of CM1-positive neurons among TH-positive neurons (32.4 ± 3.6%) than control cultures (13.1 ± 3.3%) (P = 0.004) whereas neuronal cell loss represented only 3.8% at this time point. After 24 hr, this percentage of CM1-positive neurons was lower but still significantly higher (P = 0.015) in the MPP+-treated cultures (13.9 ± 1.2%) than in the control cultures (8.4 ± 1.4%) whereas neuronal cell loss represented 18.8%. Finally, at 72 hr after treatment, this percentage was even higher in the control cultures (7.9 ± 1.7%) than in the MPP+-treated cultures (4.6 ± 3.0%). At this time, neuronal loss reached 78.5%. Concomitant Hoechst 33258 staining did not allow DNA condensation to be detected in CM1-positive DA neurons after 12 hr of MPP+ treatment (Fig. 5 A–C). In contrast, DNA condensation within DA neurons was observed after 72 hr and accompanied by CM1 staining (Fig. 5 D–F).
Figure 5.

Immunofluorescent dopaminergic neurons in primary cultures of rat mesencephalic neurons fixed after 12 hr of 1-μM MPP+ treatment stained with a TH monoclonal mouse antibody revealed by FITC (A) and a rabbit polyclonal antibody recognizing activated caspase-3 (CM1) revealed by tetramethylrhodamine B isothiocyanate (B). Hoechst 33258 staining reveals an intact nucleus in this neuron (C, arrow). (Bar = 20 μm.) In contrast, in a TH-positive neuron (D) with CM1 staining (E), DNA condensation assessed by Hoechst 33258 can be detected after 72 hr of MPP+ treatment (F, arrow). Note that a higher magnification was used to show DNA condensation. (Bar = 10 μm.)
Discussion
Caspase-3 Is Expressed in Postmitotic DA Neurons.
Activation of caspase-3 has been reported in several models of neuronal apoptosis in vitro or in vivo (11–13), but only one study has reported caspase-3 expression in the human central nervous system (21). In the present study, almost half of the pigmented DA neurons expressed caspase-3, indicating that proapoptotic effectors indeed are present in a latent state and do not require to be newly synthesized should a proapoptotic pathway be engaged. This is in agreement with recent in vitro studies showing that caspase-3 activation in various neuronal cell death paradigms is posttranscriptional (26, 27).
Caspase-3 May Be a Vulnerability Factor for Pigmented DA Neurons.
A relationship between the presence of caspase-3 in DA neurons and their sensitivity to PD is supported by the analysis of the different catecholaminergic cell groups in the control mesencephalon. Indeed, a positive correlation between the estimated percentage of caspase-3-positive neurons among pigmented neurons in the CGS, VTA, and SNpc in control subjects and the percentage loss of pigmented neurons in these regions in PD was shown in our study (19). Such a relationship also was observed in the subregions of the SNpc, but only partially. Whereas the highest percentage of caspase-3-positive neurons was observed in the lateroventral part of the structure (68.4%), where neuronal loss is most severe in PD [91% according to Fearnley and Lees (25)], it is higher in the dorsal part of the SNpc (47.2%) than in the ventromedial part (30.4%), where neuronal loss is 56% and 71%, respectively. This apparent discrepancy may be due either to differences in delineation of the SNpc subregions or to the presence of additional factors in one of these subregions (28). Of course, these speculative arguments leave the possibility open that, within the SNpc, the distribution of caspase-3 does not contribute to regional vulnerability. Alternatively, the regional vulnerability of DA neurons in the parkinsonian SNpc probably observes a more complex distribution (29) than reported initially by Fearnley and Lees (25). The relationship between the presence of caspase-3 in DA neurons and their sensitivity to PD is supported further by a 76% decrease in melanized SNpc neurons that are caspase-3-positive in PD patients compared with controls. A significant, 22% decrease of DA neurons expressing caspase-3 also was observed in mice subchronically intoxicated with MPTP. Yet, this decrease was less pronounced than in the human sample. This is probably due to the less severe degeneration of DA neurons in the SNpc in mice (20%) compared with that observed in PD patients (85%). Furthermore, the percentage of caspase-3-positive neurons was increased 3.9-fold in Lewy body-containing DA neurons as compared with DA neurons without Lewy bodies. Given the suggestion that the presence of Lewy bodies represents an indicator of neuronal suffering and/or damage (30) and the fact that incidental Lewy body disease is considered by some authors to be a presymptomatic form of PD (31), the increased expression of caspase-3 in Lewy body-containing neurons is in line with the hypothesis that caspase-3 is a probable effector of apoptotic cell demise. Taken together, these findings suggest that caspase-3-expressing neurons are particularly prone to degenerate in PD if caspase-3 is activated during the course of the disease. However, it should be emphasized that term “vulnerability factor” is based on a probabilistic rather than deterministic definition. Because about 50% of neurons that degenerate in PD do not express caspase-3 in control SNpc neurons—at least on a detectable scale—our assumption is that the presence or absence of caspase-3 does not dictate cell fate but may, however, be a necessary cofactor for eventual cell death. Furthermore, the presence in control subjects of caspase-3 in DA cell groups that are more susceptible in PD and its absence in cell groups that are preserved in PD also reinforces this notion. However, this argument is purely correlative and does not exclude that another effector caspase, such as caspase-7, may substitute for caspase-3 in a subset of DA neurons during the apoptotic process.
The Percentage of Activated Caspase-3-Positive Nigral DA Neurons Is Increased in PD.
CM1-positive DA neurons were detected both in PD and in control SNpc. Their absolute number per section was higher in control than in PD mesencephalons. However, if the mean estimated total number of CM1-positive DA neurons per section was corrected for the mean estimated total number of TH-positive neurons, the relative percentage of CM1-positive neurons was about five times higher in the PD group. In both groups, these numbers probably reflect a perimortem phenomenon related to hypoxia secondary to the patients' agonal state. Indeed, in vitro data show that hypoxia is a potent stimulator of apoptotic death in neurons (32). The number of activated caspase-3 neurons is in agreement with previous studies showing that the number of DA neurons undergoing apoptosis at the time of death is around 1–2% in control and 5–6% in PD brains (1, 2, 33, 34). Given the slow rate of neuronal degeneration in PD, especially at an advanced stage of the disease, as encountered in our sample, it is unlikely that this relatively high percentage reflects a primary disease process. Thus, it is likely that nigral DA neurons in parkinsonian brains already are altered and have a higher vulnerability to apoptosis in response to deleterious stimuli, e.g., hypoxia. In this context, one recent human postmortem study has shown that in control brains, the number of terminal deoxynucleotidyltransferase-mediated dUTP-biotin 3′ end labeling (TUNEL)-positive neurons was correlated with tissue pH, taken as an index of perimortem hypoxia in control subjects (2). Furthermore, it should be noted that if CM1 immunoreactivity reflects caspase-3 activation, the latter does not necessarily correlate with an eventual apoptotic cell death (35).
Caspase-3 Activation Probably Precedes and Does Not Cooccur with Chromatin Condensation During Apoptosis.
A key question related to the increased proportion of neurons expressing activated caspase-3 in the parkinsonian SNpc is to know whether caspase-3 activation is associated with cell death or a consequence of it. This issue was addressed by experiments performed on postmortem material and cell cultures. Using the CM1 antibody, electron microscopy did not reveal any staining in DA neurons exhibiting classic morphological features of apoptosis such as chromatin condensation. In contrast, those DA neurons that were immunoreactive for CM1 typically showed a dense perinuclear ER suggestive of increased protein synthesis. DA neurons negative for CM1 and without apoptotic features did not display dense perinuclear ER aggregation. Because protein synthesis has been shown to be a common although not necessary feature of neuronal apoptosis (36–38), we suggest that caspase-3 activation in these neurons marks the beginning of the effector phase of apoptosis. However, once the program has been completed, activated caspase-3 can no longer be detected, possibly because of degradation of the protease itself.
Low-Dose MPP+ Activates Caspase-3, Which Precedes Chromatin Condensation During Apoptosis.
The ultrastructural findings in the human postmortem brain are supported by the present in vitro data showing that caspase-3 is activated early in the course of DA cell death after treatment with a low concentration of MPP+. Concomitant DNA condensation as assessed by Hoechst 33258 staining can be observed only in late stages of cell demise and probably reflects the transition from caspase activation to cell death. The reason why CM1 staining still can be detected in cells displaying signs of nuclear fragmentation in DA cell cultures compared with the ultrastructural postmortem analysis may be related to the mode of identification of DA neurons: in culture, TH positivity is required for phenotypical identification, which implies at least basic cell viability and, thus, overall enzymatic activity, whereas human DA neurons are identified by their neuromelanin content, the presence of which is not related to cell viability. DA neurons positive for CM1 in the control condition constitute, independently of the time of analysis, around 10% of all DA neurons. This caspase-3 activation probably reflects spontaneous cell death related to astrocytic factors, which results in the death of about 80% of all DA neurons within the first 10 days of culture, representing a mean loss of 8% of cells per day (39) and, therefore, is in agreement with the present data on CM1 immunoreactivity. The finding that caspase-3 activation precedes DNA condensation suggests that it is not a consequence of cell death but associated with this process. Thus, caspase-3 activation after MPP+ intoxication strengthens the changes observed postmortem in a PD model open to experimental manipulation. This also appears important because caspase-3 activation in experimental PD models so far has relied on inhibitor studies, which are unable to demonstrate unambiguously specific caspase-3 activation (14, 16). The caspase inhibitor most widely used to block caspase-3, zDEVD-CHO, also inhibits caspase-7 potently (40).
In conclusion, although activation of several distinct upstream pathways may lead to degeneration of nigral DA neurons, caspase-3 may represent a common integration point and, thus, may constitute an attractive target for antiapoptotic therapy in PD. In this context, it is interesting to note that caspase-3 activation precedes chromatin condensation and the final breakdown of the cell, a stage at which neuroprotective strategies are likely to fail. However, at present, potential side effects induced by the use of caspase inhibitors, such as neoplasia formation and the induction of autoimmune disorders, must be taken into consideration unless cell-specific drug targeting can be achieved. Furthermore, the long-term viability and functionality of DA neurons potentially rescued by caspase inhibitors remain to be determined.
Acknowledgments
This study was supported by Institut National de la Santé et de la Recherche Médicale and the National Parkinson Foundation, Inc., Miami. A.H. is a postdoctoral fellow of the Deutsche Forschungsgemeinschaft. S.H. is a fellow of the Fondation pour la Recherche Médicale. P.P.M. is supported by Pierre Fabre Laboratories, Paris. B.A.F. is supported by the Association Claude Bernard pour la Recherche Biologique et Médicale dans les Hôpitaux de l'Assistance Publique à Paris.
Abbreviations
- CGS
central gray substance
- DA
dopaminergic
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson's disease
- TH
tyrosine hydroxylase
- SNpc
substantia nigra pars compacta
- VTA
ventral tegmental area
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040556597.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040556597
References
- 1.Anglade P, Vyas S, Javoy-Agid F, Herrero M T, Michel P P, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch E C, Agid Y. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 2.Kingsbury A E, Mardsen C D, Foster O J. Mov Disord. 1998;13:877–884. doi: 10.1002/mds.870130604. [DOI] [PubMed] [Google Scholar]
- 3.Tatton N A, Mallean-Fraser A, Tatton W G, Perl D P, Olanow C W. Ann Neurol. 1998;44,Suppl. 1:142–148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 4.Spooren W P J M, Gentsch C, Wiessner C. Neuroscience. 1998;85:649–651. doi: 10.1016/s0306-4522(97)00640-4. [DOI] [PubMed] [Google Scholar]
- 5.Tatton N A, Kish S. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- 6.Burke R E, Kholodilov N C. Ann Neurol. 1998;44,Suppl. 1:126–133. doi: 10.1002/ana.410440719. [DOI] [PubMed] [Google Scholar]
- 7.Cohen G M. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicholson D W, Ali A, Thornberry N A, Vaillancourt J P, King C K, Gallant M, Gareau Y, Griffin P R, Labelle M, Lazebnik Y A, et al. Nature (London) 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 9.Tewari M, Quan L T, O'Rourke K, Desnoyers S, Zeng Z, Beidler D R, Poirier G G, Salvesen G S, Dixit V M. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 10.Kuida K, Zheng T S, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell R A. Nature (London) 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 11.Bergeron L, Yuan J. Curr Opin Neurobiol. 1998;8:55–63. doi: 10.1016/s0959-4388(98)80008-1. [DOI] [PubMed] [Google Scholar]
- 12.Pettmann B, Henderson C E. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 13.Schulz J B, Weller M, Moskowitz M A. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 14.Dodel R C, Du Y, Bales K R, Ling Z D, Carvey P M, Paul S M. Neuroscience. 1998;86:701–707. doi: 10.1016/s0306-4522(98)00154-7. [DOI] [PubMed] [Google Scholar]
- 15.Dodel R C, Du Y, Bales K R, Ling Z, Carvey P M, Paul S M. Brain Res Mol Brain Res. 1999;64:141–148. doi: 10.1016/s0169-328x(98)00318-0. [DOI] [PubMed] [Google Scholar]
- 16.Lotharius J, Dugan L L, O'Malley K L. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirsch E C, Graybiel A M, Agid Y. Nature (London) 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan A, Roth K A, Sayers R O, Shindler K S, Wong A M, Fritz L C, Tomaselli K J. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 19.Hunot S, Brugg B, Ricard D, Michel P P, Muriel M P, Ruberg M, Faucheux B A, Hirsch E C. Proc Natl Acad Sci USA. 1997;94:7531–7536. doi: 10.1073/pnas.94.14.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michel P P, Agid Y. J Neurochem. 1996;67:1633–1642. doi: 10.1046/j.1471-4159.1996.67041633.x. [DOI] [PubMed] [Google Scholar]
- 21.Krajewska M, Wang H G, Krajewski S, Zapata J, Shabaik A, Gascoyne R, Reed J C. Cancer Res. 1997;57:1605–1613. [PubMed] [Google Scholar]
- 22.Mouatt-Prigent A, Karlsson J O, Agid Y, Hirsch E C. Neuroscience. 1996;73:979–987. doi: 10.1016/0306-4522(96)00100-5. [DOI] [PubMed] [Google Scholar]
- 23.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, Agid Y, Dugas B, Hirsch E C. J Neurosci. 1999;19:3440–3447. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anglade P, Mouatt-Prigent A, Agid Y, Hirsch E C. Neurodegeneration. 1996;5:121–128. doi: 10.1006/neur.1996.0018. [DOI] [PubMed] [Google Scholar]
- 25.Fearnley J M, Lees A J. Brain. 1991;14:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 26.Du Y, Bales K R, Dodel R C, Hamilton-Byrd E, Horn J W, Czilli D L, Simmons L K, Ni B, Paul S M. Proc Natl Acad Sci USA. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon B S, Kholodilov N G, Oo T F, Kim S Y, Tomaselli K J, Srinivasan A, Stefanis L, Burke R E. J Neurochem. 1999;73:322–333. doi: 10.1046/j.1471-4159.1999.0730322.x. [DOI] [PubMed] [Google Scholar]
- 28.Yamada T, McGeer P L, Baimbridge K G, McGeer E G. Brain Res. 1990;526:303–307. doi: 10.1016/0006-8993(90)91236-a. [DOI] [PubMed] [Google Scholar]
- 29.Damier P, Hirsch E C, Agid Y, Graybiel A M. Brain. 1999;122:1437–1448. doi: 10.1093/brain/122.8.1437. [DOI] [PubMed] [Google Scholar]
- 30.Lang A E, Lozano A M. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 31.Jenner P, Olanow C W. Ann Neurol. 1998;44,Suppl. 1:72–84. [Google Scholar]
- 32.Nath R, Probert A, Jr, McGinnis K M, Wang K K W. J Neurochem. 1998;71:186–195. doi: 10.1046/j.1471-4159.1998.71010186.x. [DOI] [PubMed] [Google Scholar]
- 33.Anglade P, Vyas S, Hirsch E C, Agid Y. Histol Histopathol. 1997;12:603–610. [PubMed] [Google Scholar]
- 34.Tompkins M M, Basgal E J, Zamrini E, Hill W D. Am J Pathol. 1997;150:119–131. [PMC free article] [PubMed] [Google Scholar]
- 35.Cosulich S C, Savory P J, Clarke P R. Curr Biol. 1999;9:147–150. doi: 10.1016/s0960-9822(99)80068-2. [DOI] [PubMed] [Google Scholar]
- 36.Schulz J B, Weller M, Klockgether T. J Neurosci. 1996;15:4696–4706. doi: 10.1523/JNEUROSCI.16-15-04696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Armstrong R C, Aja T J, Hoang K D, Gaur S, Bai X, Alnemri E S, Litwack G, Karanewsky D S, Fritz L C, Tomaselli K J. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du Y, Dodel R C, Bales K R, Hamilton-Byrd E, Paul S M. J Neurochem. 1997;69:1382–1388. doi: 10.1046/j.1471-4159.1997.69041382.x. [DOI] [PubMed] [Google Scholar]
- 39.Michel P P, Marien M, Ruberg M, Colpaert F, Agid Y. J Neurochem. 1999;72:2074–2082. doi: 10.1046/j.1471-4159.1999.0722074.x. [DOI] [PubMed] [Google Scholar]
- 40.Garcia-Calvo M, Peterson E P, Leiting B, Ruel R, Nicholson D W, Thornberry N. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]