

Abstract
After liver injury, transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF) regulate the activation of hepatic stellate cells (HSCs) and tissue remodeling. Mechanisms of PDGF signaling in the TGF-β-triggered cascade are not completely understood. TGF-β signaling involves phosphorylation of Smad2 and Smad3 at linker and C-terminal regions. Using antibodies to distinguish Smad2/3 phosphorylated at linker regions from those phosphorylated at C-terminal regions, we investigated Smad2/3-mediated signaling in rat liver injured by CCl4 administration and in cultured HSCs. In acute liver injury, Smad2/3 were transiently phosphorylated at both regions. Although linker-phosphorylated Smad2 remained in the cytoplasm of α-smooth muscle actin-immunoreactive mesenchymal cells adjacent to necrotic hepatocytes in centrilobular areas, linker-phosphorylated Smad3 accumulated in the nuclei. c-Jun N-terminal kinase (JNK) in the activated HSCs directly phosphorylated Smad2/3 at linker regions. Co-treatment of primary cultured HSCs with TGF-β and PDGF activated the JNK pathway, subsequently inducing endogenous linker phosphorylation of Smad2/3. The JNK pathway may be involved in migration of resident HSCs within the space of Disse to the sites of tissue damage because the JNK inhibitor SP600125 inhibited HSC migration induced by TGF-β and PDGF signals. Moreover, treatment of HSCs with both TGF-β and PDGF increased transcriptional activity of plasminogen activator inhibitor-1 through linker phosphorylation of Smad3. In conclusion, TGF-β and PDGF activate HSCs by transmitting their signals through JNK-mediated Smad2/3 phosphorylation at linker regions, both in vivo and in vitro.
Hepatic stellate cells (HSCs) characterized by retinoid droplets in the cytoplasm, are present in the space of Disse. After acute and chronic liver tissue damage, HSCs acquire an activated phenotype, which includes increased synthesis of extracellular matrix (ECM) components, proliferation, and migration. Several soluble factors, including growth factors, cytokines, chemokines, and oxidative stress products, play a role in the activation of HSCs.1 After liver injury, these factors directed at specific cell targets are simultaneously active in the tissue and are responsible for tissue remodeling.
The biological action of transforming growth factor-β (TGF-β) on HSCs is primarily related to the profibrogenic role of this factor.1 TGF-β is the most potent stimulus for production of ECM by HSCs.2 Accumulation of ECM proteins in the activated HSCs can be explained not only by increased ECM synthesis, but also by decreased degradation. In particular, plasminogen activator inhibitor-1 (PAI-1) and tissue inhibitor of metalloproteinase, two inhibitors of ECM-degrading enzyme, are strongly up-regulated by TGF-β in HSCs.3,4 PAI-1 inactivates urokinase plasminogen activator, but it also has a role in cell migration.5,6 Among other polypeptide growth factors potentially involved in the activation of HSCs, platelet-derived growth factor (PDGF) is the most potent mitogen for cultured HSCs isolated from liver.1,4 In addition, PDGF promotes HSC migration from the space of Disse to the sites of tissue repair.1
Throughout the past decade, the major intracellular signaling pathways elicited by these factors have been elucidated. TGF-β initiates signaling by binding to type I and type II receptor serine/threonine kinases on the cell surface. Receptor-activated Smads (R-Smads) such as Smad2 and Smad3 are phosphorylated by the activated TGF-β type I receptor (TβRI) on the C-terminal SXS motif. The activated R-Smads form the complex with the common partner Smad4. The complexes translocate to the nucleus, where they regulate target gene expression both by direct DNA binding and by interaction with other transcription factors, co-activators, and co-repressors.7–9 One of these TGF-β-induced transcriptions involves the Smad7 gene. Smad7 interacts stably with the activated TβRI receptor to inhibit TGF-β-mediated phosphorylation of Smad2 and Smad3.10
Signaling by PDGF begins by interaction with transmembrane receptor tyrosine kinases.11 Multiple signaling pathways originating from these receptors have been identified. The most prominent pathways are mediated by members of the mitogen-activated protein kinase (MAPK) family, which includes the extracellular signal-regulated protein kinase (ERK) pathway and two stress-activated protein kinase (SAPK) pathways: the c-Jun N-terminal kinase (JNK) and the p38 pathway.12 MAPK is capable of phosphorylating transcription factors, which are important in initiating cell proliferation, migration, and apoptosis.12 Notably, TGF-β also induces activation of MAPK pathways through the upstream mediators Ras, RhoA, and TGF-activated kinase 1 (TAK1).13 To investigate the roles of Smad2 and Smad3 phosphorylation in TGF-β signal transduction, we developed four types of polyclonal antibodies (Abs) in our laboratory that specifically recognized the phosphorylated linker regions and the phosphorylated C-terminal SXS regions in Smad2 and Smad3.14,15 Studies using the Abs demonstrated that JNK and/or p38 MAPK activated on TGF-β treatment could directly phosphorylate Smad2 and Smad3 at linker regions.
Although TGF-β- and PDGF-mediated signals are an important component of HSC activation, proof has been lacking that the event occurs in vivo. Additionally, cellular distribution of phosphorylated Smad2/3 has not been studied in normal or injured tissues. Lack of antibodies (Abs) able to distinguish different phosphorylation sites of Smad2/3 has impeded determination of phosphorylation sites in tissues. Using the Abs against phosphorylated Smad2/3, we investigated Smad2/3-mediated signaling in a rat model of liver injury and in cultured HSCs. Our results indicated that after injury in vivo, rapid JNK activation occurred in HSCs accompanied by Smad2/3 phosphorylation at linker regions. Moreover, co-treatment of primary cultured HSCs with TGF-β and PDGF activated the JNK pathway, leading to phosphorylation of Smad2/3 at linker regions. JNK activation induced by TGF-β and PDGF could be involved in migration of resident HSCs within the space of Disse to the area of tissue damage because treatment of HSCs with a JNK inhibitor SP600125 significantly inhibited HSC migration triggered by TGF-β and/or PDGF stimulation. Finally, the PDGF signaling pathway alone activated PAI-1 transcriptional activity in primary cultured HSCs. In addition, PDGF showed potent synergy with TGF-β signaling through Smad3 phosphorylation at the linker region. Collectively, JNK-mediated Smad2/3 phosphorylation at linker regions played an important role in transmitting the signals from TGF-β and PDGF in the activated HSCs after acute liver injury.
Experimental Procedures
Animals
Male Wistar rats were obtained from Nihon Dobutsu Co. (Osaka, Japan) and were used in all experiments. All procedures were performed in accordance with the Declaration of Helsinki and the Guiding Principles on the Care and Use of Animals. To establish acute CCl4 intoxication, rats weighting ∼200 g were given a single intragastric dose of 0.5 ml of a 1:1 (v/v) mixture of CCl4 in olive oil per 100 g of body weight. These animals were sacrificed at 0, 36, and 72 hours after administration of the agent. Animals treated only with mineral oil were used as controls.
Cell Separation Procedure
HSCs were isolated from normal rat livers or injured livers at the indicated times after CCl4 administration. HSCs were obtained using collagenase and pronase-E digestion.16 The liver was perfused in a noncirculating system for 2 minutes at 37°C (40 ml/minute). The perfusion solution was calcium-free Hanks’ buffer. The liver was then circulated with the perfusion solution for 5 minutes. A solution (200 ml) consisting of 0.1% collagenase (Wako, Osaka, Japan) in 10 mmol/L HEPES, pH 7.5, with 5 mmol/L calcium without EGTA was recirculated for 5 to 10 minutes. The liver was cut into pieces and incubated in 100 ml of Gey’s balanced salt solution with 0.1% pronase-E (Sigma, St. Louis, MO) on a magnetic stirrer at 37°C for 30 minutes. At the end of the incubation period, the suspension was filtered through nylon gauze and the filtrate was centrifuged at 450 × g for 7 minutes. The collected cells were centrifuged on a two-layer density cushion of Nycodenz (Nycomed, Oslo, Norway) (Gey’s balanced salt solution/8% Nycodenz) at 1400 × g for 17 minutes. The HSC fraction was collected from the upper layer. The purity of the HSCs was greater than 90% as assessed by typical morphological features, mainly the presence of vitamin A droplets, and the immunological staining of desmin.
Immunoprecipitation and Immunoblotting
HSCs freshly isolated after intoxication with CCl4, or primary cultured HSCs treated with 10 pmol/L TGF-β1 (R&D Systems, Minneapolis, MN) and/or 400 pmol/L PDGF (R&D Systems) for 30 minutes, were extracted with cell lysis buffer. Cell extracts were subjected to immunoprecipitation with anti-Smad2/3 Ab (BD Bioscience, San Jose, CA), followed by adsorption to protein G-Sepharose (Pharmacia, Peapack, NJ). The phosphorylation levels of Smad2/3 were analyzed using rabbit polyclonal anti-pSmad2L (Ser 249/254) Ab, anti-pSmad2C (Ser 465/467) Ab, anti-pSmad3L (Ser 207/212) Ab, and anti-pSmad3C (Ser423/425) Ab, as described previously.14,15 Immunoblots also were analyzed using rabbit polyclonal anti-phosphorylated JNK1/2 Ab (Promega, Madison, WI) and rabbit polyclonal anti-JNK1/2 Ab (Cell Signaling Technology, Beverly, MA) as primary Abs.
In Vitro Kinase Assays
Bacterial expression and purification of GST-Smad2 and GST-Smad3 were performed according to the manufacturer’s instructions (Amersham Biosciences, Piscataway, NJ). Primary cultured HSCs were starved for 15 hours in serum-free medium, and then were incubated with 10 pmol/L TGF-β1 and/or 400 pmol/L PDGF for 15 minutes. HSCs freshly isolated after intoxication with CCl4 or primary cultured HSCs were extracted with cell lysis buffer. Endogenous kinases were isolated from the cell extracts using anti-phospho-JNK1/2 Ab (Promega). The immune complex was collected with protein G-Sepharose. The samples were washed with kinase assay buffer (25 mmol/L Tris-HCl, pH 7.5, 5 mmol/L β-glycerophosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4, and 10 mmol/L MgCl2). Pellets were resuspended in 50 μl of kinase assay buffer supplemented with 100 μmol/L ATP, 2 μg of GST-Smad2, or GST-Smad3. Reactions were performed at 30°C for 30 minutes and then were stopped with Laemmli sample buffer. The phosphorylation sites of Smad2/3 were determined by immunoblot using each anti-phospho-Smad2/3 Ab.
Immunohistochemical Analyses
After freshly harvested livers from normal and CCl4-treated rats were fixed in 3% formalin for 2 to 3 days, the livers were dehydrated through a graded alcohol series, embedded in paraffin, and sectioned in mirror image at a thickness of 3 μm. The sections then were deparaffinized in xylene and rehydrated. Nonenzymatic antigen retrieval was performed by heating the sections to 121°C in 0.01 mol/L sodium citrate buffer (pH 6.0) for 10 minutes. After cooling, sections were rinsed in Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated in methanol-3% H2O2 for 30 minutes to quench endogenous peroxidase activity. Sections then were rinsed with TBST. Primary Abs were incubated overnight at 4°C in a humid chamber. Primary Abs used in this study included mouse monoclonal anti-α-smooth muscle actin (SMA) Ab (clone 1A4, 1.7 μg/ml; DAKO, Glostrup, Denmark), mouse monoclonal anti-PCNA Ab (clone PC10, 2.4 μg/ml; DAKO), anti-pSmad2L (0.5 μg/ml), anti-pSmad2C (0.5 μg/ml), anti-pSmad3L (1 μg/ml), and anti-pSmad3C (1 μg/ml). Anti-pSmad3C Ab weakly cross-reacted with the C-terminally phosphorylated Smad2. To block the binding of anti-Smad3C Ab to the phosphorylated domains in Smad2, anti-pSmad3C Ab was adsorbed with 1 μg/ml C-terminally phosphorylated Smad2 peptide. After sections were rinsed in TBST, peroxidase-labeled polymer conjugated to goat anti-mouse or anti-rabbit immunoglobulin (DAKO) was incubated for 1 hour at room temperature and rinsed in TBST. Finally, the sections were developed with 3,3′-diaminobenzidine tetrahydrochloride (Vector Laboratories, Burlingame, CA) counterstained with hematoxylin (Merck, Darmstadt, Germany) and mounted under coverslips.
Migration Assay
To mimic the microenvironment of the space of Disse, the membranes with 8-μm pores covered with Matrigel (BD Biosciences, Bedford, MA) on the upper side were coated with type I collagen on the lower side. HSCs (2 × 104/well) were added into the upper chamber, and were serum-starved in the absence or presence of 10 μmol/L JNK inhibitor SP600125 for 6 hours. The bottom wells of the chamber were filled with Dulbecco’s modified Eagle’s medium containing 10 pmol/L TGF-β1 and/or 400 pmol/L PDGF. The Boyden chamber was incubated for 12 hours to allow possible migration of HSCs through the membrane into the lower chamber. The chambers were then immersed in 100% methanol for 1 minute for fixation, and all cells were then stained by hematoxylin. The cells remaining on the top surface of the membrane were completely removed with a cotton swab, and the membrane was removed from the chamber and mounted on a glass slide. The number of infiltrating cells was counted in five regions selected at random, and the extent of invading cells was determined by the mean count. Duplicate filters were used, and the experiments were repeated three times.
Transcriptional Response Assay
HSCs were seeded at 2 × 105 cells per well in six-well clusters. Twelve hours later, cells were subjected to transfection with LipofectAMINE (Invitrogen, Carlsbad, CA), 0.4 μg of reporter plasmid, and the indicated constructs, or with empty vector alone. After changing the medium, the cells were incubated for another 30 hours in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. After washing the cells with the medium, the cells were incubated for 12 hours in serum-free Dulbecco’s modified Eagle’s medium in the absence or presence of 10 pmol/L TGF-β1 and/or 400 pmol/L PDGF. Finally, they were lysed, and the luciferase activity of the cell extracts was measured by a luminometer (Berthold, Bad Wildbad, Germany) using the dual-luciferase reporter assay system (Promega). The luciferase activities were normalized on the basis of the Renilla luciferase activity.
Statistical Analysis
All data are expressed as the mean (SD). Experimental and control values were compared using the unpaired Student’s t-test and analysis of variance. P < 0.05 was considered to indicate significance.
Results
Differential Localization of pSmad2L and pSmad3L in α-SMA-Immunoreactive Mesenchymal Cells in the Injured Liver
Administration of a single dose of CCl4 resulted in steatosis and necrosis of centrilobular hepatocytes within 2 days. The centrilobular area in the rat liver 36 hours after CCl4 intoxication was characterized by necrotic hepatocytes, acidophilic bodies, and a dense cellular infiltrate containing macrophages and lymphocytes. At this time point, TGF-β1 and ECM constitutions including PAI-1 and α2(I) procollagen are highly expressed in the activated HSCs.17 Necrotic material tended to decrease at 72 hours when TGF-β1- and ECM-related transcripts have decreased.
Smad2 and Smad3 have two major phosphorylation sites at linker regions and at C-terminal regions.18,19 To investigate Smad2/3-mediated signaling in vivo, we generated four Abs specific to each phosphorylation site (Figure 1).15 Using these Abs, we revealed that Smad3 phosphorylated at the linker region and that phosphorylated at the C-terminal region existed as separate molecules and transmitted distinct signals (unpublished data). Accordingly, each molecule including Smad2/3 phosphorylated at C-terminal regions (pSmad2C and pSmad3C) and Smad2/3 phosphorylated at linker regions (pSmad2L and pSmad3L), can be localized in different cell types, and can be detected in different intracellular locations, even in the same cell.
Figure 1.

Antibodies selectively distinguish Smad2/3 phosphorylation at linker regions and C-terminal SSXS regions. Smad2 and Smad3 proteins as well as domain-specific Abs are shown schematically. Anti-pSmad2L (Ser 249/254) Ab and anti-pSmad3L (Ser 207/212) Ab recognize SAPK-dependent phosphorylation sites in Smad2 and Smad3, whereas anti-pSmad2C (Ser 465/467) Ab and anti-pSmad3C (Ser 423/425) Ab recognize phosphorylated C-terminal SXS sites in Smad2/3 activated by TβRI.
Figure 2 presents the distribution of pSmad2L and pSmad3L in normal rat liver and injured liver. We tried to confirm the nuclear and cytoplasmic location of pSmad2L and pSmad3L and identify hepatocytes and mesenchymal cells in normal rat liver and injured liver. For this, the sections stained using anti-pSmad2L Ab and anti-pSmad3L Ab paired with mirror image sections using anti-α-SMA Ab and anti-proliferating cell nuclear antigen (PCNA) Ab. α-SMA was up-regulated in the activated HSCs, displaying the typical cytoplasmic localization.20,21 PCNA staining was used as a nuclear marker.22
Figure 2.

Differential localization of pSmad2L and pSmad3L in α-SMA-immunoreactive mesenchymal cells in the injured liver. Formalin-fixed, paraffin-embedded sections of normal rat liver (top) and injured liver at 36 hours after CCl4 intoxication (middle and bottom) were stained with anti-pSmad2L Ab (α pSmad2L) and anti-pSmad3L Ab (α pSmad3L). These sections paired with mirror image sections stained using anti-α-SMA Ab (α α-SMA) and anti-PCNA Ab (α PCNA). The Abs then were bounded by goat anti-mouse immunoglobulins conjugated with peroxidase-labeled polymer. Peroxidase activity was detected by 3,3′-diaminobenzidine tetrahydrochloride. Representative combinations are shown for: A, pSmad2L and α-SMA; B, pSmad2L and PCNA; C, pSmad3L and α-SMA; D, pSmad3L and PCNA. Bottom: Higher magnification of the boxed areas in middle. All sections were counterstained with hematoxylin (blue). Brown indicates specific Ab reactivity. In normal rat liver, Smad2 and Smad3 were minimally phosphorylated at linker regions (top panels in α pSmad2L and α pSmad3L). At 36 hours, pSmad2L was predominantly localized in the cytoplasm of α-SMA-immunoreactive mesenchymal cells adjacent to necrotic hepatocytes in centrilobular areas (A and B, middle and bottom). In contrast, pSmad3L was located in nuclei of the α-SMA-immunoreactive mesenchymal cells (C and D, middle and bottom). Scale bars, 50 μm.
Immunostaining of normal rat liver with specific Abs to pSmad2L or pSmad3L showed scant phosphorylation of Smad2 and Smad3 throughout the liver (Figure 2, top panels in α pSmad2L and α pSmad3L columns). The phosphorylation was induced in the injured liver at 36 hours after CCl4 treatment (Figure 2, middle panels in α pSmad2L and α pSmad3L columns). Simultaneously, marked α-SMA immunoreactivity was observed in groups of mesenchymal cells adjacent to necrotic hepatocytes in centrilobular areas in the injured liver (Figure 2, A and C; middle panels in α α-SMA columns). Hepatocytes were constantly α-SMA-negative. The distributions of pSmad2L and pSmad3L fitted well with the pattern obtained by α-SMA immunolabeling in mirror image sections (Figure 2, A and C; middle). Surprisingly, pSmad2L and pSmad3L were undetectable in the majority of hepatocytes. pSmad2L and pSmad3L were differentially localized in α-SMA-immunoreactive mesenchymal cells (Figure 2, A and C; bottom). In contrast to focal or diffuse cytoplasmic localization of pSmad2L (Figure 2, A and B; bottom), pSmad3L accumulated in nuclei of the cells (Figure 2, C and D; bottom). Amounts of such phosphorylation apparent in mesenchymal cells surrounding centrilobular areas decreased at 72 hours after CCl4 intoxication (unpublished data).
We further investigated the distribution of pSmad2C and pSmad3C in normal rat liver and injured liver. Normal rat liver showed slight phosphorylation of Smad2/3 at C-terminal regions (Figure 3, top). In contrast to scantly phosphorylated states of Smad2/3L in hepatocytes, amounts of the C-terminal phosphorylation were increased in the nuclei of hepatocytes at 36 hours after CCl4 intoxication (Figure 3, middle and bottom). Mesenchymal cells surrounding centrilobular areas also showed an increased pSmad2/3C in the nuclei. The C-terminal phosphorylation decreased at 72 hours after CCl4 intoxication (unpublished data).
Figure 3.

Both pSmad2C and pSmad3C are localized in nuclei of hepatocytes and mesenchymal cells in injured liver. Formalin-fixed, paraffin-embedded sections of normal rat liver and injured liver were stained with the Abs indicated, which in turn were bounded by goat anti-rabbit immunoglobulins conjugated to peroxidase-labeled polymer. Peroxidase activity then was detected by 3,3′-diaminobenzidine tetrahydrochloride. Representative examples are shown for pSmad2C (α pSmad2C) and pSmad3C (α pSmad3C) immunostaining in normal liver (top), and injured liver at 36 hours after CCl4 intoxication (middle and bottom). Bottom: Higher magnification of the boxed areas in the middle. All sections were counterstained with hematoxylin (blue). Brown indicates specific Ab reactivity. In normal rat liver, Smad2 and Smad3 were slightly phosphorylated at C-terminal regions (top). Amounts of the C-terminal phosphorylation in the nuclei of hepatocytes and mesenchymal cells surrounding centrilobular areas increased at 36 hours after CCl4 intoxication (middle and bottom). Scale bars, 50 μm.
Smad2 and Smad3 Are Transiently Phosphorylated Not Only at C-Terminal Regions but Also at Linker Regions in the Activated HSCs after CCl4 Intoxication
To quantify phosphorylation of each domain in Smad2 and Smad3, we isolated HSCs from normal and CCl4-injured livers, and performed immunoblotting of cell extracts with domain-specific anti-phospho-Abs (Figure 4). Neither the linker regions nor the C-terminal regions of Smad2/3 were phosphorylated in the quiescent HSCs. When cells were obtained from livers 36 hours after treatment with CCl4, Smad2/3 in the activated HSCs were phosphorylated not only at C-terminal regions but also at linker regions. Phosphorylation had decreased in HSCs by 72 hours. Taken together with the results obtained from immunohistochemical analyses (Figures 2 and 3), these findings indicated that phosphorylation of each domain in Smad2 and Smad3 occurred transiently within α-SMA-immunoreactive HSCs during the process of liver regeneration after a single chemical insult.
Figure 4.

Smad2 and Smad3 are phosphorylated not only at C-terminal regions but also at linker regions in the activated HSCs after CCl4 intoxication. HSCs were obtained from normal rat liver and injured livers at 36 and 72 hours after CCl4 intoxication. Cell lysates were subjected to anti-Smad2/3 immunoprecipitation (IP), and were immunoblotted with each anti-pR-Smad Ab (top). Relative amounts of endogenous Smad2/3 were determined by immunoblotting (IB) using anti-Smad2/3 Ab (bottom).
JNK in the Activated HSCs from CCl4-Injured Liver Directly Phosphorylates Smad2/3 at Linker Regions
Recently, activated SAPK has been implicated in a wide range of physiological responses of cells to various injuries.12 In particular, SAPK activation is involved in migration and proliferation of HSCs.23 Moreover, anti-pSmad2L Ab and anti-pSmad3L Ab recognize SAPK-dependent phosphorylation sites in Smad2 and Smad3.15 Accordingly, we focused on SAPK activities in the activated HSCs after a single chemical insult. We investigated activation of JNK by immunoblotting of HSCs at various time points after CCl4 intoxication, using Abs against the active, phosphorylated form of JNK (Figure 5, top). Neither the linker regions nor the C-terminal regions of Smad2/3 were phosphorylated in quiescent HSCs. JNK showed a high degree of phosphorylation in the activated HSCs at 36 hours after CCl4 intoxication. JNK phosphorylation had decreased in HSCs by 72 hours. We also investigated activation of p38 MAPK after a single chemical insult. As with JNK, p38 MAPK was phosphorylated transiently in the activated HSCs at 36 hours (unpublished data). Because the profile of Smad2/3 phosphorylation in HSC after a single chemical insult resembled that of JNK activation, these events may be causally linked.
Figure 5.
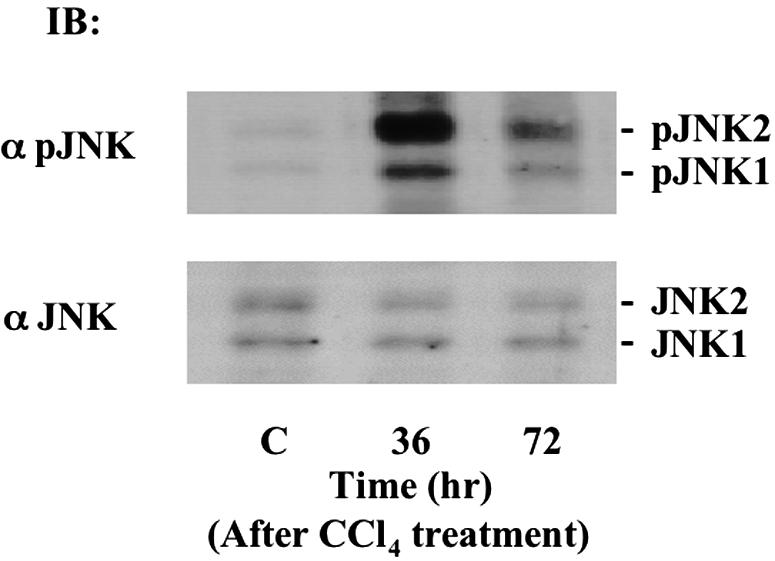
JNK is phosphorylated in the activated HSCs after CCl4 intoxication. HSCs were obtained from normal rat liver and injured livers at 36 and 72 hours after CCl4 intoxication. Phosphorylation of endogenous JNK1/2 was analyzed by immunoblotting with anti-pJNK1/2 Ab (α pJNK1/2; top). Expression of endogenous JNK1/2 was monitored by immunoblotting using anti-JNK1/2 Ab (α JNK1/2; bottom).
To address the functional relationship between activated SAPK and Smad2/3 phosphorylation in the activated HSCs, we performed an in vitro kinase assay. Phosphorylation of Smad2/3 was assessed by immunoblotting using anti-pSmad2L Ab or pSmad3L Ab. JNK in the activated HSCs obtained 36 hours after CCl4 poisoning could directly phosphorylate Smad2 and Smad3 at linker regions (Figure 6, top). As JNK activation declined by 72 hours, the extent of Smad2/3 phosphorylation at linker regions decreased. On the other hand, p38 MAPK specifically phosphorylated the linker region of Smad3 (unpublished data). Collectively, each SAPK showed distinct substrate specificity. Taken together with the nuclear localization of pSmad3L in the activated HSCs (Figure 2, C and D; bottom), SAPK directly phosphorylated the linker region of Smad3, leading to translocation of the protein into the cell nucleus.
Figure 6.

JNK in the activated HSCs after CCl4 intoxication directly phosphorylates Smad2/3 at linker regions. HSCs were extracted from normal rat liver and injured livers at 36 and 72 hours after CCl4 intoxication. Immunoprecipitate of cell extracts with Ab against phosphorylated JNK were mixed with bacterially expressed GST-Smad2 (A) and GST-Smad3 (B). Phosphorylation of Smad2 and Smad3 was analyzed by immunoblotting (IB) using each anti-pSmad2/3 Ab (top). Total Smad2 and Smad3 were monitored by immunoblotting (IB) using anti-Smad2/3 Ab (bottom).
Both TGF-β and PDGF Signals Phosphorylate Linker Regions of Smad2/3 in HSCs
Both TGF-β and PDGF are expressed locally in the activated HSCs after acute liver injury.16,24 To investigate how TGF-β- and PDGF-mediated signals participate in phosphorylation of Smad2/3, we isolated HSCs from normal liver by density gradient centrifugation and cultured them on plastic dishes. We monitored the phosphorylation state of Smad2/3 by immunoblot analyses with anti-phosphospecific Smad2/3 Abs in cell extracts prepared from HSCs stimulated with TGF-β and/or PDGF. TGF-β and PDGF treatment induced phosphorylation of Smad2/3 within 10 minutes, reaching a maximum at 30 minutes, and then decreasing (unpublished data). TGF-β treatment enhanced Smad2/3 phosphorylation at linker regions as well as at C-terminal regions. On the other hand, the PDGF signal specifically phosphorylated their linker regions (Figure 7). Smad2 phosphorylation at the linker region was more sensitive to PDGF than to TGF-β, whereas the effect of TGF-β on Smad3 phosphorylation at the linker region was nearly equal to that observed with PDGF. Smad2/3 phosphorylation at linker regions was stimulated additionally by combined TGF-β and PDGF treatment. Taken together, these results suggested that molecules activated in common by TGF-β and PDGF signals might phosphorylate the linker regions of Smad2 and Smad3.
Figure 7.

Both TGF-β and PDGF signals phosphorylate linker regions of Smad2/3 in HSCs. HSCs were incubated with or without TGF-β1 and PDGF for 30 minutes as indicated. Followed by immunoprecipitation (IP) of cell lysates with anti-Smad2/3 Ab, phosphorylation of Smad2 and Smad3 was analyzed by immunoblotting (IB) using each anti-pSmad2/3 Ab. Expression of endogenous Smad2 and Smad3 was monitored by immunoblotting using anti-Smad2/3 Ab (bottom).
TGF-β and PDGF Signals in JNK Phosphorylation Show Collaborative Interactions
MAPK pathways were shown to be activated in response to TGF-β and PDGF.12,25 We therefore investigated the time course of JNK activation in response to TGF-β stimulation. Both TGF-β and PDGF rapidly induced JNK phosphorylation in primary cultured HSCs, phosphorylation had occurred at 5 minutes after TGF-β or PDGF treatment, reaching a maximum at 15 minutes, and then gradually declining (unpublished data). We next investigated whether PDGF could activate JNK with TGF-β signaling at 15 minutes after simultaneous treatment with TGF-β and PDGF (Figure 8). As previously mentioned TGF-β or PDGF treatment resulted in a clear increase in JNK phosphorylation. Moreover, phosphorylation was stimulated additionally by treatment with both TGF-β and PDGF.
Figure 8.

Both TGF-β and PDGF signals stimulate JNK phosphorylation in HSCs. HSCs were incubated with TGF-β1 and/or PDGF for 15 minutes. Phosphorylation of endogenous JNK1/2 was analyzed by immunoblotting using anti-pJNK1/2 Ab (α pJNK1/2; top). Expression of endogenous JNK1/2 was monitored by immunoblot using anti-JNK1/2 Ab (α JNK1/2; bottom).
JNK Activated by TGF-β and PDGF Signals in HSCs Directly Phosphorylates Smad2/3 at Linker Regions
Because TGF-β and PDGF signals resulted in phosphorylation of JNK (Figure 8) followed by Smad2/3 phosphorylation at linker regions (Figure 7), we further investigated whether or not endogenous MAPK activated by TGF-β and PDGF signals in primary cultured HSCs could directly phosphorylate Smad2 and Smad3 in vitro. The serum-starved HSCs were extracted at 15 minutes after TGF-β and/or PDGF treatment, when JNK phosphorylation had reached a maximum (Figure 8). Degree of Smad2/3 phosphorylation was monitored by immunoblotting using anti-pSmad2L Ab or pSmad3L Ab. JNK additionally activated by both TGF-β and PDGF signals could directly phosphorylate Smad2 and Smad3 at linker regions in vitro (Figure 9, top). However, JNK failed to phosphorylate Smad2 or Smad3 at C-terminal regions on TGF-β or PDGF treatment (unpublished data). Collectively, these data indicated that both Smad2 and Smad3 served as substrates for JNK in vitro after TGF-β and PDGF treatment.
Figure 9.

JNK activated by TGF-β and PDGF signals in HSCs directly phosphorylates Smad2/3 at linker regions. HSCs were extracted at 15 minutes after TGF-β1 and/or PDGF treatment, and cell extracts were immunoprecipitated with anti-pJNK1/2 Ab. In vitro kinase assays were performed using GST-Smad2 (A) and GST-Smad3 (B) as substrates. Phosphorylation states of Smad2/3 were analyzed by immunoblotting (IB) using each anti-pSmad2/3 Ab (top). Total Smad2 and Smad3 were monitored by immunoblot (IB) using anti-Smad2/3 Ab (bottom).
A JNK Inhibitor SP600125 Treatment Causes the Reduction of Migratory Response of HSCs to TGF-β and PDGF Stimulation
Migration of resident HSCs within the space of Disse to the area of tissue damage is considered important for tissue remodeling in response to liver injury.26,27 To study the migratory properties of the activated HSCs, we used a two-compartment Boyden chamber system that partially mimics in vivo space of Disse in healthy and injured livers.28 The upper chamber represented the normal space of Disse that contained a basement membrane-like matrix (Matrigel), which was rich in type IV collagen and anchors resident HSCs. The lower chamber represented the diseased space of Disse, containing growth factors and type I collagen. Without migratory stimuli from the lower chamber, only a few activated HSCs migrated through pores of the membrane (Figure 10). When the medium in the lower compartment was supplemented with either TGF-β or PDGF, the activated HSCs showed an increased migratory response. Additional treatment of PDGF with TGF-β increased the migration greater than that of TGF-β or PDGF treatment alone. Addition of a JNK inhibitor SP600125 to the supernatant in the upper chamber significantly inhibited migration of HSCs triggered by TGF-β and/or PDGF stimulation, suggesting a direct role of JNK pathway in facilitating HSC migration in response to TGF-β and PDGF stimulation.
Figure 10.

A JNK inhibitor SP600125 treatment causes the reduction of migratory response of HSCs to TGF-β and PDGF stimulation. HSCs were cultured on Matrigel for 12 hours with TGF-β1 and/or PDGF in the absence or presence of SP600125. After fixation with 100% methanol, the cells were stained by hematoxylin. The number of infiltrating cells was counted in five regions selected at random, and the extent of invading cells was determined by the mean count. Duplicate filters were used, and the experiments were repeated three times.
Both TGF-β and PDGF Signals Stimulate PAI-1 Transcriptional Activity through pSmad3L in HSCs
The processes of cellular migration are characterized, in part, by altered local proteolysis and stimulating cell growth. PAI-1, the main inhibitor of the urokinase-type plasminogen activator system, conducts the cells to migration and invasion by blocking cellular adhesion and by promoting basement membrane degradation.5,6 We therefore chose the PAI-1 gene response as an indicator of the phosphorylated Smad2/3 effects on HSC migration. For this we used PF1-Luc, because this segment in the PAI-1 promoter was sufficient to obtain TGF-β-dependent induction.29 HSCs transfected with PF1-Luc alone showed a low degree of transcriptional activity (Figure 11). TGF-β induced transcriptional activity, and PDGF treatment led to a twofold increase in the activity. Additional treatment with PDGF plus TGF-β increased transcriptional activity beyond that seen with TGF-β treatment alone. In cells co-transfected of PF1-Luc with Smad3WT, treatment with TGF-β increased transcriptional activity. PDGF treatment alone also activated transcription. Moreover, treatment with PDGF plus TGF-β led to a potent increase in transcriptional activity beyond that seen with TGF-β treatment alone. Co-transfection with Smad3EPSM, which lacked phosphorylation sites in the linker region, induced an increase of transcriptional activity triggered by TGF-β stimulation, but could not significantly modify transcriptional activity in HSCs treated with PDGF alone. Similarly, combined treatment with TGF-β and PDGF cells expressing Smad3EPSM did not lead to an additional increase in transcriptional activity. Co-transfection with Smad3(3S-A) as a control, which lacked C-terminal serine residues, induced an increase of the transcriptional activities triggered by either TGF-β or PDGF stimulation. Moreover, a combined treatment with TGF-β and PDGF of the cells expressing Smad3(3S-A) led to an additional increase in the activity. These results were similar to those in co-transfection with Smad3WT. Thus, the PDGF signal alone could activate PAI-1 transcriptional activity through pSmad3L. In addition, PDGF showed potent synergy with the TGF-β signal at this phosphorylation site.
Figure 11.

Both TGF-β and PDGF signals stimulate PAI-1 transcriptional activity through pSmad3L in HSCs. HSCs were transiently co-transfected of PF1-Luc with Smad3WT, Smad3EPSM, Smad3(3S-A), or empty control vector (pcDNA3). After changing the medium, cells were incubated for another 30 hours. Under serum-free conditions, the cells were incubated for 12 hours with TGF-β1, PDGF, or a combination of both. Luciferase activity was determined and normalized to transfection efficiency. Values of samples from cells transfected with PF1-Luc alone and those left untreated were set arbitrarily at 1. Results shown are the mean ± SD (n = 4) from a representative experiment. *, P < 0.05; **, P < 0.01, by one-way analysis of variance.
Discussion
We demonstrated here for the first time that pSmad2/3L mediated signaling in vivo. In the presence of TGF-β and PDGF after a single CCl4 administration, Smad2/3 in the activated HSCs were phosphorylated transiently at linker regions. Further, JNK activated by TGF-β and PDGF signals could directly phosphorylate the linker regions. pSmad3L was localized predominantly in the nuclei of α-SMA-positive cells within fibrotic septa in CCl4-treated rats. Moreover, PDGF treatment together with TGF-β enhanced PAI-1 transcription through pSmad3L. Stimulation of PAI-1 production might lead to an increase in migratory capacity of the activated HSCs, because PAI-1 conducts the cells to migration by blocking cellular adhesion and by promoting basement membrane degradation.5,6 JNK pathway is also involved in TGF-β and/or PDGF-induced migration of the activated HSCs. Together, these results indicated that TGF-β and PDGF could transmit the migratory signals of the activated HSCs through JNK-mediated Smad2/3 phosphorylation at linker regions.
Current evidence suggests that regulation of ECM production in acute and chronic liver disease may involve different mechanisms, although HSCs are the principal effector in both cases.3 HSCs undergo progressive activation to myofibroblast (MFB)-like cells in a state of chronic liver damage. On the other hand, considerable complexity of Smad2/3-mediated signaling can result from temporal variation of SAPK. Our current in vivo model reveals that SAPK activation results in phosphorylation of both Smad2L and Smad3L, particularly in situations in which SAPKs are activated transiently in HSCs after acute liver injury. When sustained activation of SAPK in MFBs is achieved through constitutive TGF-β signal during chronic liver injury, SAPK-dependent Smad3 phosphorylation alone promotes nuclear accumulation of Smad3, in which it stimulates ECM production in MFB.14 Additional complexity of signaling can be derived from the variety of Smad2/3 differentially phosphorylated by JNK and p38 MAPK. JNK phosphorylates both Smad2L and Smad3L, but p38 MAPK phosphorylates only Smad3L (unpublished data).
TGF-β regulates particularly rapid physiological processes. Because TGF-β-dependent induction of Smad7 ensures swift elimination of its signal, Smad7 is considered to act as an important physiological regulator of the TGF-β signal.10 We revealed that pSmad3L and pSmad3C transmitted their signals independently (unpublished data). Although pSmad3C can activate the Smad7 promoter, pSmad3L cannot transactivate the Smad7 gene. In contrast, pSmad3L and pSmad3C can enhance transcriptional activation of ECM genes such as PAI-1 and d2(I) procollagen genes.14 In HSCs after acute liver injury, TβRI activated by endogenous TGF-β signal phosphorylated Smad3C, further up-regulating Smad7 transcription. Subsequently, Smad7 terminates fibrogenic signals mediated by signal-transducing Smads, and could be involved in the transient response to the autocrine TGF-β signal after acute liver injury.17,30 In contrast, pSmad3L leads to constitutive fibrogenic signaling, owing to loss of responsiveness of the phosphorylated Smad3 to the Smad7 transcript in MFBs during chronic liver injury.14,17
Compensatory growth of the liver to regain mass lost by partial hepatectomy or chemical damage is orchestrated by the interplay of positive and negative polypeptide cytokines and growth factors.31 In addition to its potent role in promotion of ECM production in the activated HSCs, the TGF-β signal transduction system has been implicated in negative regulation of the growth response in hepatocytes. Our current evidence, however, suggests that activation of SAPK/Smads cascade is strictly restricted to mesenchymal cells including HSCs in vivo, and contributes to promote HSC migration through pSmad3L. Surprisingly, pSmad2L and pSmad3L were undetectable in hepatocytes (Figure 2). In contrast, both pSmad2C and pSmad3C were located predominantly in the nuclei of hepatocytes after the chemical insult (Figure 3). Taken together with the findings that TGF-β secreted by mesenchymal cells including HSCs is present within hepatocytes,32 TGF-β in hepatocytes mainly transmits the signal through pSmad2C and pSmad3C. In most normal epithelial cell types, TGF-β arrests cell-cycle progression in the G1 phase by up-regulating expression of cyclin-dependent kinase inhibitors (CDKIs), p21WAF1/CIP1, and/or p15INK4B.33,34 Induction of CDKI expression requires the C-terminal phosphorylation of Smad2 and Smad3.35,36 Accordingly, pSmad2/3C-mediated signaling could take part in growth inhibition through up-regulation of CDKI expression in the hepatocytes after acute liver injury.
Emerging evidence indicates that TGF-β and PDGF participate not only in a restoration of the normal hepatic structure if triggered by a single liver injury, but also in development of pathological fibrotic states.1–3 Our present findings concerning phosphorylation sites of Smad2/3 should help elucidate the molecular mechanisms by which Smad2/3 can mediate synergetic signals from serine kinase receptors and tyrosine kinase receptors. Future studies using our Abs also should enhance our understanding of TGF-β signal modification by JNK pathway in such pathological conditions as fulminant hepatitis and liver fibrosis. Ultimately, Smad2/3 phosphorylation might represent a potential target for therapeutic intervention.37
Acknowledgments
We thank Dr. R. Derynck (University of California at San Francisco, San Francisco, CA) for providing us with cDNAs of human Smad2 and Smad3.
Footnotes
Address reprint requests to Koichi Matsuzaki, M.D., Third Department of Internal Medicine, Kansai Medical University, 10-15 Fumizonocho, Moriguchi, Osaka 570-8507, Japan. E-mail: matsuzak@takii.kmu.ac.jp.
Supported by a grant-in-aid for scientific research from the Ministry of Education, Science, and Culture of Japan.
References
- Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis. 2001;21:397–416. doi: 10.1055/s-2001-17554. [DOI] [PubMed] [Google Scholar]
- Bissell DM, Roulot D, George J. Transforming growth factor β and the liver. Hepatology. 2001;34:859–867. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- Fibbi G, Pucci M, D’Alessio S, Grappone C, Pellegrini G, Salzano R, Casini A, Milani S, Del Rosso M. Transforming growth factor beta-1 stimulates invasivity of hepatic stellate cells by engagement of the cell-associated fibrinolytic system. Growth Factors. 2001;19:87–100. doi: 10.3109/08977190109001078. [DOI] [PubMed] [Google Scholar]
- Gutierrez LS, Schulman A, Brito-Robinson T, Noria F, Ploplis VA, Castellino FJ. Tumor development is retarded in mice lacking the gene for urokinase-type plasminogen activator or its inhibitor, plasminogen activator inhibitor-1. Cancer Res. 2000;60:5839–5847. [PubMed] [Google Scholar]
- Hirashima Y, Kobayashi H, Suzuki M, Tanaka Y, Kanayama N, Terao T. Transforming growth factor-1 produced by ovarian cancer cell line HRA stimulates attachment and invasion through an up-regulation of plasminogen activator inhibitor type-1 in human peritoneal mesothelial cells. J Biol Chem. 2003;278:26793–26802. doi: 10.1074/jbc.M212187200. [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, Feng XH. Smads: transcriptional activators of TGF-β responses. Cell. 1998;95:737–740. doi: 10.1016/s0092-8674(00)81696-7. [DOI] [PubMed] [Google Scholar]
- Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–192. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, Dijke PT. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki Y, Nishizawa M, Fujisawa J, Inoue K. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M, Fujisawa J, Okazaki K. TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- Date M, Matsuzaki K, Matsushita M, Tahashi Y, Furukawa F, Inoue K. Modulation of transforming growth factor β function in hepatocytes and hepatic stellate cells in rat liver injury. Gut. 2000;46:719–724. doi: 10.1136/gut.46.5.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. Differential regulation of TGF-β signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology. 2002;35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFβ/ Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macías-Silva M, Abdollah S, Hoodless PA, Pirone R, Attisano L, Wrana JL. MADR2 is a substrate of the TGFβ receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell. 1996;87:1215–1224. doi: 10.1016/s0092-8674(00)81817-6. [DOI] [PubMed] [Google Scholar]
- Rockey DC, Housset CN, Friedman SL. Activation-dependent contractility of rat hepatic lipocytes in culture and in vivo. J Biol Chem. 1993;92:1795–1804. doi: 10.1172/JCI116769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittel T, Kobold D, Saile B, Grundmann A, Neubauer K, Piscaglia F, Ramadori G. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 1999;117:1205–1221. doi: 10.1016/s0016-5085(99)70407-5. [DOI] [PubMed] [Google Scholar]
- Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am J Physiol. 1992;263:579–585. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
- Schnabl B, Bradham CA, Bennett BL, Manning AM, Stefanovic B, Brenner DA. TAK/JNK and p38 have opposite effects on rat hepatic stellate cells. Hepatology. 2001;34:953–963. doi: 10.1053/jhep.2001.28790. [DOI] [PubMed] [Google Scholar]
- Pinzani M, Milani S, Grappone C, Weber FL, Jr, Gentilini P, Abboud HE. Expression of platelet-derived growth factor in a model of acute liver injury. Hepatology. 1994;19:701–707. doi: 10.1002/hep.1840190323. [DOI] [PubMed] [Google Scholar]
- Mulder KM. Roles of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- Pinzani M. Liver fibrosis. Springer Semin Immunopathol. 2000;21:475–490. doi: 10.1007/s002810000037. [DOI] [PubMed] [Google Scholar]
- Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K, Xu L, Eng F, Afdhal N, Kalluri R. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology. 2003;124:147–159. doi: 10.1053/gast.2003.50012. [DOI] [PubMed] [Google Scholar]
- Hua X, Liu X, Ansari DO, Lodish HF. Synergistic cooperation of TFE3 and Smad proteins in TGF-β-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev. 1998;12:3084–3095. doi: 10.1101/gad.12.19.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S, Hamzavi J, Breitkopf K, Wiercinska E, Said HM, Lorenzen J, Dijke PT, Gressner AM. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology. 2003;125:178–191. doi: 10.1016/s0016-5085(03)00666-8. [DOI] [PubMed] [Google Scholar]
- Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Carr BI, Scott CD. Modulation of insulin-like growth factor-II/mannose 6-phosphate receptors and transforming growth factor-β1 during liver regeneration. J Biol Chem. 1991;266:22444–22450. [PubMed] [Google Scholar]
- Koff A, Ohtsuki M, Polyak K, Roberts JM, Massagué J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-β. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-β-induced cell cycle arrest. Nature. 1994;371:257–260. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J. 2000;19:5178–5193. doi: 10.1093/emboj/19.19.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Kardassis D. Regulation of the human p21/WAF1/Cip1 promoter in hepatic cells by functional interactions between Sp1 and Smad family members. Proc Natl Acad Sci USA. 1998;95:6733–6738. doi: 10.1073/pnas.95.12.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin Liver Dis. 2001;21:437–451. doi: 10.1055/s-2001-17558. [DOI] [PubMed] [Google Scholar]