

Abstract
The epothilones are naturally occurring antimitotic drugs that share with the taxanes a similar mechanism of action without apparent structural similarity. Although photoaffinity labeling and electron crystallographic studies have identified the taxane-binding site on β-tubulin, similar data are not available for epothilones. To identify tubulin residues important for epothilone binding, we have isolated two epothilone-resistant human ovarian carcinoma sublines derived in a single-step selection with epothilone A or B. These epothilone-resistant sublines exhibit impaired epothilone- and taxane-driven tubulin polymerization caused by acquired β-tubulin mutations (β274Thr→Ile and β282Arg→Gln) located in the atomic model of αβ-tubulin near the taxane-binding site. Using molecular modeling, we investigated the conformational behavior of epothilone, which led to the identification of a common pharmacophore shared by taxanes and epothilones. Although two binding modes for the epothilones were predicted, one mode was identified as the preferred epothilone conformation as indicated by the activity of a potent pyridine-epothilone analogue. In addition, the structure–activity relationships of multiple taxanes and epothilones in the tubulin mutant cells can be fully explained by the model presented here, verifying its predictive value. Finally, these pharmacophore and activity data from mutant cells were used to model the tubulin binding of sarcodictyins, a distinct class of microtubule stabilizers, which in contrast to taxanes and the epothilones interact preferentially with the mutant tubulins. The unification of taxane, epothilone, and sarcodictyin chemistries in a single pharmacophore provides a framework to study drug–tubulin interactions that should assist in the rational design of agents targeting tubulin.
The clinical success of paclitaxel (PTX) and docetaxel has stimulated a search for compounds with a similar mode of action and has resulted in the identification of three nontaxane chemical classes of natural products: the soil bacteria-derived epothilones A and B (Epo A and Epo B) (1), the marine sponge-derived discodermolide (2, 3), and the coral-derived eleutherobins/sarcodictyins (4, 5). All three classes stabilize microtubules (MTs) and competitively inhibit the binding of PTX to tubulin polymers, indicating overlap of binding sites (1–3, 6, 7). The epothilones, however, display some superior qualities: they are water soluble, can be produced in large quantities through bacterial fermentation, and retain activity against multidrug-resistant (MDR) cell lines and tumors (5, 8, 9).
The PTX-binding site on β-tubulin has been identified both by electron crystallography, showing PTX bound on the of αβ-tubulin dimer (10), and by photoaffinity labeling, which has identified amino acids β1–31 and β217–233 as important areas for PTX binding (11, 12). Similar studies, however, for epothilones are not available. In this study, we present a report of epothilone-resistant (EpoR) human cancer cell lines with acquired β-tubulin mutations. The residues involved, β274 and β282, both map near the taxane-binding site in the atomic model of tubulin (10). These mutations affect the ability of epothilones to induce tubulin polymerization as well as inhibit cell growth. Using molecular modeling and guided by the mutation data and the activity profile of several MT-stabilizing agents against the cell lines with mutant tubulins, we were able to identify a common pharmacophore shared by taxanes and epothilones and in turn model Epo binding onto tubulin. In addition, we have identified an MT-active agent of the sarcodictyin family which, in contrast to taxanes and epothilones, is preferentially active against the tubulin mutants, and we have modeled the way it binds tubulin.
The unification of taxane, epothilone, and sarcodictyin chemistries in a single pharmacophore, and the fact that their structure–activity relationship profile against both the parental and mutant tubulins can be successfully explained by our modeling and docking studies, provide a framework to study drug–tubulin interactions that should assist in the rational design of agents targeting tubulin.
Materials and Methods
Materials.
PTX and docetaxel were obtained from the Drug Synthesis and Chemistry
Branch of the National Cancer Institute, Bethesda, MD. Epo A and B
(13), Epo B pyridine analogue,†† and
sarcodictyin A analogue (14) were prepared by chemical
synthesis.

Baccatin and mouse monoclonal anti-β-tubulin antibody (Tub2.1) were from Sigma, and horseradish peroxidase-conjugated sheep anti-mouse IgG antibody was from Amersham.
Cell Culture and Cytotoxicity Assay.
The EpoR cell lines, 1A9/A8 and 1A9/B10, were isolated in a single step after exposure of the human ovarian carcinoma cell line A2780 (1A9) to lethal concentrations (IC99) of either Epo A or Epo B (IC99 concentrations: 6 nM for Epo A and 0.5 nM for Epo B). After an initial expansion, the concentration of epothilones in the culture medium was gradually increased to 30 nM for Epo A and 5 nM for Epo B. Cells were maintained in drug, in a 5% CO2 humidified atmosphere at 37°C in RPMI medium 1640 (GIBCO/BRL) containing 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) (GIBCO/BRL). Before an experiment, the 1A9/A8 and 1A9/B10 cells were cultured for at least 7–10 days in drug-free medium. Cytotoxicity assays using the protein-staining sulforhodamine B method were performed in 96-well plates as described previously (15), by seeding 500 cells per well and incubating with cytotoxic agents for 4 days.
Tubulin Polymerization Assay.
Quantitation of the degree of in vivo tubulin polymerization in response to MT-stabilizing agents was performed as previously described (15). Briefly, cells were plated in 24-well plates and the following day were exposed to increasing concentrations of PTX or epothilones for 5 h. Then, cells were lysed in a hypotonic buffer (1 mM MgCl2/2 mM EGTA/0.5% Nonidet P-40/2 mM phenylmethylsulfonyl fluoride/200 units/ml aprotinin/100 μg/ml soybean trypsin inhibitor/5.0 mM ɛ-aminocaproic acid/1 mM benzamidine/20 mM Tris⋅HCl, pH 6.8), the cytoskeletal and cytosolic fractions containing polymerized (p) and soluble (s) tubulin, respectively, were separated by centrifugation, resolved by electrophoresis through SDS/10% polyacrylamide gels, and immunoblotted with an antibody against β-tubulin.
PCR and Sequencing of β-Tubulin.
PCR amplification and sequencing of the predominant β-tubulin isotype (protein class I/gene M40) from the EpoR clones (1A9/A8 and 1A9/B10) were performed with overlapping sets of primers as previously described (15).
Molecular Modeling Methods.
For molecular modeling methods and docking of epothilones and sarcodictyins on the αβ tubulin crystal structure, please refer to the supplemental data on the PNAS web site, www.pnas.org.
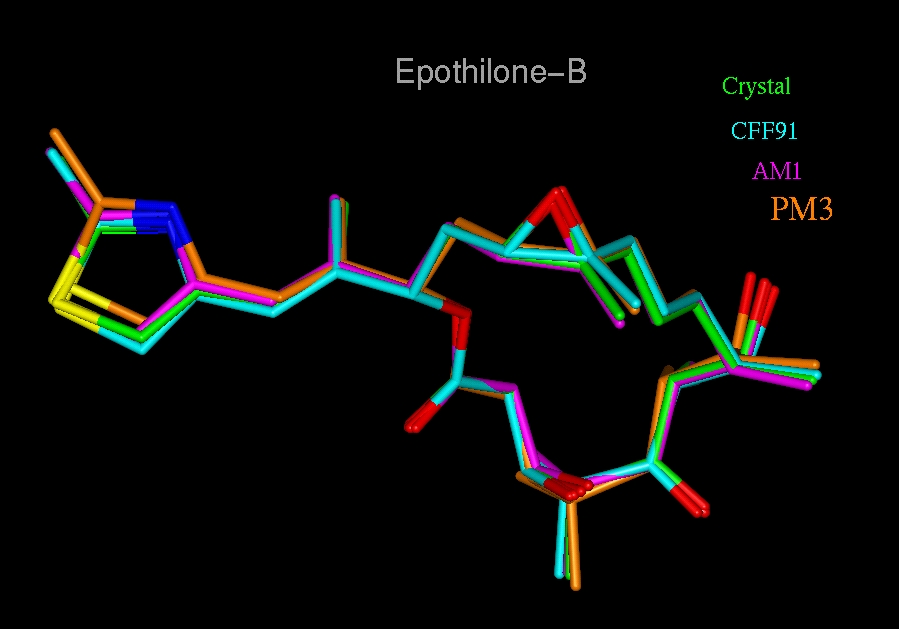
In brief, the initial model of Epo B was the crystallographic coordinates (16) and was optimized (PM3 Hamiltonian) (rms deviation = 0.087 Å). Molecular mechanics potentials (CFF91 force field) (17) were selected to optimize as close as possible to the crystal structure. High-temperature (1000 K) molecular dynamics (MD) was used to generate Epo conformations. Conformers of Epo were analyzed by tracking distances between the epoxide O and other atoms to identify a common pharmacophore with taxanes. The Epo/docetaxel overlap model was formed by using template forcing. The deposited tubulin structure (10) (Refcode 1TUB) was energy refined by using tethered minimizations. Epo was docked into the binding site formerly occupied by docetaxel and optimized. Docetaxel was completely ligated by water to the tubulin structure and optimized.
Results and Discussion
Two EpoR clones were isolated by selecting 1A9 human ovarian carcinoma cells with Epo A or Epo B. The two clones, designated 1A9/A8 and 1A9/B10, are 25- to 57-fold resistant to Epo A and B and 5- to 10-fold cross-resistant to PTX, docetaxel, and their precursor, baccatin (Table 1). Neither of the EpoR clones express MDR1 mRNA (data not shown), showing that epothilones do not select for an multidrug resistance phenotype, as the selection of the resistant clones was performed in the absence of any P-glycoprotein (Pgp) modulator.
Table 1.
Cytotoxicity profile of 1A9/EpoR cells to drugs acting on MTs
| Drug | IC50,* nM
|
||
|---|---|---|---|
| 1A9 | 1A9/A8 (β274) | 1A9/B10 (β282) | |
| PTX | 2 ± 0.23 | 19.5 ± 1.14 (10) | 13 ± 0.58 (6.5) |
| Docetaxel | 1 ± 0.15 | 7 ± 0.45 (7) | 5 ± 0.25 (5) |
| Baccatin | 550 ± 70 | 3950 ± 1060 (7) | 3000 ± 870 (5.4) |
| Epo A | 3.2 ± 0.42 | 127 ± 14.7 (40) | 182 ± 6.25 (57) |
| Epo B | 0.22 ± 0.03 | 5.4 ± 0.64 (25) | 5.2 ± 0.35 (24) |
| B-pyridine | 0.1 ± 0.015 | 3 ± 0.25 (30) | 6 ± 0.2 (60) |
| Sarc. A analogue† | 3.75 ± 0.02 | 1.18 ± 0.12 (0.3)‡ | 0.45 ± 0.018 (0.12)‡ |
IC50 values (±SEM) represent the mean of 3–13 independent 96-h growth inhibition assays. Values in parentheses are fold-resistance factors, defined as the IC50 value obtained for the β-tubulin mutant line divided by that obtained for the parental line.
† Sarcodictyin A analogue is methylated at the C-3 oxygen atom of the parent compound. This analogue is shown as an active member representative of the sarcodictyin family.
‡ The fold resistance factors in parentheses are less than 1 because the IC50 values for the β-tubulin mutant cells are lower than the IC50 for the 1A9 parental cells.
Compared with parental cells, both resistant clones demonstrated impaired in vivo epothilone-driven tubulin polymerization (Fig. 1). In parental cells, 150 nM Epo A resulted in the polymerization of 98% of tubulin. In contrast, even 5,000 nM Epo A was unable to induce substantial tubulin polymerization in the EpoR clones (see Fig. 1, 10% P in 1A9/A8 and 21% P in 1A9/B10 cells). Similarly, PTX-driven polymerization was impaired. Treatment with 1,500 nM PTX resulted in the polymerization of only 25% and 40% of tubulin in clones 1A9/A8 and 1A9/B10, respectively, compared with 100% in parental cells. These in vivo polymerization results concur with the cytotoxicity profile of the two EpoR clones, showing higher resistance to Epo A than to PTX (see Table 1). These observations suggested that the resistant phenotype most likely resulted from impaired Epo interaction with tubulin and prompted us to sequence tubulin in the EpoR cells.
Figure 1.

1A9/A8 and 1A9/B10 cells exhibit impaired in vivo tubulin polymerization. Drug-sensitive parental 1A9 and the EpoR clones, 1A9/A8 and 1A9/B10, were treated for 5 h with or without (0) various concentrations of PTX and Epo A as indicated. After cell lysis, the polymerized (P) and the soluble (S) protein fractions were separated by centrifugation, resolved by SDS/PAGE, and immunoblotted with an antibody against β-tubulin. The percent of polymerized tubulin (% P) was determined by dividing the densitometric value of polymerized tubulin by the total tubulin content (the sum of P plus S). The results shown are from a representative experiment of four independent observations.
We sequenced the predominant β-tubulin isotype (protein class I/gene M40) in both EpoR clones and the 1A9 cells from which they were derived. Class I β-tubulin is one of six different β-tubulin isotypes expressed in human cells, and it accounts for 90% of total β-tubulin mRNA in 1A9 cells (15) and the EpoR clones (data not shown). A different point mutation was identified in each clone: β274Thr→Ile (ACC→ATC) in clone 1A9/A8 and β282Arg→Gln (CGA→CAA) in clone 1A9/B10. Both β274 and β282 are evolutionarily conserved in all vertebrate β-tubulins and all known β-tubulin isotypes in these organisms (18). Furthermore, they cluster in space with two β-tubulin mutations previously identified in PTX-resistant cell lines (β270Phe→Val and β364Ala→Thr) (15). This spatial clustering on the region identified in the crystal structure of αβ-tubulin as the taxane-binding site, together with the cross-resistance patterns, reinforces the idea that epothilones share a common binding site with the taxanes. The above observations raised the possibility of a common pharmacophore; the flexible nature of the epothilones, however, required consideration of many epothilone conformations and their relationships with the relatively rigid taxane structure. We therefore postulated that the common pharmacophore between taxanes and epothilones should (i) be observable within a set of low-energy conformational isomers of epothilones, (ii) consistently overlap in conformational space with the taxanes, and (iii) sterically fit into the taxane–tubulin binding site model in a manner that is consistent with the relevant mutation data. These three criteria reduced the number of possible conformational Epo isomers to two.
Investigation of possible taxane–Epo superimpositions started with the use of molecular dynamics to sample Epo conformations (see Molecular Modeling Methods). This procedure resulted in a sample of 100 low-energy, conformationally distinct structures. Next, because the C12,C13 epoxide is the most rigid portion on the Epo ring, we used its oxygen as a reference to track distances to other features of the Epo molecule. Such measurements revealed that the centroid of the 3-OH, 7-OH, and 4-gem-dimethyl groups of Epo fell consistently at mean distance of 6.93 Å (SD = 1.18 Å) from its epoxide oxygen. Interestingly, this feature corresponds to the centroid of the part of the baccatin ring system of the taxanes consisting of the 1-OH, 9-carbonyl, and 15-gem-dimethyl group, which is 6.95 Å from its oxetane oxygen. These common groups were then used as a template to fit the Epo molecule onto the baccatin ring system of taxanes. Fig. 2 A and B illustrate the two-dimensional pharmacophoric overlap of Epo B and PTX. Two Epo conformations are presented. Fig. 2 C and D display the three-dimensional overlaps corresponding to the pharmacophores in 2 A and B, respectively. In these three-dimensional figures, docetaxel, which has a C5′-tert-butoxycarbonyl moiety in place of the C5′-benzoyl group of PTX, is the taxane depicted. To reiterate, these two conformations were the only ones that met the intramolecular pharmacophore requirements and at the same time fit the docetaxel–tubulin binding site model in a manner consistent with the tubulin mutation data. We emphasize that in the absence of additional biological or crystallographic data, the two different conformations of epothilones presented here appear equally likely.
Figure 2.

Proposed common overlaps of PTX and Epo B. (A and B) Atoms shown in red represent the pharmacophore shared by the two drugs. X is a pseudoatom at the centroid of the 4-gem-dimethyl, 3-OH, 7-OH system of Epo B, as well as the corresponding groups in PTX. The average distance of X from the epoxide O of Epo B is 6.93 Å (SD = 1.18 Å) as measured in the 100 lowest-energy conformations extracted from 10 separate 100-ps molecular dynamics simulations under 10 different dielectric conditions. Displayed in blue are atoms overlapping as a result of a template-forced minimization of Epo B onto PTX. (C and D) Three-dimensional overlap of the common substituents of Epo B (carbon atoms shown in cyan) with those of docetaxel (carbon atoms shown in yellow). For both molecules, oxygen atoms are displayed in red and nitrogen atoms are displayed in blue. Functional groups have been identified in white for docetaxel and black for Epo B. The two Epo conformations presented here reflect the two potential binding modes for Epo interaction with tubulin. Binding mode I in A and C and binding mode II in B and D.
Using this pharmacophore and guided by the mutations in the EpoR cells, we succeeded in docking Epo on an energy-refined model of the 3.7-Å density map of the docetaxel-binding site on β-tubulin (Fig. 3). In agreement with the cross-resistance data and the in vivo polymerization studies, the model predicts that the change in residue β274 from Thr to Ile should have a greater impact on the binding of epothilones as compared with that of taxanes. The model predicts that the reason for this is that epothilones use their C7-OH group to hydrogen bond in the vicinity of the Thr-β274; when Thr-β274 is mutated to Ile, this hydrogen bond is disrupted. In contrast, taxanes, which have hydrogen bond donors or acceptors at the C10, C9, and C7 positions, can form alternate hydrogen bonds.
Figure 3.

Space-filling model of the Epo B conformation shown in Fig. 2 A and C (carbon atoms in cyan) docked into an energy-refined model of tubulin. The backbone ribbon structure of tubulin is in light green and the M loop is in yellow. Two residues that confer resistance against epothilones are displayed as stick models. The carbon atoms of these residues are shown in light magenta for the wild type and in white for the mutants.
The β282Arg→Gln mutation sits on the M loop (Fig. 3), identified as the major region in lateral contacts between protofilaments (19), and is located near the common binding site of taxanes and epothilones. As the N terminus of the M loop is part of the taxane ring-binding region, the mutation of Arg-β282 could directly affect the binding of taxanes and epothilones. In addition, the mutation could cause a disruption of lateral contacts. This is in agreement with our findings that in the absence of drug the Epo B-selected clone, 1A9/B10, harboring the β282Arg→Gln mutation, has a significantly slower growth rate than the parental 1A9 cells and the Epo A-selected clone, 1A9/A8. The doubling times for 1A9, 1A9/A8, and 1A9/B10 cells are 24 h, 30 h, and 50 h, respectively. A combined effect on both drug binding and lateral interactions is also possible.
The data presented thus far do not allow one to determine a preference between the two modes of Epo binding shown in Fig. 2. However, drug sensitivity data in 1A9 cells and the previously described PTX-selected cell line (PTX10), containing a β270Phe→Val mutation (15), provided us with a tool to test this conformational hypothesis. Specifically, drug sensitivity data using an Epo B analogue†† with a pyridine moiety in place of the thiazole ring of the parent compound (Table 1) allowed us to identify binding mode II (Fig. 2) as the preferred conformation of epothilones. This preference is because the pyridine ring is bulkier than the thiazole side chain, which in binding mode II is in close proximity to Phe-β270. If binding mode II is preferred, one would predict that the β270Phe→Val mutation would have a greater impact on the sensitivity of the pyridine-containing Epo B derivative compared with the thiazole-containing molecule. This prediction is confirmed by the cytotoxicity data, which show that the β270Phe→Val mutation has only a 3-fold effect on the sensitivity of the thiazole containing Epo B (15) compared with a 10-fold change for the pyridine Epo B (IC50 values: 0.1 nM for 1A9 and 1 nM for the β270Phe→Val mutant cells). While these data, together with the fact that the Epo B-pyridine analogue is equipotent to Epo B, provide strong evidence in support of binding mode II, further empirical crystallographic evidence will be required to determine which binding mode is more likely.
The model presented here is in agreement with studies of both taxane and Epo structure–activity relationships (SAR). For example, SAR studies have shown that the C4–C5 oxetane ring of taxanes is absolutely required for activity (20), whereas in epothilones replacement of the C12,C13 epoxide with a cyclopropane ring results in total loss of activity both in vitro with purified tubulin and in whole cells (21). In the proposed Epo/tubulin pharmacophore model, the oxygen atoms contained in the oxetane of taxanes and the epoxide of epothilones overlap (Fig. 2). Both the epoxide and the oxetane are restricted ethers containing conformationally rigid oxygens that are likely to function as hydrogen bond acceptors. In the tubulin structure, the C4–C5 oxetane of docetaxel is located near a cluster of polar backbone atoms from tubulin residues 273, 275, and 276, with the hydroxyl side chain of Thr-β274 also located in this hydrophilic area. In the Epo/tubulin model the epoxide is similarly disposed. In addition, studies have shown that with the C12,C13 epoxide present, some substitutions at C12 lead to analogues with increased biological activity (13). Included among these is the naturally occurring Epo B, which differs from Epo A by a single methyl group substitution at C12, a modification that increases its potency by 14-fold (see Table 1). According to the model presented here, the C12 methyl group stabilizes a favorable hydrophobic interaction in the vicinity of the side chains of Leu-β273, Leu-β215, Leu-β228, and Phe-β270; this may account for the increased potency of Epo B compared with Epo A. Furthermore, Epo analogues in which the C12,C13 epoxide is removed and replaced by a double bond (deoxy-12,13-olefinic derivative), display intact enhancement of in vitro tubulin polymerization (21, 22). This result further supports the pharmacophore model described here, because the C12–C13 double bond results in a very favorable hydrophobic interaction in a similar manner to the methyl group in Epo B, while at the same time, the projection of its electron-rich π cloud toward a water molecule may serve as a weak hydrogen bond acceptor.
One way to conceptualize these observations is to compare the epothilones with baccatin, the “inactive” taxane precursor. Baccatin is a structurally rigid molecule, with poor in vitro tubulin-polymerization-enhancing activity, likely resulting from its weak binding to tubulin. Enhanced baccatin binding has been achieved in the taxanes with the introduction of the C13 and the C2 side chains, the importance of which is underscored by the electron crystallographic data showing the docetaxel side chains in contact with β-tubulin (10). Although epothilones lack the overall bulk of taxanes, their macrolide ring system provides a larger hydrophobic core compared with the diterpene ring system of baccatin. This, coupled with the occupancy of epothilones' side chain in the vicinity of the C13 side chain of taxanes and the critical hydrogen bonds formed by the C1-OH, C7-OH, and epoxide oxygens, enables epothilones to interact with high affinity with tubulin even though they have significantly less molecular volume than taxanes. A recent report of a common pharmacophore between nonataxel (a nonaromatic PTX analogue) and epothilones proposes that baccatin is a nonessential component of these drugs (23). This conclusion was based on a relatively inactive analogue that was designed before data on the taxane-binding site, available from the crystal structure of α/β tubulin dimer (10), could be considered. Thus, our modeling in combination with the mutational data reinforces our belief that baccatin is an essential moiety for tubulin binding and should not be viewed as a passive scaffold holding functional groups. We would emphasize that our proposed conformation of the binding of Epo to the docetaxel-binding site of tubulin is distinct from both the crystal structure (16) and the reported solution conformations (24). This difference is not surprising, given the finding that ligands frequently undergo conformational changes upon binding to proteins (25).
The identification of a common pharmacophore between taxanes and epothilones prompted us to examine whether sarcodictyins, a distinct class of coral-derived MT-stabilizing agents, would fit in the same model. However, in contrast to taxanes and epothilones, which have reduced activity in the mutant cells compared with parental cells, the activities of sarcodictyins A and B (not shown) and a methyl ketal analogue of sarcodictyin A (7) were enhanced (Table 1). This favorable interaction of sarcodictyins with the EpoR cells harboring mutant tubulins can be explained according to our pharmacophore model. The sarcodictyin A analogue was modeled into the taxane-binding site in a manner similar to that described for the epothilones (Molecular Modeling Methods) (Fig. 4). According to the pharmacophore model, the isopropyl group at C14 of sarcodictyin corresponds to the gem-dimethyl groups of the epothilones and the taxanes. Furthermore, the restricted ether oxygen attached to C4 and C7 of sarcodictyins is within 2 Å from the epoxy and oxetane oxygens of the epothilones and the taxanes, respectively, and is also favorably placed to accept the same hydrogen bond from the single water molecule. The methylimidazole side chain of sarcodictyins lies in the vicinity of Phe-β270 (Fig. 4). This location is similar to those of the aromatic side chain in binding mode II of the epothilones and the C13 side chain of taxanes. The sarcodictyins contain a methyl group at C7, which makes an unfavorable yet tolerable hydrophobic–polar interaction with the alcohol side chain of Thr-β274 (Fig. 4). However, in the β274Thr→Ile mutant, this same interaction becomes a highly favorable hydrophobic interaction between the C7 methyl group of sarcodictyin and the Ile-β274 aliphatic side chain. This explains why the potency of sarcodictyins is substantially increased in the 1A9/A8 cells with the β274Thr→Ile mutation compared with parental 1A9 cells with a Thr at position 274.
Figure 4.

Stick model of sarcodictyin A analogue (carbon atoms in yellow) docked into an energy-refined model of tubulin. The backbone ribbon structure of tubulin is in green. Arrows point at two residues that confer Epo and taxane resistance. For reference some of the carbons have been labeled in black.
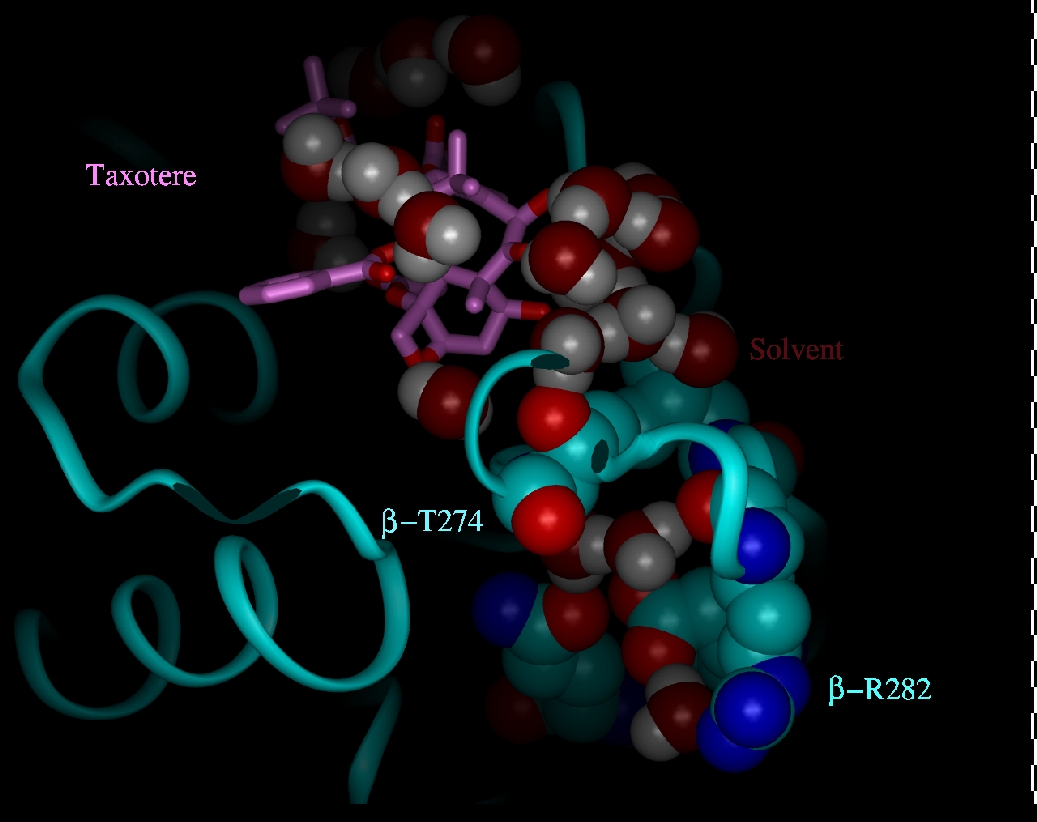
To further understand the molecular mechanism of drug binding, we utilized an energy-refined model of the 3.7-Å crystal structure of the docetaxel-binding site on β-tubulin that includes water interactions with the protein (Fig. 5). In this energy-refined model, water molecules were added to portions of tubulin that were sterically and electrostatically feasible as evidenced by their energy-minimized coordinates. The model must be interpreted with caution and may provide a possible explanation for the mutational analysis. We postulate that, in the apo-structure of tubulin (unbound tubulin), solvent may occupy a large portion of the binding pocket, forming a hydrogen-bonded network that is coordinated by the spatial disposition of neighboring polar amino acid side chains and backbone residues. For example, in such a model, Arg-β282 is linked to this solvent network by a salt bridge with Glu-β288 (Fig. 5). At the apex of this water network, the OH group of Thr-β274 controls the location of water molecules that form a critical hydrogen bond with the C7-OH of Epo. According to this model, the β274Thr→Ile mutation is disruptive because it shifts the location of these waters, resulting in the loss of the C7-OH hydrogen bond with tubulin. The β282Arg→Gln mutation may also be disruptive because loss of the salt bridge with Glu-β288 enhances the anionic charge of the latter. In an attempt to screen this anionic charge, more water may enter the area, encouraging the OH side chain of Thr-β274 and its hydrogen-bonded waters to shift to a new location. This finding is further supported by the enhanced activity of sarcodictyin in the β282Arg→Gln mutant cells. As before, the shift of the OH group of the Thr-274 side chain causes the simultaneous shift of the methyl group of the Thr side chain in the location previously occupied by the OH moiety. This rearrangement explains the decreased activity of epothilones caused by the unfavorable hydrophobic–polar interaction between the C7-OH of epothilones and the methyl group of the Thr-274 side chain. At the same time it also explains the increased activity of sarcodictyin caused by the favorable hydrophobic–hydrophobic interaction between the C7-methyl group of sarcodictyins and the methyl group of the Thr-274 side chain.
Figure 5.

Shown with its carbon atoms displayed in light magenta, docetaxel is hydrogen bonded to waters that form a contiguously hydrogen-bonded solvent network. According to the proposed model, the amino acids illustrated (in stick figure with labels) are residues that are principally involved in determining the location of these solvent molecules. The backbone ribbon structure of tubulin is in cyan, water molecules are in white (H) and red (O), and hydrogen bonds are yellow dashed lines.
The clinical success of taxanes has made tubulin a very attractive target for cancer chemotherapy (26) and has prompted a worldwide search for compounds with similar mechanism of action. Here we present acquired β-tubulin mutations in human carcinoma cells that confer resistance to epothilones and taxanes by impairing their binding to tubulin. The biological importance of this finding is underscored in a recent study reporting that patients with non-small-cell lung cancer not responding to PTX harbored β-tubulin mutations near the taxane-binding site (27). The identification of a common pharmacophore for taxanes and epothilones presented here is a valuable example of molecular modeling guided by mutational/biological data that helps us to better understand the interaction of these compounds with their common intracellular receptor, tubulin. We believe our data should assist in the engineering of MT-stabilizing molecules with improved characteristics, such as the Epo B-pyridine analogue and the sarcodictyin A analogue, setting the stage for advances in cancer chemotherapy.
Supplementary Material
Abbreviations
- PTX
paclitaxel
- MT
microtubule
- Epo A
epothilone A
- Epo B
epothilone B
- EpoR
epothilone-resistant
Footnotes
The synthesis of this Epo B analogue will be reported elsewhere.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040546297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040546297
References
- 1.Bollag D M, McQueney P A, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods C M. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 2.ter Haar E, Kowalski R J, Hamel E, Lin C M, Longley R E, Gunasekera S P, Rosenkranz H S, Day B W. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 3.Kowalski R J, Giannakakou P, Gunasekera S P, Longley R E, Day B W, Hamel E. Mol Pharmacol. 1997;52:613–622. [PubMed] [Google Scholar]
- 4.Long B H, Carboni J M, Wasserman A J, Cornell L A, Casazza A M, Jensen P R, Lindel T, Fenical W, Fairchild C R. Cancer Res. 1998;58:1111–1115. [PubMed] [Google Scholar]
- 5.Nicolaou K C, Winssinger N, Vourloumis D, Ohshima T, Kim S, Pfefferkorn J, Xu J Y, Li T. J Am Chem Soc. 1998;120:10814–10826. [Google Scholar]
- 6.Kowalski R J, Giannakakou P, Hamel E. J Biol Chem. 1997;272:2534–2541. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 7.Hamel E, Sackett D L, Vourloumis D, Nicolaou K C. Biochemistry. 1999;38:5490–5498. doi: 10.1021/bi983023n. [DOI] [PubMed] [Google Scholar]
- 8.Cowden C J, Paterson I. Nature (London) 1997;387:238–239. doi: 10.1038/387238a0. [DOI] [PubMed] [Google Scholar]
- 9.Service R F. Science. 1996;274:2009–2010. doi: 10.1126/science.274.5295.2009. [DOI] [PubMed] [Google Scholar]
- 10.Nogales E, Wolf S G, Downing K H. Nature (London) 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 11.Rao S, Krauss N E, Heerding J M, Swindell C S, Ringel I, Orr G A, Horwitz S B. J Biol Chem. 1994;269:3132–3134. [PubMed] [Google Scholar]
- 12.Rao S, Orr G A, Chaudhary A G, Kingston D G, Horwitz S B. J Biol Chem. 1995;270:20235–20238. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou K C, Winssinger N, Pastor J, Ninkovic S, Sarabia F, He Y, Vourloumis D, Yang Z, Li T, Giannakakou P, Hamel E. Nature (London) 1997;387:268–272. doi: 10.1038/387268a0. [DOI] [PubMed] [Google Scholar]
- 14.Nicolaou K C, Xu J Y, Kim S, Pfefferkorn J, Ohshima T, Vourloumis D, Hosokawa S. J Am Chem Soc. 1998;120:8661–8673. [Google Scholar]
- 15.Giannakakou P, Sackett D L, Kang Y K, Zhan Z, Buters J T, Fojo T, Poruchynsky M S. J Biol Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 16.Hofle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem Int Ed Engl. 1996;35:1567–1569. [Google Scholar]
- 17.Maple J R, Hwang M J, Jalkanen K J, Stockfisch T P, Hagler A T. J Comp Chem. 1998;19:430–458. [Google Scholar]
- 18.Sullivan K F, Cleveland D W. Proc Natl Acad Sci USA. 1986;83:4327–4331. doi: 10.1073/pnas.83.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nogales E, Whittaker M, Milligan R A, Downing K H. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 20.Kingston D G. Trends Biotechnol. 1994;12:222–227. doi: 10.1016/0167-7799(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 21.Nicolaou K C, Finlay M R, Ninkovic S, King N P, He Y, Li T, Sarabia F, Vourloumis D. Chem Biol. 1998;5:365–372. doi: 10.1016/s1074-5521(98)90070-9. [DOI] [PubMed] [Google Scholar]
- 22.Su D S, Meng D F, Bertinato P, Balog A, Sorensen E J, Danishefsky S J, Zheng Y H, Chou T C, He L F, Horwitz S B. Angew Chem Int Ed Engl. 1997;36:757–759. [Google Scholar]
- 23.Ojima I, Chakravarty S, Inoue T, Lin S, He L, Horwitz S B, Kuduk S D, Danishefsky S J. Proc Natl Acad Sci USA. 1999;96:4256–4261. doi: 10.1073/pnas.96.8.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor R E, Zajicek J. J Org Chem. 1999;64:7224–7228. [Google Scholar]
- 25.Nicklaus M C, Wang S, Driscoll J S, Milne G W. Bioorg Med Chem. 1995;4:411–428. doi: 10.1016/0968-0896(95)00031-b. [DOI] [PubMed] [Google Scholar]
- 26.Jordan M A, Wilson L. Curr Opin Cell Biol. 1998;10:123–130. doi: 10.1016/s0955-0674(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 27.Monzo M, Rosell R, Sanchez J J, Lee J S, O'Brate A, Gonzalez-Larriba J L, Alberola V, Lorenzo J C, Nunez L, Ro J Y, Martin C. J Clin Oncol. 1999;17:1786–1793. doi: 10.1200/JCO.1999.17.6.1786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}