

Abstract
Hyperphosphorylation of microtubule-associated proteins such as tau and neurofilament may underlie the cytoskeletal abnormalities and neuronal death seen in several neurodegenerative diseases including Alzheimer's disease. One potential mechanism of microtubule-associated protein hyperphosphorylation is augmented activity of protein kinases known to associate with microtubules, such as cdk5 or GSK3β. Here we show that tau and neurofilament are hyperphosphorylated in transgenic mice that overexpress human p25, an activator of cdk5. The p25 transgenic mice display silver-positive neurons using the Bielschowsky stain. Disturbances in neuronal cytoskeletal organization are apparent at the ultrastructural level. These changes are localized predominantly to the amygdala, thalamus/hypothalamus, and cortex. The p25 transgenic mice display increased spontaneous locomotor activity and differences from control in the elevated plus-maze test. The overexpression of an activator of cdk5 in transgenic mice results in increased cdk5 activity that is sufficient to produce hyperphosphorylation of tau and neurofilament as well as cytoskeletal disruptions reminiscent of Alzheimer's disease and other neurodegenerative diseases.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by loss of cognitive function. One of the neuropathological hallmarks in AD and other neurodegenerative diseases such as Pick's disease is neurofibrillary tangles (NFTs). NFTs are composed of the microtubule-binding protein tau that is hyperphosphorylated (reviewed in ref. 1).
Although many protein kinases phosphorylate tau at AD-relevant epitopes in vitro (reviewed in ref. 2), only two have been copurified with microtubules, GSK3β and cdk5 (3, 4). To our knowledge, only these two kinases will phosphorylate tau in a cellular environment (e.g., refs. 5 and 6). We chose to focus on cdk5 because it is active predominantly in neurons whereas GSK3β plays a role in energy metabolism and is active in all cells. cdk5 is a member of the cyclin-dependent protein kinase gene family. Rather than cyclins, cdk5 associates with the positive allosteric regulators p35 (7), amino-terminal proteolytic fragments of p35 (e.g., p25; ref. 8), and p39 (9). These proteins share minimal amino acid sequence homology to cyclins, but the mechanism of activation of cdk5 by p25/35 may be similar to the activation of cdk2 by cyclin A (10). p25/35 is expressed predominantly in neurons, implying that most cdk5 activity is concentrated in neuronal structures (7, 8). cdk5 plays a pivotal role in neuronal development as evidenced by the abnormal corticogenesis and perinatal lethality of cdk5 knockout mice (11) and the disturbances in neuronal migration and early death in p35 knockout mice (12). A number of potential cdk5 substrates have been identified and most are consistent with a putative role in neurite outgrowth and plasma membrane dynamics. These include cytoskeletal proteins such as tau and neurofilament (e.g., refs. 13 and 14) and synaptic vesicle proteins (15, 16). To clarify the potential role of cdk5 in neurodegenerative diseases in vivo, we overexpressed human p25 in the brains of transgenic mice to determine whether increased cdk5 activity would lead to hyperphosphorylation of tau and neurofilament and/or cytoskeletal disturbances.
Materials and Methods
Animal Handling.
All experimentation was performed under protocols approved by the Pfizer Institutional Animal Care and Use Committee.
Production of the p25 Transgenic Mouse.
The human p25 fragment was constructed by PCR amplification from a full-length human p35 clone (David Auperin, Pfizer). To produce an in-frame p25 fragment, a PCR fragment with NotI restriction site ends and a start methionine codon ATG was engineered upstream of the beginning of the GCC for alanine at amino acid residue 99 of p35. The p25 NotI fragment was cloned into plasmid pSP72 (Promega) with an SV40 polyadenylation signal sequence. The rat neuron-specific enolase (NSE) promoter (obtained from J. G. Sutcliffe of the Research Institute of Scripps Clinic) was blunt-end-ligated 5′ of the p25 human cDNA and SV40 poly(A) sequences in the pSP72 backbone. The 3.2-kb NSE-p25 transgene was end-purified for microinjection in Elutip columns (Schleicher & Schuell). The production of transgenic founders by pronuclear microinjection was achieved by using well established procedures (17). Pronuclear stage embryos were microinjected with the NSE-p25 transgene at ≈3.0 ng/ml in 10 mM Tris, pH 7.4/0.1 mM EDTA. Microinjected embryos were transferred to pseudopregnant CD-1 females for continued development to term. At weaning, genomic DNA was isolated from the tail tissue by phenol and chloroform extraction. Founder transgenics were identified by a PCR that specifically amplified DNA sequences from the human p25 cDNA. Founder mice were bred to FVB/N mates, and transgene positive offspring were used to maintain the individual transgenic lines.
Biochemistry.
Brains from 4-month-old mice were removed and snap-frozen in liquid nitrogen. Preparation of brain lysates, Western blots (20 μg protein per lane), and immunoprecipitation/phosphorylation experiments (150–800 μg protein) were performed as described previously (18). cdk5 and p35 antibodies were obtained from Santa Cruz Biotechnology and provided by L. H. Tsai (Harvard University), respectively.
For immunoblots, snap-frozen brains were thawed and the amygdala was removed and homogenized in 0.5 ml of lysis buffer containing 250 mM NaCl, 50 mM Tris, 5 mM EDTA, 0.1% NP-40, protease inhibitor mixture (Boehringer Mannheim), and 1 μM okadaic acid (Calbiochem). The samples were centrifuged for 30 min at 12,500 × g. Five micrograms of supernatant (lysate, L) and 25 μl of solubilized pellets (P) were separated and transferred to ProBlott membrane paper (Applied Biosystems) as described (19). Blots were probed with AT8 (1:400), Tau-1 (1:10,000), PHF-13 (1:10,000), Tau-5 (1:200), SMI-31 (1:1,000), and SMI-33 (1:1,000). Protein bands were visualized by using ECL reagents (Amersham Pharmacia).
Immunocytochemistry, Histology, and Electron Microscopy.
Brain tissue was prepared for immunohistochemical analysis as described in ref. 20. For AT8 and PHF13, sections were incubated overnight at 4°C in primary antibody, processed, and visualized by using horseradish peroxidase/diaminobenzidine (DAB) according to the kit manufacturer (Vector Laboratories).
For AT8/glial fibrillary acidic protein (GFAP) double labeling, brains were perfusion-fixed in situ with 10% neutral-buffered formalin. Trimmed tissues were dehydrated through graded alcohols and embedded in paraffin. Sections (6 μm) first were labeled with GFAP (Dako) and visualized with DAB (Dako), labeled with AT8 (Innogenetics, Zwijnkrecht, Belgium), and visualized with Vector VIP (Vector Laboratories).
For silver staining and SMI 34 immunohistochemistry, brains were perfusion-fixed in situ with 10% neutral-buffered formalin or 4% paraformaldehyde. Trimmed tissues were dehydrated and embedded as above, and sections (6 μm) were stained with the modified Bielschowsky stain (21, 22).
Separate mice were perfused as above for transmission electron microscope. Brain sections were placed in 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 M NaPO4 buffer overnight, postfixed in Dalton's osmium (23), and embedded in Spurr's resin. Grids were stained and examined by using a Hitachi 7100 TEM.
Antibodies.
The following are antibodies that were used: AT-8, Tau phospho-Ser 202 and Thr 205 (24) (Innogenetics); PHF-13, Tau phospho-Ser 396 and Thr 404 (25) (gift from V. Lee, Univ. of Pennsylvania); SMI-34, 31, and 33, phosphorylated (SMI-31, 33) and total (SMI-34) heavy neurofilament (26) (Sternberger–Meyer, Jarrettsville, MD); Tau-1, Tau dephospho-Ser202 and Thr 205 (27) (Boehringer Mannheim); Tau-5, total tau (28) (NeoMarkers, Fremont, CA); and anti-GFAP, GFAP associated with cells of astrocytic origin (29) (Dako).
Behavioral Methods.
Mice were observed weekly for gross neurological deficits (30) in clear Plexiglas open fields (height, 45.7 cm2 × 17.8 cm), in which the floor was demarcated into nine squares (15.2 cm2). Locomotor activity (LMA) was measured as the number of square crossings in 5 min. Automated LMA was measured in photocell chambers (21 cm3) for 60 min. Elevated plus-maze testing occurred in a gray Plexiglas maze with two open arms and two closed arms (each 8-cm width × 17-cm length, with walls at 14.5-cm height) at a height of 36 cm. Mice were placed into the center of the maze, and time spent on and number of entries into the arms were measured. ANOVA followed by Newman–Keuls and Dunnett's t tests was used to analyze data from all behavioral testing.
Results
Biochemical Characterization.
Human p25 cDNA was expressed in transgenic mice under the control of the neuron-specific enolase promoter (31) (Fig. 1A). Brain lysates prepared from the amygdala, thalamus/hypothalamus, cerebral cortex, and cerebellum of p25 transgenic and wild-type mice were compared for protein expression of p25 by immunoblotting (Fig. 1B). Consistent with other experiments performed in rodents (e.g., ref. 7), no constitutive p25 was detected in wild-type animals. In contrast, transgenic animals displayed robust expression of p25. No alterations in the expression levels of either p35 (Fig. 1B) or cdk5 (Fig. 1C) were apparent. However, relative to constitutive p35, expression of p25 was substantially lower in the cerebellum compared with the other regions (Fig. 1B; the ratio of p35:p25 expression, quantitated by image analysis, in amygdala, thalamus/hypothalamus, cortex, and cerebellum was 1.1 ± 0.1, 1.0 ± 0.1, 1.2 ± 0.1, and 2.3 ± 0.3, mean ± SEM., respectively; n ≥ 3). To determine whether the increased expression of p25 resulted in an increase in the catalytic activity of cdk5, immunoprecipitation/phosphorylation experiments were performed. As seen in Fig. 1D, the degree of cdk5-mediated histone phosphorylation after immunoprecipitation with an antibody that recognizes cdk5 was increased by approximately 2-fold in all areas except cerebellum. Similar results were observed with an antibody that recognizes p25/35 (data not shown).
Figure 1.

(A) NSE-p25 transgene. Expression construct for human p25 cDNA sequences under the control of the rat NSE promoter. (B) Expression of p25 in transgenic mice. Western blots of wild-type (lanes 1–4) and transgenic (lanes 5–8) brain lysates from the amygdala (lanes 1 and 5), thalamus/hypothalamus (lanes 2 and 6), cerebral cortex (lanes 3 and 7), and cerebellum (lanes 4 and 8) using an antibody that recognizes both p35 and p25. (C) Expression of cdk5 in transgenic mice. Immunoblots of wild-type (lanes 1–4) and transgenic (lanes 5–8) brain lysates from the amygdala (lanes 1 and 5), thalamus/hypothalamus (lanes 2 and 6), cerebral cortex (lanes 3 and 7), and cerebellum (lanes 4 and 8) using an antibody that recognizes cdk5. (D) Increased cdk5 activity in transgenic mice. cdk5/p25/p35 complexes from wild-type (■) and transgenic (○) amygdala, thalamus/hypothalamus, cerebral cortex, and cerebellum were immunoprecipitated with an antibody that recognizes cdk5, then assessed for kinase activity by incubating with [32P]ATP and histone. The y axis represents the total cpm incorporated into histone minus a blank (no primary antibody) for three mice in each group (mean ± SEM). Similar results were obtained for an antibody that recognizes p35/p25 (not shown).
Histological Characterization.
Brains from 4-month-old transgenic and wild-type mice were compared for the presence of tau and neurofilament phosphoepitopes by immunohistochemistry. AT-8 and PHF-13, mAbs that recognize phosphorylated Ser-202, Thr-205, and Ser-396, Ser-404, respectively, labeled neurons in amygdala of transgenic animals but not wild-type animals (Fig. 2 A–D). Neurons in thalamus/hypothalamus and cerebral cortex adjacent to the external capsule also were labeled, but the cerebellum of transgenic animals and corresponding regions of wild-type animals were not (not shown). The AT8 staining was restricted to cells of neuronal morphology because double-labeling experiments using GFAP and AT8 displayed a nonoverlapping pattern of distribution (Fig. 3). Similar to AT8 and PHF13, a mAb that recognizes phosphorylated neurofilament H (SMI34) showed increased immunostaining in these three brain regions of transgenic mice (amygdala staining shown in Fig. 2 E and F). Immunostaining with antibodies that recognize either tau (Anti-Tau; BioMakor, Rehovot, Israel) or neurofilament (SMI-33) independent of phosphorylation state displayed no significant differences in pattern or intensity of staining in wild type vs. transgenic (data not shown).
Figure 2.

Abnormal phosphorylation epitopes in p25 transgenic vs. wild-type brains. Brain sections from p25 transgenic animals (A, C, and E) were immunostained in parallel with corresponding regions from wild-type animals (B, D, and F). Immunostaining of AT8 in transgenic (A) and wild-type (B) amygdala. Many cells in this region had densely stained cell bodies with well defined immunopositive axons and axonal hillocks (arrow). Inset shows an increased magnification of these cells from a different transgenic animal. Other immunopositive structures were more lightly stained and had diffuse borders (arrowheads). Immunostaining of PHF13 in transgenic (C) and wild-type (D) amygdala is shown. Background staining of this antibody was routinely higher in transgenic brain sections and represents greater immunoreactivity in the neuropil. Immunostaining with SMI34 in cortex adjacent to external capsule in transgenic (E) and wild-type (F) mice also is shown. Note the constitutive staining of external capsule axons (ec) by SMI34 in both transgenic and wild-type sections. This pattern of staining was seen in 14 of 15 4-month-old transgenic mice and never seen in 6 wild-type mice of the same age. (Bar = 50 μ.)
Figure 3.

Comparison of AT8 and GFAP immunostaining. Amygdala sections from transgenic (A) and wild-type mice (B) were immunostained with AT8 (blue) and an anti-GFAP antibody (brown). Note the nonoverlapping distribution of GFAP and AT8 staining in the transgenic section (A) and the lack of AT8 staining in the wild-type section (B). (Bar = 100 μ.)
Silver-based staining methods have been used to identify pathological changes in neurons containing altered tau proteins in AD (e.g., Fig. 4A). In p25 transgenic mice, neurons in the cerebral cortex adjacent to the external capsule (Fig. 4 B and C) and in the thalamus/hypothalamus and amygdala (not shown) displayed positive labeling when the modified Bielschowsky silver stain was used. Spinal cord axons were also positive for AT8, PHF13, SMI34, and Bielschowsky staining (not shown). In the amygdala, 4.7% of neuronal somata were argyrophilic as assessed by cell counting. This staining was absent in wild-type animals (Fig. 4).
Figure 4.
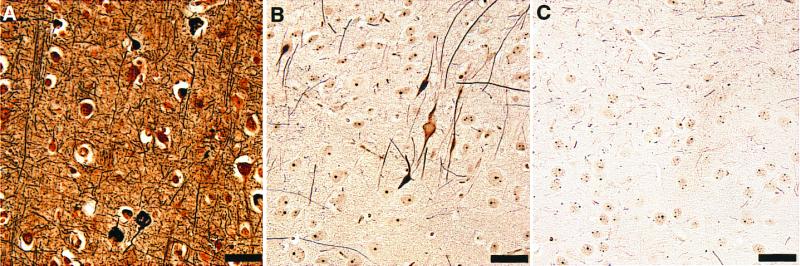
Argyrophilic structures from p25 transgenic brain. A section from cortex of an Alzheimer's brain (A) is compared with sections from p25 transgenic cortex (B) and wild-type cortex (C) silver-stained by using the modified Bielschowsky method. Unlike wild type, p25 transgenic brain contained argyrophilic cell bodies and axons. This pattern of staining was seen in 14 of 15 4-month-old transgenic and never seen in 15 wild-type mice of the same age. (Bar = 50 μ.)
Ultrastructurally, axonal swelling was observed in the amygdala (Fig. 5 A and B) and spinal cord (not shown) of 3- to 6-month-old p25 transgenic mice. Affected axoplasm was filled with numerous, abnormally clustered mitochondria and lysosomes, and the cytoskeletal components were disorganized. These changes were absent in age-matched, wild-type mice (Fig. 5 C and D).
Figure 5.

Electron microscopy of amygdala from p25 transgenic and wild-type brains. Axons within the amygdala of transgenic mice were markedly dilated (A). Compare abnormal dilated axons (arrows) with axons of normal size (arrowheads). Mitochondria (M) and lysosomes (L) were clustered together abnormally and were interspersed among disorganized cytoskeletal components (B, *). Axons from the same location in age-matched, wild-type mice were normal (C and D). Axonal changes were observed ultrastructurally in five of six transgenic mice aged 3–6 months and not observed in any of five age-matched, wild-type mice. [Bars = 4 μ (A and C) or 500 nm (B and D).]
Tau and Neurofilament Immunoblots.
To determine whether overexpression of p25 altered the expression of tau and neurofilament, immunoblots were performed by using antibodies that recognize tau and neurofilament independent of the phosphorylation state (Tau-5 and SMI-33, respectively). No differences between transgenic and wild-type mice were observed (Fig. 6). In addition, no differences in the amount of phospho- and dephospho-Ser-202, Thr-205 tau (detected with AT8 and Tau-1, respectively) and phospho-neurofilament (detected with SMI-31) were observed (Fig. 6). Concentrations of all antibodies used in immunoblot experiments were 2- to 3-fold greater than those used in immunohistochemistry experiments and identified only proteins that migrated with molecular masses consistent with either tau (60–66 kDa) or neurofilament (ca. 200 kDa) (data not shown).
Figure 6.

Immunoblots for tau and neurofilament. Immunoblots containing tissue lysates (L) and pellets (P) derived from the amygdala from wild-type (WT) and p25 transgenic (P) mice were probed with antibodies specific for phospho- (AT8) and dephospho-Thr-202, Ser-205 of tau (Tau-1), phospho-tau Ser-396 (PHF-13), total tau (Tau-5), phospho-neurofilament (SMI-31), and total neurofilament (SMI-33). No differences in any of these epitopes between transgenic and wild-type mice were detected.
Neurobehavioral Evaluation of p25 Transgenic Mice.
At 4–9 weeks of age, whole-body exertion tremors were observed in the transgenic mice [96.7% and 100% of male (n = 30) and female mice (n = 16), respectively]. In addition, transgenic mice displayed increases in open-field LMA beginning at 5–7 weeks of age that reached 2-fold greater than wild type by 16 weeks (Fig. 7A).
Figure 7.

(A) p25 transgenic mice exhibit increased locomotor activity in an open field. p25 transgenic mice exhibited significantly more square crossings than wild-type littermates in an open field (*, P < .05; **, P < .01). Females (data not shown) showed the same effect beginning at 5 weeks of age. Numbers next to symbols represent the number of animals tested at each time point. (B) p25 transgenic mice spend increased time on the open arms of an elevated plus maze. p25 transgenic mice exhibited significantly greater time than wild-type littermates on the open arms of an elevated plus maze (*, P < .05; **, P < .01). Numbers within bars represent the number of animals in each group.
Subsequent behavioral experiments were based on immunohistochemical results. For example, lesioning of the amygdala in rats results in increases in spontaneous locomotor activity and “anxiolytic” effects (e.g., refs. 32 and 33). Accordingly, 9- to 12-week-old mice were tested further for locomotor changes and in the elevated plus maze to evaluate behavior in an anxiety-producing environment. Consistent with results in the open field, transgenic mice exhibited significantly increased locomotor activity in automated activity chambers (not shown). In the elevated plus-maze test, both male and female transgenic mice spent significantly more time than wild-type mice on the open arms of the maze (Fig. 7B). No differences in the number of arm entries were observed.
Discussion
The p25 transgenic mice display hyperphosphorylation of tau and neurofilament by immunostaining. These results are consistent with in vitro and whole-cell data suggesting that tau and neurofilament are authentic substrates for cdk5/p25. However, we cannot rule out the possibility that overexpression of p25 results in increased cdk5 activity by a mechanism that is independent of association with cdk5 or that cdk5 regulates the activity of a protein kinase or phosphatase that is responsible for the observed results. In addition, the antibody we used to detect phospho-neurofilament, SMI34, has been reported to cross-react with phospho-tau (e.g., ref. 34). However, the dilution of SMI34 necessary to visualize phospho-tau (1:300; ref. 35) is 100-fold lower than that used in the current experiments (1:30,000) and makes SMI34 recognition of phospho-tau in these experiments unlikely.
The reasons for the restricted localization of phospho-tau, phospho-neurofilament, and silver staining in the transgenic animals are unclear but likely related to the 2-fold augmentation of cdk5 activity in these regions. Consistent with these results, no increase in cdk5 activity or histopathological changes were detected in the cerebellum. The ratio of p25 to p35 expression was 50% lower in the cerebellum relative to the affected areas, raising the possibility that the level of p25 expression required to produce an increase in cdk5 activity was not achieved in this region. Thus, although we cannot exclude the possibility that neurons in the positive-staining regions are more sensitive to the sequelae of increased p25 expression, the results are consistent with the conclusion that increased cdk5 activity, and not merely overexpression of p25, is responsible for the observed phenotype.
The fraction of neurons positive for silver staining in the transgenic mice was relatively low, approaching 5%, and probably explains the lack of difference between transgenic and wild-type mice in the phospho-tau and phospho-neurofilament immunoblot experiments. However, based on the positive immunostaining, the local concentrations of the phospho-tau and phospho-neurofilament epitopes in immunopositive neurons are likely to be relatively high.
The morphology of cells positively stained for hyperphosphorylated tau and neurofilament ranged from healthy looking neurons to cells with condensed nuclei and cytoplasm and little remaining axonal or dendritic processes. No glial staining was detected. Hyperphosphorylated tau and neurofilament staining was most concentrated in the soma, dendrites, and axon hillock of positive neurons. This finding is consistent with in vitro data demonstrating that phosphorylation of these microtubule-associated proteins reduces their affinity for microtubules resulting in microtubule destabilization and redistribution from the axon to other cellular compartments (36, 37). The presence of hyperphosphorylated tau and neurofilament in these cellular locales is associated with neurodegenerative diseases. Furthermore, brain regions that stained positively for hyperphosphorylated tau and neurofilament were also positive for the modified Bielschowsky silver stain. Positive labeling of cytoskeletal elements in the somatodendritic regions of neurons with this stain is generally interpreted as signifying neuronal dysfunction or degeneration leading ultimately to cell death (21, 22). Braak et al. (38), using AT8, silver staining, and Alz50, an antibody that recognizes a conformation of tau common in neurodegenerative diseases (39), described a sequence of cytoskeletal changes related to the formation of neurofibrillary tangles. Although we did not utilize Alz50, the combination of AT8 and silver staining in the transgenic mice appears similar to that described as the “pretangle” (Group 1) stage of AD.
The cytoskeletal disruptions suggested by the immunohistochemical experiments were confirmed by ultrastructural studies. The most significant findings comprised axonal swelling, unusual clustering of lysosomes and mitochondria, and cytoskeletal disorganization, features consistent with the loss of a functioning microtubule network. Thus, we propose that a p25-mediated increase in cdk5 activity results in hyperphosphorylation of tau and neurofilament, which, in turn, promotes destabilization of the microtubule network and disruption of the cytoskeleton. The cytoskeletal abnormalities seen at the ultrastructural level, the positive silver staining, and the subcellular localization of the hyperphosphorylated tau and neurofilament suggest that neurons in the affected areas are under substantial cytoskeletal stress and that their function is likely to be compromised.
Similar to animals with lesions of the amygdala (32), the p25 transgenic mice showed significantly increased LMA. Because previous reports suggest that animals with lesions of the amygdala exhibit decreased fear and anxiety under a variety of experimental conditions (e.g., ref. 33), the p25 transgenic mice were tested in the elevated plus-maze task to investigate this possibility. Transgenic mice spent increased time on the open arms of the maze, consistent with decreased anxiety (40). The number of arm entries was not different, suggesting that the result is probably not due to the observed increases in LMA. Although we cannot rule out that the observed behavioral changes occur by mechanisms other than changes in amygdala function, results from these two independent behavioral measures are consistent with compromised amygdala function.
The results from other studies exploring the pathological consequences of tau phosphorylation are complementary to the present study. For example, overexpression of either the longest (41) or shortest (42) form of human tau in transgenic mice results in somatodendritic localization of tau. The reasons for the change in tau localization in these mice are unclear, but may be due to saturation of microtubule-binding sites. Calcineurin Aα knock-out mice display increased tau phosphorylation at Ser-396/404 (43), and in transgenic animals that overexpress GSK3β (44) or Mos kinase (45), increases in phosphorylated tau or neurofilament are observed. Chronic infusion of the phosphatase inhibitor okadaic acid also results in increased tau phosphorylation (46). These studies provide valuable information regarding the potential pathophysiological role of tau or tau kinase overexpression that is consistent with the present work. However, the p25 transgenic mouse displays a novel combination of immunocytochemical, histopathological, ultrastructural, and behavioral evidence that suggests that augmented cdk5 activity may play a role in the pathophysiology of neurodegenerative diseases.
In conclusion, the present study demonstrates that overexpression of the cdk5 activator p25 in mouse brain results in hyperphosphorylation of tau and neurofilament, positive silver staining, and cytoskeletal disturbances that are similar to several neurodegenerative diseases including AD. Alterations in behavioral measures consistent with compromised neuronal function in an affected area are also observed. The p25 transgenic mouse will facilitate the study of the sequelae of augmented cdk5 activity and hyperphosphorylated tau and neurofilament as well as aid in the development of therapeutic molecules meant to slow the progression of AD and other neurodegenerative diseases.
Acknowledgments
We thank B. Appel, G. Boucher, K. Steever, and J. Wolfgang of the Department of Pathology; D. Auperin, M. Boucher, and V. Guanowsky of the Department of CNS Discovery; J. Stock of the Department of Genetic Technologies; and E. Glynn, P. McGill, and B. Yanak of the Department of Comparative Medicine and Biology, Pfizer Central Research, for expert assistance. In addition, we thank Dr. J. G. Sutcliffe of Scripps Institute for the rat NSE promoters, Dr. Li-Huei Tsai (Harvard) for providing the anti-p35 antibody, and Dr. Virginia Lee (Univ. of Pennsylvania) for providing the PHF13 antibody.
Abbreviations
- AD
Alzheimer's disease
- GFAP
glial fibrillary acidic protein
- LMA
locomotor activity
- NSE
neuron-specific enolase
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040577797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040577797
References
- 1.Goedert M, Crowther R A, Spillantini M G. Neuron. 1998;21:955–958. doi: 10.1016/s0896-6273(00)80615-7. [DOI] [PubMed] [Google Scholar]
- 2.Imahori K, Uchida T. J Biochem. 1997;121:179–188. [PubMed] [Google Scholar]
- 3.Ishiguro K, Takamatsu M, Tomizawa K, Omori A, Takahashi M, Arioka M, Uchida T, Imahori K. J Biol Chem. 1992;267:10897–10901. [PubMed] [Google Scholar]
- 4.Hosoi T, Uchiyama M, Okumura E, Saito T, Ishiguro K, Uchida T, Okuyama A, Kishimoto T, Hisanaga S. J Biochem. 1995;117:741–740. doi: 10.1093/oxfordjournals.jbchem.a124771. [DOI] [PubMed] [Google Scholar]
- 5.Wagner U, Utton M, Gallo J M, Miller C C. J Cell Sci. 1996;109:1537–1543. doi: 10.1242/jcs.109.6.1537. [DOI] [PubMed] [Google Scholar]
- 6.Michel G, Mercken M, Murayama M, Noguchi K, Ishiguro K, Imahori K, Takashima A. Biochim Biophys Acta. 1998;1380:177–182. doi: 10.1016/s0304-4165(97)00139-6. [DOI] [PubMed] [Google Scholar]
- 7.Tsai L H, Delalle I, Caviness V S, Jr, Chae T, Harlow E. Nature (London) 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 8.Lew J, Huang Q-Q, Qi Z, Winkfein R J, Aebersold R, Hunt T, Wang J H. Nature (London) 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 9.Tang D, Lee K Y, Qi Z, Matsuura I, Wang J H. Biochem Cell Biol. 1996;74:419–429. doi: 10.1139/o96-046. [DOI] [PubMed] [Google Scholar]
- 10.Tang D, Yeung J, Lee K Y, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang J H. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 11.Ohshima T, Ward J M, Huh C G, Longenecker G, Veeranna, Pant H C, Brady R O, Martin L J, Kulkarni A B. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chae T, Kwon Y T, Bronson R, Dikkes P, Li E, Tsai L H. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 13.Paudel H K, Lew J, Ali Z, Wang J H. J Biol Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- 14.Sun D, Leung C L, Liem R K H. J Biol Chem. 1996;271:14245–14251. doi: 10.1074/jbc.271.24.14245. [DOI] [PubMed] [Google Scholar]
- 15.Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- 16.Shuang R, Zhang L, Fletcher A, Groblewski G E, Pevsner J, Stuenkel E L. J Biol Chem. 1998;273:4957–4966. doi: 10.1074/jbc.273.9.4957. [DOI] [PubMed] [Google Scholar]
- 17.Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 18.Tsai L H, Takahashi T, Caviness V S, Jr, Harlow E. Development. 1993;119:1029–1040. doi: 10.1242/dev.119.4.1029. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli U K. Nature (London) 1970;227:680–684. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Rhodes K, Monaghan M M, Barrezueta N X, Nawaschik S, Bekele-Arcuri Z, Matos M F, Nakahira K, Schechter L E, Trimmer J S. J Neurosci. 1996;16:4846–4860. doi: 10.1523/JNEUROSCI.16-16-04846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fix A S, Ross J F, Stitzel S R, Switzer R C. Toxicol Pathol. 1996;24:291–303. doi: 10.1177/019262339602400305. [DOI] [PubMed] [Google Scholar]
- 22.Powers R E, Wilson R D. J Histotechnol. 1996;19:235–240. [Google Scholar]
- 23.Dalton A J. Anat Rec. 1955;121:281. [Google Scholar]
- 24.Biernat J, Mandelkow E-M, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M, et al. EMBO J. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann, R., Lee, V. M.-Y., Leight, S., Varga, I. & Otvos, L. Biochemistry 36, 8114–8124. [DOI] [PubMed]
- 26.Sternberger L A, Sternberger N H. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szendrei G I, Lee V M, Otvos L., Jr J Neurosci Res. 1993;334:243–249. doi: 10.1002/jnr.490340212. [DOI] [PubMed] [Google Scholar]
- 28.LoPresti P, Szuchet S, Papasozomenos S C, Zinkowski R P, Binder L I. Proc Natl Acad Sci USA. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viale G, Gambacorta M, Coggi G, Dell'Orto P, Milani M, Doglioni C. Virchows Arch A Pathol Anat Histopathol. 1991;418:339–448. doi: 10.1007/BF01600164. [DOI] [PubMed] [Google Scholar]
- 30.Forss-Petter S, Danielson P E, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe J G. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 31.Irwin S. Psychopharmacologia. 1968;13:222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- 32.Burns L H, Annett L, Kelley A E, Everitt B J, Robbins T W. Behav Neurosci. 1996;110:1. doi: 10.1037//0735-7044.110.1.60. , 60–73. [DOI] [PubMed] [Google Scholar]
- 33.Swartzwelder H S. Physiol Behav. 1981;26:323–326. doi: 10.1016/0031-9384(81)90030-5. [DOI] [PubMed] [Google Scholar]
- 34.Ksiezak-Reding H, Dickson D W, Davies P, Yen S-H. Proc Natl Acad Sci USA. 1987;84:3410–3414. doi: 10.1073/pnas.84.10.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Sternberger N H, Rubinstein L J, Herman M M, Binder L I, Sternberger L A. Proc Natl Acad Sci USA. 1989;86:8045–8049. doi: 10.1073/pnas.86.20.8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biernat J, Gustke N, Drewes G, Mandelkow E M, Mandelkow E. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 37.Bramblett G T, Goedert M, Jakes R, Merrick S E, Trojanowski J Q, Lee V M. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 38.Braak E, Braak H, Mandelkow E-M. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 39.Carmel G, Mager E M, Binder L I, Kuret J. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 40.Lister R G. Psychopharmacology. 1987;92:180–185. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- 41.Gotz J, Probst A, Spillantini M G, Schafer T, Jakes R, Burki K, Goedert M. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brion J-P, Tremp G, Octave J-N. Am J Pathol. 1999;154:255–270. doi: 10.1016/S0002-9440(10)65272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kayyali U S, Zhang W, Yee A G, Seidman J G, Potter H. J Neurochem. 1997;68:1668–1678. doi: 10.1046/j.1471-4159.1997.68041668.x. [DOI] [PubMed] [Google Scholar]
- 44.Brownlees J, Irving N G, Brion J-P, Gibb B J M, Wagner U, Woodgett J, Miller C C J. NeuroReport. 1997;8:3251–3255. doi: 10.1097/00001756-199710200-00013. [DOI] [PubMed] [Google Scholar]
- 45.James N D, Davis D R, Sindon J, Hanger D P, Brion J-P, Miller C C J, Rosenberg M P, Anderton B H, Propst F. Neurobiol Aging. 1996;17:235–241. doi: 10.1016/0197-4580(95)02068-3. [DOI] [PubMed] [Google Scholar]
- 46.Arendt T, Holzer M, Fruth R, Bruckner M K, Gartner U. Neuroscience. 1995;69:691–698. doi: 10.1016/0306-4522(95)00347-l. [DOI] [PubMed] [Google Scholar]