

Abstract
A major receptor for nitric oxide (NO) is the cGMP-synthesizing enzyme, soluble guanylyl cyclase (sGC), but it is not known how this enzyme behaves in cells. In cerebellar cells, NO (from diethylamine NONOate) increased astrocytic cGMP with a potency (EC50 ≤ 20 nM) higher than that reported for purified sGC. Deactivation of NO-stimulated sGC activity, studied by trapping free NO with hemoglobin, took place within seconds (or less) rather than the minute time scale reported for the purified enzyme. Measurement of the rates of accumulation and degradation of cGMP were used to follow the activity of sGC over time. The peak activity, occurring within seconds of adding NO, was swiftly followed by desensitization to a steady-state level 8-fold lower. The same desensitizing profile was observed when the net sGC activity was increased or decreased or when cGMP breakdown was inhibited. Recovery from desensitization was relatively slow (half-time = 1.5 min). When the cells were lysed, sGC desensitization was lost. Analysis of the transient cGMP response to NO in human platelets showed that sGC underwent a similar desensitization. The results indicate that, in its natural environment, sGC behaves much more like a neurotransmitter receptor than had been expected from previous enzymological studies, and that hitherto unknown sGC regulatory factors exist. Rapid sGC desensitization, in concert with variations in the rate of cGMP breakdown, provides a fundamental mechanism for shaping cellular cGMP responses and is likely to be important in decoding NO signals under physiological and pathophysiological conditions.
Nitric oxide (NO) performs numerous physiological functions, including relaxation of smooth muscle, inhibition of platelet aggregation, and neural communication in the brain (1, 2). A major receptor for NO is the enzyme, soluble guanylyl cyclase (sGC), which catalyzes the production of the effector molecule, cGMP from GTP (3, 4).
Compared with neurotransmitter receptors or related adenylyl and guanylyl cyclases (5, 6), the NO receptor enzyme appears rather unremarkable. It is composed of two different subunits (α and β), but only two isoforms have been shown to exist at the protein level: the α1β1 isoform, which is expressed widely, and the α2β1 isoform present in human placenta (7–9). Also, sGC appears to lack the functional complexity exhibited by related enzymes or receptors. For example, there is no established mechanism for regulation of the enzyme (e.g., by phosphorylation) and, on activation by NO, purified sGC generates cGMP at a constant rate for long periods of time (10, 11). Furthermore, the two naturally occurring sGC isoforms possess very similar functional and pharmacological properties (9).
How sGC responds to NO in living cells, however, has not been investigated, nor is it understood why different cells display very different patterns of NO-stimulated cGMP accumulation ranging from a transient spike-like response (12) to a more slowly developing plateau (13). Here we have analyzed the kinetics of NO-stimulated sGC activity in intact cells. The results show that, in its natural environment, sGC performs differently from what had been assumed from previous biochemical studies. In particular, within seconds (or less), the enzyme undergoes substantial desensitization. In concert with cGMP-metabolizing enzymes, sGC desensitization enables diverse patterns of cGMP responses to NO to be generated by different cells.
Materials and Methods
Cell Preparations.
Cerebellar cells from 8-day-old postnatal rats were prepared and incubated as before (14) except that hydroxyurea pretreatment, used to deplete dividing cells, was used in the initial experiments only (Fig. 2 a–d), and the incubation medium contained 100 μM nitroarginine (to prevent endogenous NO formation) but not BSA. The cells, incubated at 20 × 106 cells/ml, were exposed to the NO donor, diethylamine NONOate (DEA/NO). Decomposed DEA/NO (10 μM), on its own or in the presence of 1 μM DEA/NO, did not influence cGMP accumulation. Washed human platelets were prepared and incubated as described (15). When used, phosphodiesterase (PDE) inhibitors were added 10 min before DEA/NO. cGMP was quantified by radioimmunoassay and protein by the bicinchoninic acid method. Extracellular cGMP was measured after filtering the suspensions through 0.6-μm (cerebellar cells) or 0.22-μm (platelets) cut-off filters. As found before (13, 14), the amount of cGMP in the medium from cerebellar cells stimulated with DEA/NO was negligible (1–2% of total). Values for extracellular cGMP in the platelet suspensions are given in the main text and Fig. 6 legend. NO concentrations were measured by using an electro-chemical probe (World Precision Instruments, Stevenage, Herts, U.K.; Iso-NO electrode). Data are given as means ± SEM (n = 3–6).
Figure 2.

Characteristics of NO-stimulated cGMP accumulation in cerebellar cells. The concentration–cGMP response curve for DEA/NO (a) was obtained by using a 2-min exposure. Deactivation of sGC was determined by addition of Hb (50 μM) 15 s (b) or 125 s (c) after DEA/NO (1 μM) or by addition of ODQ (10 μM, d). ●, controls; □, Hb or ODQ added 5 s before DEA/NO; ○, Hb or ODQ added at arrows; solid lines fit the decline in cGMP to the Michaelis–Menten equation by using identical parameters (Kp and Vp).
Figure 6.

Kinetics of cGMP synthesis and degradation in human platelets. The accumulation of cGMP induced by DEA/NO (1 μM) in the presence (○) and absence (●) of PDE inhibitors (100 μM sildenafil + 100 μM erythro-9-(2-hydroxy-3-nonyl)adenine) is in a. (Inset) Decay of cGMP levels after addition of 10 μM Hb in the presence of the PDE inhibitors, fitted by the Michaelis–Menten equation. At the 2-min time-point, extracellular cGMP accounted for 52 ± 8 pmol/mg protein (about 3% of total), which was subtracted from each data point. The analysis of vs and vd in the presence of the PDE inhibitors is shown in b, together with the raw data. In c, the cGMP response in control platelets (●) is shown; the parameters, Vp and Kp, describing cGMP breakdown in the presence of the PDE inhibitors were altered by increasing Vp or decreasing Kp as indicated. In d are depicted the different shapes of cGMP response (scaled to the peaks) that would be produced by a combination of a desensitizing vs and increases in Vp (both derived from cerebellar cells stimulated with DEA/NO). Reductions in Kp have broadly similar effects, except that with a low Kp, the late phase becomes truncated (cf. solid line in c).
sGC Activity in Lysed Cerebellar Cells.
Cells were lysed in 10 mM Tris/1 mM DTT (pH 7.4) to give the equivalent of 20 × 106 cells/ml, and sGC activity was assayed in the presence of 1 μM DEA/NO as before (16) except that Mg2+ replaced Mn2+ as cofactor, and PDE activity was inhibited by 1 mM EGTA and 1 μM sildenafil. Under these conditions, a cGMP concentration (2 μM) similar to that ordinarily produced after 2-min stimulation with DEA/NO was stable over 5 min (i.e., PDE activity was zero).
Immunocytochemistry.
Cerebellar cells exposed to DEA/NO (1 μM) were fixed (4% paraformaldehyde in 0.1 M phosphate buffer) for 30 min at room temperature, resuspended in buffer, dried onto gelatin-coated slides, rehydrated with distilled water, and permeabilized with 1% Triton X-100 in Tris-buffered saline for 15 min. After rinsing (0.1% Triton X-100/0.2% BSA in Tris-buffered saline), the slides were incubated with normal rabbit and horse sera for 30 min and then with the primary antibodies, sheep anti-cGMP (1:8,000, a gift from J. de Vente, University of Maastricht, the Netherlands) and mouse antiglial fibrillary acidic protein (1:800, Chemicon) overnight at 4°C. After rinsing and incubation with secondary antibodies (anti-mouse FITC and anti-sheep TRITC) for 1 h at room temperature, the slides were mounted and viewed under differential interference contrast and fluorescence optics.
Determination of vd and vs.
The decline in cGMP levels occurring after deactivation of sGC in cerebellar cells and platelets was fitted to the integrated Michaelis–Menten equation:
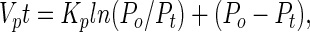 |
where Po = starting cGMP level, Pt = cGMP level at time t, and Vp and Kp are the apparent Michaelis–Menten constants (Vmax and Km); Vp, Kp, and Po were found by iteration (17). The rate of degradation, vd = (VpP)/(Kp + P).
The time courses of cGMP accumulation in cerebellar cells and platelets treated with PDE inhibitors were described by a generalized hyperbola.
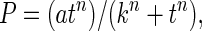 |
where P = cGMP level, a = maximum cGMP level, k is a constant defining the steepness of the hyperbola, and n is a second constant required to give a gradient of zero at t = 0, thereby accommodating the brief lag because of mixing and NO dissociation from the donor. Differentiating and inserting the result into the expression, dP/dt = vs − vd, gives:
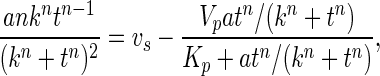 |
from which vs was found with respect to t.
Results
cGMP Accumulation in Cerebellar Cells.
In the first instance, cell suspensions from the postnatal rat cerebellum were used because this brain region has low PDE activity (18), and the dispersed cell preparation eliminates the diffusional constraints inherent in tissue slices (19). DEA/NO, which dissociates with a half-life of 2.1 min, was used as the source of NO. Consistent with previous observations (13), DEA/NO caused large increases in cGMP levels. Although a mixture of cell types is present, heterogeneity of target cell cGMP responses to NO is unlikely to create complications because immunocytochemistry indicated that the increase in cGMP was localized to a discrete subpopulation of cells, the astrocytes. This applied whether the exposure to DEA/NO was 2 min (Fig. 1 a–c) or 10 s (not illustrated) and agrees with results obtained from cerebellar slices from rats of the same age (14, 20).
Figure 1.

Location of NO-stimulated cGMP accumulation in cerebellar cell suspension. The same field is shown under differential interference contrast optics (a) and after immunofluorescent staining for glial fibrillary acidic protein (b) and cGMP (c). The cells were fixed after 2-min exposure to DEA/NO (1 μM). Arrows indicate examples of colocalized staining in individual cells. Bar = 50 μm.
The maximal effect occurred at 0.3 μM DEA/NO and the EC50 was about 0.1 μM (Fig. 2a). The NO concentration produced in the cell suspension by 0.1 μM DEA/NO was below the detection limit of an electrochemical probe, indicating that it was less than 20 nM.
Kinetic Analysis of cGMP Formation and Degradation.
The rate of cGMP accumulation is simply the difference between its rate of formation by sGC (vs) and its rate of degradation (vd) by PDEs (or another route). If the rate of cGMP accumulation and vd are measured, vs can be determined.
To find vd, DEA/NO was added to the cells at a supramaximal concentration (1 μM) to prevent decay of NO being limiting and cGMP accumulation allowed to proceed. Then sGC activity was arrested and the fall in cGMP levels followed with time. Arrest of sGC was achieved in two ways. The first was by trapping free NO with hemoglobin (Hb) (21) at a concentration (10–50 μM) that, if added 5 s beforehand, abolished the cGMP response to DEA/NO (Fig. 2 b and c). Whether added after 15-s (Fig. 2b) or 125-s (Fig. 2c) exposure to DEA/NO, Hb caused an abrupt cessation of cGMP accumulation and, thereafter, cGMP levels declined progressively. The rate of decline followed Michaelis–Menten-type kinetics, so that vd could be described operationally by the expression: vd = VpP/(Kp + P). Significantly, the parameters Kp and Vp determined by adding Hb after 125 s also described the rate of degradation at lower starting cGMP levels (Hb added at 15 s), indicating that PDE behaves in a simple substrate-linked fashion.
To check the validity of the use of Hb, we tested another strategy: inhibition of sGC with 1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-l-one (ODQ) (22). The results were the same (Fig. 2d); moreover, the parameters determined by using the Hb method also described the decay after addition of ODQ, showing that the Hb method for determining vd is reliable.
Detailed time courses of the DEA/NO-stimulated accumulation of cGMP showed a steep rate of rise over the initial 15 s that tailed off, giving a quasi-plateau after about 30 s (Fig. 3a). At the plateau, by definition, the rate of cGMP degradation equals the rate of cGMP synthesis. The initial rate of cGMP degradation during the plateau was about 0.07 pmol/106 cells/s, implying that sGC was generating cGMP at the same rate. Inspection of the initial rising phase of the cGMP response, however, suggested a rate about 7-fold faster (Fig. 3a), pointing to a large decline in enzyme activity with time. The decline could not be attributed to depletion of NO, because the NO concentration was well sustained over 2 min (0.22 ± 0.03 μM; n = 4) and because a second dose of DEA/NO (1 μM) added after 2 min did not alter cGMP levels (data not shown).
Figure 3.

Kinetics of cGMP synthesis and degradation in intact (a and b) and lysed (c) cerebellar cells. Filled symbols (a–c) represent cGMP levels after addition of DEA/NO (1 μM). Open symbols (a) chart the decline in cGMP after addition of Hb (at arrow); the data are fitted to the Michaelis–Menten equation (solid line). Broken lines (a) indicate the differing initial rates of cGMP accumulation and degradation. In b, the progress curve for cGMP accumulation (data from a) is fitted by a generalized hyperbola and vd and vs determined as described in Materials and Methods. In lysed cells (c), the linear cGMP accumulation is equivalent to an sGC activity of 4,140 pmol/mg protein/min; vd was eliminated by PDE inhibition.
To extract the kinetics of sGC quantitatively and more accurately, the accumulation of cGMP with time was fitted by a generalized hyperbola (Fig. 3b). Differentiation of this expression and correcting for vd gives vs, the rate of cGMP synthesis (see Materials and Methods). Analysis of the data in this way confirmed that vs in the intact cells varied dramatically with time after addition of DEA/NO (Fig. 3b). Initially, it increased sharply to reach a mean peak of 0.95 ± 0.11 pmol/106 cells/s after 3.8 ± 0.8 s and thereafter declined rapidly to an 8-fold lower rate (0.12 ± 0.08 pmol/106 cells/s) at 2 min (n = 6).
In contrast to this pattern of sGC activity in intact cells, when the enzyme was assayed in lysed cells, the NO-stimulated activity was linear with time (3.5 pmol/106 cells/s; Fig. 3c). To test the validity of the analysis of sGC activity in intact cells, therefore, and to check the possible influence of substrate depletion and/or end-product inhibition on the kinetics of sGC, the two determinants of cGMP accumulation, vs and vd, were deliberately altered.
Effect of Varying sGC Activity.
To reduce vs, ODQ was used at a concentration (0.3 μM) causing a 70% reduction in the maximum cellular cGMP response to DEA/NO. Under these conditions, the shape of the response was unchanged (Fig. 4a). The parameters governing cGMP degradation (Kp and Vp) were also the same (results not shown), indicating a lack of effect of ODQ on PDE activity. Accordingly, the derived kinetic profile of vs showed exactly the same increase and decrease seen in control cells, despite a peak activity that was 65% lower (Fig. 4b). To increase vs, we used the allosteric sGC activator, YC-1 (23). In the presence of YC-1 (100 μM), the cGMP response to DEA/NO was 3-fold higher than in control cells, but the time courses, again, were similar (Fig. 4a). Because YC-1 stabilizes the binding of NO to sGC (24) and can inhibit PDE activity (25), vd could not be determined. Therefore, vs was derived for the extreme cases of PDE activity being normal (using the vd found in control cells) and zero. In both cases, vs increased to a peak rate double that of control cells and then decreased (Fig. 3b).
Figure 4.

Pharmacological manipulation of vs in cerebellar cells. In a, addition of 0.3 μM ODQ (■) decreased cGMP relative to the control (C1, ●); 100 μM YC-1(□) increased cGMP relative to the control (C2, ○). In all cases, the cells were stimulated with 1 μM DEA/NO and the time courses were fitted by a generalized hyperbola. The derived profiles of vs from these data are shown in b. For YC-1-treated cells, profiles assuming control PDE activity (dashed line) and zero PDE activity (dotted line) are both shown. In Insets, the profiles of vs are scaled by peak height; solid lines are the controls.
Thus, over a wide range of sGC activity resulting in a 10-fold variation in the accumulation of cGMP, the amplitude of vs changed in a predictable way, but its kinetic profile stayed the same.
Effect of Reducing PDE Activity.
The aim was to eliminate vd and so determine vs directly from the experimental data. An effective way of inhibiting cGMP breakdown in the cerebellar cells was with a combination of sildenafil and rolipram. In the presence of these PDE inhibitors, the cGMP response progressively exceeded that of control cells (Fig. 5a). Because cGMP degradation was negligible over the relevant time intervals (Fig. 5b), vd can be taken as zero. Differentiation of the cGMP response then gives vs. The profile of vs obtained in this way superimposed accurately on the profile determined by using the usual method (Fig. 5c).
Figure 5.

Effect of PDE inhibition in cerebellar cells. In a, addition of PDE inhibitors (100 μM sildenafil + 1 μM rolipram) (○) increased the cGMP response to DEA/NO (1 μM) relative to control (●). The control response was fitted by the usual hyperbolic function (solid line). In b, decay of cGMP levels is shown after addition of 10 μM Hb in the presence (○) and absence (●) of the PDE inhibitors. Solid lines fit the data to the Michaelis–Menten equation. In c are the profiles of vs generated by differentiating the cGMP data obtained in the presence of the PDE inhibitors (○) and by the usual analysis of the control cGMP accumulation data (solid line). Differentiation was carried out by using microcal origin software, Ver. 4.10.
Recovery from Desensitization.
This was studied by first stimulating the cells with DEA/NO (1 μM) for 2 min to induce desensitization. Then, Hb (10 μM) was added and, after various intervals, DEA/NO was reapplied at a concentration (100 μM) sufficient to overcome the effect of Hb. Analysis of the mean initial rate of rise in cGMP levels indicated that sGC was able to recover fully from desensitization within 10 min, the half-time being about 1.5 min (results not shown).
Kinetics of cGMP in Platelets.
To investigate whether sGC desensitization might be a general property of the enzyme, experiments were conducted by using human platelets. Consistent with a previous study (26), addition of DEA/NO (1 μM) led to a transient increase in cGMP, peaking after 15 s and then falling to a much lower steady state (Fig. 6 a and c). Under control conditions, cGMP breakdown in platelets was too fast to be measured. A combination of the PDE inhibitors, sildenafil and erythro-9-(2-hydroxy-3-nonyl)adenine greatly reduced the rate of cGMP degradation such that it was similar to the uninhibited rate observed in cerebellar cells (Fig. 6a Inset). Under these conditions, the platelet cGMP response was transformed from a small brief transient into one that was 10-fold larger and that had a hyperbolic shape resembling that found normally in cerebellar cells (Fig. 6a). Accordingly, deconvolution indicated that vs rose sharply and then fell, much as in cerebellar cells (Fig. 6b).
A prediction is that if vd is increased (i.e., inhibition is removed), the transient cGMP response normally observed in platelets in the absence of PDE inhibitors should be regained. PDE catalytic efficiency, as determined in the presence of the inhibitors, was augmented by increasing Vp or by decreasing Kp. In both cases, a transient cGMP response similar to the measured one was obtained (Fig. 6c), the main difference being that an increased Vp (47-fold) apparently matched better the later falling and steady-state phases. The latter phase of the measured response is artifactual, however, because the cGMP in the platelet suspension after 2 min appeared to be all extracellular (equivalent to 24.0 ± 1.2 pmol cGMP/mg protein). The simulation obtained by decreasing Kp (210-fold) should be more applicable, because it corresponds to removal of competitive inhibition (27), and it appeared to be so because it undershot the later phases.
Discussion
Analysis of NO-stimulated sGC activity in intact cells has revealed several properties that were unexpected from the behavior of the enzyme in tissue homogenates or in its purified form.
First, the potency of NO in cells is greater than has been reported for purified sGC (the α1β1 isoform), where an EC50 of 250 nM was estimated (28). However, other studies that used DEA/NO found an EC50 of about 100 nM (29), suggesting a greater potency, though the resulting NO concentration was not measured directly. In the cerebellar cells, which we have found to express α1 and β1 mRNA and protein (B. Gibb & J.G., unpublished observations), NO is at least an order of magnitude more potent. This finding, which is consistent with the potency of NO to relax vascular smooth muscle (EC50 = 10 nM) (30), helps define the physiologically relevant range of NO concentrations and suggests that current thinking about the distance over which an NO source exerts biological effects (31) will need revision.
Second, an important determinant of the dynamics of NO–sGC signaling is the rate at which sGC deactivates. Measurements of the off-rate of NO from purified sGC, assuming this represents the deactivation rate, have yielded confusing results. When Hb or myoglobin was used to trap NO, the half-life of the NO–sGC complex was estimated to be about 2 min at 37°C (32, 33). From a physiological perspective, this is very slow and difficult to reconcile with the rate at which smooth muscle recovers from NO-induced relaxation (34). A later investigation concluded that, with GTP and Mg2+ present, the half-life fell to about 5 s (35), but a different study claimed a 3-min half-life under these conditions (32). A particularly relevant experiment was when Hb was added during the early steeply rising phase of cGMP accumulation in the cerebellar cells (Fig. 2b). Just 5 s later, cGMP was not only significantly less than in control cells but was at the level predicted should the amount of cGMP at this time be governed solely by PDE activity (i.e., residual sGC activity was zero). Other experiments in which Hb was added later gave concordant results. Hence, in living cells, sGC deactivation occurs with a half-life of no more than a few seconds and possibly much less, allowing sGC to respond dynamically to fluctuations in NO concentration.
The third and most surprising result was that NO-stimulated sGC activity, both in rat cerebellar cells and in human platelets, underwent rapid desensitization. This phenomenon is unlikely to be peculiar to exogenously added NO, because the kinetics of cGMP accumulation observed in the cerebellar cells in the present study is very similar to those occurring in the same cells when endogenous NO production is provoked (13, 14, 19). Consistent with the type of desensitization we describe also existing in vivo is the finding that the hypotensive effects of nitrovasodilators are enhanced when endogenous NO formation is inhibited acutely, an effect attributed from in vitro experiments, to reflect alterations in the sensitivity of smooth muscle sGC to NO (36).
Desensitization appeared to curtail sGC activity within a few seconds of addition of the NO source but because these very early kinetics will also be influenced by the rate of dissociation of NO from the donor, desensitization may well set in earlier. Recovery from desensitization, by comparison, was slow, taking several minutes to be complete. Fast entry into and slow exit from the desensitized state are characteristics classically associated with neurotransmitter receptors (37). The kinetics of onset and recovery clearly distinguishes sGC desensitization recorded in our experiments from sGC down-regulation (sometimes also called “desensitization”) typically seen after chronic (hours–days) exposure to NO-releasing agents and which probably reflects destabilization of subunit mRNA and/or protein (38–40).
The mechanism of sGC desensitization remains unknown. Depletion of substrate (GTP) or direct end-product inhibition (by cGMP) appear not to participate because the desensitization kinetics was little changed despite variations of 6-fold in sGC activity and 10-fold in cGMP levels. That sGC failed to desensitize when assayed in lysed cerebellar cells agrees with numerous previous studies (10, 11, 16) and suggests the existence of a cellular factor that loses its activity (possibly by dilution) in cell-free preparations. Because sGC activity in the lysed cells was about 4-fold higher than the peak activity recorded in intact cells (Fig. 3 b and c), the endogenous factor could function as an inhibitor. A previous study has suggested that there is a protein inhibitor of sGC in bovine lung (41), although it remains to be characterized. Recently, Ca2+ was found to inhibit sGC (42), but only at concentrations orders of magnitude higher than those normally found free in the cytosol. Accordingly, sGC desensitization in cerebellar cells was unaffected by removal of extracellular Ca2+ or by intracellular loading with Ca2+ chelators (unpublished observations), suggesting that Ca2+ is not involved.
Functionally, desensitization of sGC works in concert with PDE activity to shape the cellular cGMP response to NO. That is, as PDE activity differs (43), sGC densitization imposes varying patterns of cGMP accumulation, ranging from a brief low-amplitude transient (with high PDE activity) to a large sustained plateau (with low PDE activity) or a mixture of the two (Fig. 6d). This range of behavior encompasses all the shapes of cGMP response to NO that we have encountered in the literature. The varying temporal and amplitude characteristics are expected to be important in the differential selection of downstream transduction pathways [e.g., high-affinity cGMP-dependent protein kinases vs. low affinity cGMP-gated ion channels (44)].
In conclusion, NO-stimulated sGC activity within a cellular environment is more complex than previously assumed, implying the existence of hitherto unknown regulatory factors. The rapid on- and off-kinetics and the desensitizing profile of activity indicate that the properties of sGC are much more akin to those of a neurotransmitter receptor than previous evidence had suggested. The possibility that alterations in sGC regulation contribute to clinical conditions associated with abnormal tissue responsiveness to NO, such as vascular disorders (45) and erectile dysfunction (46), merits investigation.
Acknowledgments
This work was supported by The Wellcome Trust and a Medical Research Council (U.K.) Studentship (to T.C.B.). Our thanks to Dr. M. P. Gordge for expert help with platelet experiments and to Dr. J. de Vente (University of Maastricht, the Netherlands) for the kind gift of cGMP antiserum.
Abbreviations
- DEA/NO
diethylamine NONOate
- Hb
hemoglobin
- NO
nitric oxide
- ODQ
1-H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-l-one
- PDE
phosphodiesterase
- sGC
soluble guanylyl cyclase
References
- 1.Moncada S, Palmer R M, Higgs E A. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 2.Garthwaite J, Boulton C L. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 3.Waldman S A, Murad F. Pharmacol Rev. 1987;39:163–196. [PubMed] [Google Scholar]
- 4.Ignarro L J. Biochem Pharmacol. 1991;41:485–490. doi: 10.1016/0006-2952(91)90618-f. [DOI] [PubMed] [Google Scholar]
- 5.Sunahara R K, Dessauer C W, Gilman A G. Annu Rev Pharmacol Toxicol. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- 6.Drewett J G, Garbers D L. Endocr Rev. 1994;15:135–162. doi: 10.1210/edrv-15-2-135. [DOI] [PubMed] [Google Scholar]
- 7.Hobbs A J. Trends Pharmacol Sci. 1997;18:484–491. doi: 10.1016/s0165-6147(97)01137-1. [DOI] [PubMed] [Google Scholar]
- 8.Denninger J W, Marletta M A. Biochim Biophys Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 9.Russwurm M, Behrends S, Harteneck C, Koesling D. Biochem J. 1998;335:125–130. doi: 10.1042/bj3350125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katsuki S, Arnold W, Mittal C, Murad F. J Cyclic Nucleotide Res. 1977;3:23–35. [PubMed] [Google Scholar]
- 11.Gerzer R, Hofmann F, Schultz G. Eur J Biochem. 1981;116:479–486. doi: 10.1111/j.1432-1033.1981.tb05361.x. [DOI] [PubMed] [Google Scholar]
- 12.Saito M, Deguchi T. Biochim Biophys Acta. 1979;586:473–480. doi: 10.1016/0304-4165(79)90037-0. [DOI] [PubMed] [Google Scholar]
- 13.Garthwaite J, Charles S L, Chess Williams R. Nature (London) 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- 14.Garthwaite J, Garthwaite G. J Neurochem. 1987;48:29–39. doi: 10.1111/j.1471-4159.1987.tb13123.x. [DOI] [PubMed] [Google Scholar]
- 15.Gordge M P, Meyer D J, Hothersall J, Neild G H, Payne N N, Noronha Dutra A. Br J Pharmacol. 1995;114:1083–1089. doi: 10.1111/j.1476-5381.1995.tb13317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunn S J, Garthwaite J, Wilkin G P. Neurochem Int. 1986;8:179–185. doi: 10.1016/0197-0186(86)90162-2. [DOI] [PubMed] [Google Scholar]
- 17.Fernley H N. Eur J Biochem. 1974;43:377–378. doi: 10.1111/j.1432-1033.1974.tb03423.x. [DOI] [PubMed] [Google Scholar]
- 18.Greenberg L H, Troyer E, Ferrendelli J A, Weiss B. Neuropharmacology. 1978;17:737–745. doi: 10.1016/0028-3908(78)90088-6. [DOI] [PubMed] [Google Scholar]
- 19.Garthwaite J. Br J Pharmacol. 1985;85:297–307. doi: 10.1111/j.1476-5381.1985.tb08860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Vente J, Bol J G J M, Berkelmans H S, Schipper J, Steinbusch H M W. Eur J Neurosci. 1990;2:845–862. doi: 10.1111/j.1460-9568.1990.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 21.Gibson Q H, Roughton F J W. J Physiol (London) 1957;136:507–526. doi: 10.1113/jphysiol.1957.sp005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garthwaite J, Southam E, Boulton C L, Nielsen E B, Schmidt K, Mayer B. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 23.Koesling D. N Schm Arch Pharmacol. 1998;358:123–126. doi: 10.1007/pl00005232. [DOI] [PubMed] [Google Scholar]
- 24.Friebe A, Koesling D. Mol Pharmacol. 1998;53:123–127. doi: 10.1124/mol.53.1.123. [DOI] [PubMed] [Google Scholar]
- 25.Friebe A, Mullershausen F, Smolenski A, Walter U, Schultz G, Koesling D. Mol Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- 26.Salvemini D, Radziszewski W, Korbut R, Vane J. Br J Pharmacol. 1990;101:991–995. doi: 10.1111/j.1476-5381.1990.tb14194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corbin J D, Francis S H. J Biol Chem. 1999;274:13729–13732. doi: 10.1074/jbc.274.20.13729. [DOI] [PubMed] [Google Scholar]
- 28.Stone J R, Marletta M A. Biochemistry. 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 29.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 30.Carter T D, Bettache N, Ogden D. Br J Pharmacol. 1997;122:971–973. doi: 10.1038/sj.bjp.0701549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaughn M W, Kuo L, Liao J C. Am J Physiol. 1998;274:H1705–H1714. doi: 10.1152/ajpheart.1998.274.5.H1705. [DOI] [PubMed] [Google Scholar]
- 32.Brandish P E, Buechler W, Marletta M A. Biochemistry. 1998;37:16898–16907. doi: 10.1021/bi9814989. [DOI] [PubMed] [Google Scholar]
- 33.Kharitonov V G, Sharma V S, Magde D, Koesling D. Biochemistry. 1997;36:6814–6818. doi: 10.1021/bi970201o. [DOI] [PubMed] [Google Scholar]
- 34.Palmer R M, Ferrige A G, Moncada S. Nature (London) 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 35.Kharitonov V G, Russwurm M, Magde D, Sharma V S, Koesling D. Biochem Biophys Res Commun. 1997;239:284–286. doi: 10.1006/bbrc.1997.7470. [DOI] [PubMed] [Google Scholar]
- 36.Moncada S, Rees D D, Schulz R, Palmer R M. Proc Natl Acad Sci USA. 1991;88:2166–2170. doi: 10.1073/pnas.88.6.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones M V, Westbrook G L. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 38.Filippov G, Bloch D B, Bloch K D. J Clin Invest. 1997;100:942–948. doi: 10.1172/JCI119610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldman S A, Rapoport R M, Ginsburg R, Murad F. Biochem Pharmacol. 1986;35:3525–3531. doi: 10.1016/0006-2952(86)90622-2. [DOI] [PubMed] [Google Scholar]
- 40.Ujiie K, Hogarth L, Danziger R, Drewett J G, Yuen P S, Pang I H, Star R A. J Pharmacol Exp Ther. 1994;270:761–767. [PubMed] [Google Scholar]
- 41.Kim T D, Burstyn J N. J Biol Chem. 1994;269:15540–15545. [PubMed] [Google Scholar]
- 42.Parkinson S J, Jovanovic A, Jovanovic S, Wagner F, Terzic A, Waldman S A. Biochemistry. 1999;38:6441–6448. doi: 10.1021/bi990154v. [DOI] [PubMed] [Google Scholar]
- 43.Beavo J A. Physiol Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 44.Kramer R H, Karpen J W. Nature (London) 1998;395:710–713. doi: 10.1038/27227. [DOI] [PubMed] [Google Scholar]
- 45.Katz S D, Schwarz M, Yuen J, LeJemtel T H. Circulation. 1993;88:55–61. doi: 10.1161/01.cir.88.1.55. [DOI] [PubMed] [Google Scholar]
- 46.Christ G J, Kim D C, Taub H C, Gondre C M, Melman A. Can J Physiol Pharmacol. 1995;73:1714–1726. doi: 10.1139/y95-735. [DOI] [PubMed] [Google Scholar]