

Abstract
The biological effect of cytokines is mainly determined by the cytokine-receptor interaction, which is modulated by the concentration and the activity of cytokines and/or their receptors. Because αv-containing integrins can bind to and/or activate latent TGF-β, these integrins have been thought to be involved in the pathogenesis of fibrotic disorders. Our recent observations that αvβ5 is up-regulated in scleroderma fibroblasts and that the transient overexpression of αvβ5 increases the human α2(I) collagen gene expression in normal fibroblasts suggest the involvement of αvβ5 in the self-activation system in scleroderma fibroblasts. In this study, we established stable transfectants with αvβ5 using normal dermal fibroblasts and demonstrated that such cells differentiated into myofibroblasts by the stimulation of autocrine TGF-β. This observation is explained by 1) αvβ5 recruiting latent TGF-β1 on the cell surface, 2) endogenous active TGF-β localizing on the cell surface, and 3) αvβ5 interacting with TGF-β receptors. Furthermore, blockade of αvβ5 reversed the myofibroblastic phenotype in scleroderma fibroblasts. These data identify a novel mechanism for the establishment of autocrine TGF-β signaling in dermal fibroblasts by the up-regulation of αvβ5 and suggest the possibility of regulating fibrotic disorders, especially scleroderma, by targeting this integrin.
The transforming growth factor-β (TGF-β) superfamily mediates a broad range of biological responses.1 One of the principal effects of TGF-β on mesenchymal cells is its stimulation of extracellular matrix (ECM) deposition, which is achieved by coordinating the expression of several functionally distinct but biologically related gene products. TGF-β increases the expression of various ECM components, such as collagen types I, III, VI, VII, and X, fibronectin, and proteoglycans.2–8 It also inhibits the production of proteolytic enzymes that catalyze ECM degradation, while enhancing the expression of protease inhibitors.9 As a result of these multifaceted effects, TGF-β is believed to play a critical role in a variety of biological processes, such as wound healing and tissue remodeling.10,11 Studies of animal models and human clinical specimens strongly suggest that TGF-β is also important in the pathogenesis of several diseases, including fibrotic disorders. For example, it has been demonstrated that the dermal fibroblast activation in scleroderma may be a result of the stimulation by autocrine TGF-β.12 However, the mechanism of the initiation and maintenance of autocrine TGF-β signaling in scleroderma fibroblasts remains to be clarified.
TGF-β1 is normally secreted as a complex composed of three proteins, including the bioactive peptide of TGF-β1, latency-associated peptide-β1 (LAP-β1), and latent TGF-β binding protein-1. TGF-β1 forms a complex with LAP-β1 noncovalently, forming the small latent complex (SLC), and in this configuration, TGF-β1 is unable to bind to its receptors. SLC is joined by latent TGF-β binding protein-1, the N-terminal region of which is covalently cross-linked to ECM proteins by transglutaminase, and the complex of all three proteins is called the large latent complex.13 The constitutive secretion of latent TGF-β1 by many cell types in culture suggests that there are extracellular mechanisms to control the activity of this potent cytokine. Although these processes are not fully understood, recent reports demonstrated that cell surface molecules or secreted extracellular molecules can activate latent TGF-β1. Specifically, integrin αvβ6 and thrombospondin (TSP)-1 have been implicated in the activation of latent TGF-β1 through nonproteolytic mechanisms.14–19 In addition, plasmin has been proposed to lead to the activation of latent TGF-β1 through the proteolytic degradation of LAP-β1.20 Integrin αvβ8 has also been demonstrated to be able to activate latent TGF-β1 by the membrane type 1-matrix metalloproteinase-dependent degradation of LAP-β1.21 Thus, normal TGF-β function seems to be largely controlled by its interaction with extracellular or cell surface molecules.
LAP-β1 contains an RGD motif that is recognized by αv-containing integrins, including αvβ1, αvβ3, αvβ5, αvβ6, and αvβ8. Although all αv-containing integrins bind to LAP-β1, only αvβ6 and αvβ8 have been demonstrated to be able to activate SLC.14,21–23 In particular, the αvβ6-mediated activation of SLC was demonstrated to play an important role in response to tissue injury because epithelium-restricted β6−/− mice showed only a minor fibrotic response to bleomycin compared with wild-type mice. Although there have been no reports indicating the activation of SLC by other αv-containing integrins, such as αvβ1, αvβ3, and αvβ5, we recently demonstrated that αvβ5 is up-regulated in scleroderma dermal fibroblasts, and the transient overexpression of αvβ5 induces the increased transcriptional activity of human α2(I) collagen (COL1A2) gene in normal dermal fibroblasts.24 These findings imply the involvement of αvβ5 in the pathogenesis of scleroderma. The purpose of this study was to assess the hypothesis described above. To this end, we established stable transfectants with αvβ5 and investigated the detailed mechanism underlying the αvβ5-dependent phenotypical alteration of dermal fibroblasts.
Materials and Methods
Reagents
Recombinant human TGF-β1 and SLC were obtained from R & D Systems (Minneapolis, MN). Pan-specific rabbit anti-TGF-β antibody (AB-100-NA), which has the ability to neutralize the biological activity of TGF-β1, -2, -3, and -5, was also purchased from R & D Systems. Actinomycin D, cytochalasin D, and antibodies for β-actin and α-smooth muscle actin (α-SMA) were purchased from Sigma (St Louis, MO). Antibodies for β5-subunit, αv-subunit, β1-subunit, β3-subunit, TβRI, TβRII, Smad2/3, Smad3, and clathrin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-serine antibody was purchased from Biomeda (Foster City, CA). Functional blocking antibody against αvβ5 (P1F6) was purchased from Chemicon (San Francisco, CA). FuGENE 6 was obtained from Roche Diagnostics (Indianapolis, IN). Synthetic RGD-containing peptides (GRGDSP) and RGE-containing peptide (GRGESP) were purchased from Sigma.
Establishment of Stable Transfectants with αvβ5
Normal human dermal fibroblasts, scleroderma fibroblasts, and the expression vector of β5-subunit were prepared as described previously.24 A normal human dermal fibroblast was transfected with either this construct or empty vector (pcDNA3) by electroporation. Stable transfectants were selected in the presence of the neomycin analog G418 (0.25 mg/ml; Sigma).
Immunoblotting
Protein extracts were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. Membranes were incubated overnight with primary antibody, washed, and incubated for 1 hour with secondary antibody. After washing, visualization was performed by enhanced chemiluminescence (Amersham Pharmacia Biotech, Buckinghamshire, UK).
Immunoprecipitation
Protein extracts (1 μg) were pre-adsorbed with protein G-agarose beads (Santa Cruz) and incubated with 2 μg of appropriate antibodies and then with protein G-agarose beads. The precipitated proteins were subjected to immunoblotting.
Biotinylation and Immunoprecipitation
Confluent quiescent cells were incubated with membrane-impermeant NHS-LC-biotin (Pierce, Rockford, IL) dissolved at 0.5 mg/ml in phosphate-buffered saline (PBS) at 4°C for 30 minutes. Immunoprecipitation was performed using whole-cell lysates. Each immunoprecipitate was subjected to SDS-PAGE, and Western blots were prepared. The blots were probed with streptavidin coupled to horseradish peroxidase and visualized by enhanced chemiluminescence.
Cell Adhesion Assays
Ninety-six-well plates were coated with vitronectin or type I collagen (0.3, 1, 3, 10, and 30 μg/ml). Additional wells were coated with 1% bovine serum albumin (BSA) in PBS (minimal adhesion-negative control) and 1 mg/ml of poly-d-lysine (maximal adhesion-positive control). Cells were detached by trypsin-EDTA, washed once in Eagle’s minimum essential medium (MEM) with trypsin inhibitor, and then washed three times with serum-free medium (MEM plus 0.1% BSA). Thereafter, the cells were resuspended in serum-free medium and rocked for 1 hour at 37°C. The cells were then plated onto coated wells (3 × 104 cells/200 μl/well) and incubated for 1 hour at 37°C. Nonadherent cells were removed by centrifugation (top side down) at 48 × g for 5 minutes. The attached cells were fixed with 1% formaldehyde, stained with 0.5% crystal violet overnight, washed with PBS, and lysed in buffer containing 0.1% NaOH and 0.1% SDS. The absorbance (Abs) at 595 nm was measured. The percentage of cells attached to each well was calculated from formula as follows:
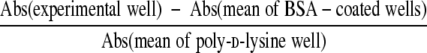 |
 |
For inhibition experiments, cells were incubated for 1 hour with or without increasing dilutions (1/200,1/100, or 1/50) of anti-αvβ5 antibody, plated on to wells coated with vitronectin or type I collagen (5μg/ml), and incubated for 1 hour at 37°C. The percentage of cells attached to each well was calculated from the following formula:
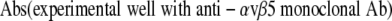 |
 |
 |
RNA Preparation and Northern Blot Analysis
Total RNA (2 μg) was extracted and subjected to electrophoresis on a 1% agarose/formaldehyde gels and blotted onto a nylon filter. The filter was hybridized with DNA probes for COL1A2, TGF-β1, and glyceraldehyde-3-phosphate dehydrogenase genes, or RNA probes for αv- and β5-subunit genes. The membrane was then washed and exposed to X-ray film.
Immunofluorescence
Cells grown in 4-well Lab-Tek chambers (Nunc, Naperville, Illinois) were fixed with 3.7% formaldehyde, permeabilized, and blocked with 10% fetal bovine serum in PBS containing 0.5% Triton X-100. Cells were stained with anti-α-SMA antibody, washed, and incubated with fluorescein isothiocyanate-conjugated secondary antibody (Sigma).
DNA Affinity Precipitation
Two double-stranded oligonucleotides, 3xCAGA oligo and 3xCAGA-M oligo, containing biotin on the 5′-nucleotide of the sense strand were prepared as described previously.25 Poly(dI-dC) competitor (5 μg) was incubated with 500 μg of nuclear protein for 30 minutes at 4°C, followed by 1 hour incubation with 500 pmol of each oligonucleotides. Then, 65 μl of streptavidin-agarose (Sigma) was added to the reaction and incubated at 4°C for overnight. The precipitated proteins were subjected to immunoblotting with anti-Smad2/3 antibody. The specific binding of Smad3 with 3xCAGA oligo was confirmed by experiments using 3xCAGA-M oligo. The binding of Smad3 with 3xCAGA-M oligo was not observed in the presence or absence of TGF-β1 (data not shown).
Plasmid Construction
A −353COL1A2/CAT construct was generated as previously described.26 Plasmids were purified twice on CsCl gradients. At least two different plasmid preparations were used for each experiment.
Transient Transfection
Cells were grown to 50% confluence in 100-mm dishes, transfected with the indicated constructs (2 μg) along with pSV-β-galactosidase (1 μg) using FuGENE6, and incubated for 48 hours. Extracts, normalized for protein content, were incubated with butyl-CoA and [14C]-chloramphenicol for 90 minutes at 37°C. Butylated chloramphenicol was extracted using an organic solvent (a 2:1 mixture of tetramethylpentadecane and xylene) and quantitated by scintillation counting.
Measurement of TGF-β1 Levels
Cells were grown to confluence in 6-well plates. The culture medium was removed, cells were washed with MEM to remove excess TGF-β1, and medium was replaced with serum-free medium. After 24 hours, the levels of active and total TGF-β1 in the supernatants were measured using a TGF-β1 enzyme-linked immunosorbent assay system (Amersham Pharmacia Biotech).27 This system is designed to measure active TGF-β1. Total TGF-β1 was assayed after acid activation by the addition of 1 N HCl to samples.
Latent TGF-β1 Binding Assay
The binding of cells to SLC was determined as described previously with minor modification.28,29 Cells were incubated at 4°C for 1 hour in binding buffer (1 ml of MEM containing 200 mmol/L HEPES [pH 7.4] and 25 mg/ml BSA) supplemented with35S-labeled SLC, which was synthesized in vitro from TGF-β1 cDNA by using the TNT Coupled Reticulocyte Lysate System (Promega, Madison, WI), and then washed twice with PBS containing 0.9 mmol/L CaCl2, 0.5 mmol/L MgCl2, and 0.1% BSA. Cell surface-bound SLC was removed by incubating once with extraction buffer containing 150 mmol/L NaCl, 0.1% acetic acid, and 2 mol/L urea, at 4°C for 3 minutes, and extracted 35S-labeled SLC was quantitated by scintillation counting. To determine RGD-specific binding, we constructed mutant SLC, in which the RGD site in LAP-β1 is changed to RGE, and performed the binding assay using this construct under the same condition. We also performed the same experiments in the presence of excessive cold SLC (R and D system) to determine the nonspecific binding of 35S-labeled SLC. Specific 35S-labeled SLC binding was calculated by subtracting nonspecific binding (measured in the presence of excessive cold SLC) from total binding (measured in the absence of unlabeled SLC).
TGF-β Bioassay
To determine the TGF-β activation, TMLC cells were co-cultured with either transfectant as described previously with minor modifications.14,21 TMLC and test cells were mixed at a ratio of 1:1 and suspended at 1 × 106 cells/ml in MEM containing 10% fetal bovine serum. These cells were plated at 200 μl/well in 12-well plates and allowed to attach for 1 hour. Medium was replaced with 200 μl/well of new medium with or without antibodies and cultured for 24 hours. Cell lysates were prepared using the Reporter Lysis buffer (Promega), and the luciferase activity was determined using the Promega luciferase assay system. Similar co-cultures were performed in 24-well plates with inserts designed for attachment-dependent cell culture (Millicell-PCF 3-μm filter, Millipore, Bedford, MA), but 1.5 × 105 TMLC and test cells were added to upper and lower chambers, respectively. In some experiments, assays were performed in the presence of anti-αvβ5 antibody, preimmune mouse IgG, anti-TGF-β antibody, or preimmune rabbit IgG.
Statistical Analysis
Data presented as bar graphs are the means ± SD of at least five independent experiments. Statistical analysis was performed using the Mann-Whitney U-test (P < 0.05 was considered significant).
Results
Generation and Characterization of Stable αvβ5-Transfectants
We transfected the β5-subunit expression vector or pcDNA3 (empty vector) into four strains of normal dermal fibroblasts. The β5-positive transfectants (β5-transfectants) were screened for increased expression of β5 mRNA (Figure 1A). Twenty positive clones were isolated that expressed higher levels of β5 mRNA than normal dermal fibroblasts or pcDNA3-transfected control cells (mock transfectants). The increased β5 mRNA expression was accompanied by increased cell surface expression of αvβ5 (Figure 1B). The levels of cell surface αv-subunit dimerized with β5-subunit also increased in β5-transfectants, but there was no significant difference in the levels of αv mRNA between these three groups (Figure 1C). This is consistent with the notion that αv-subunit is excessively expressed and cell surface expression levels of αv-containing integrins are regulated by the levels of β-subunits.30 In contrast, the levels of αvβ1 and αvβ3 were unchanged in β5-transfectants (Figure 1, D and E), excluding the possibility that the increased β5 expression might compete with other β-subunits for dimerizing with the αv-subunit. Consistent with previous reports, the expression of β6 and β8 was not observed in normal dermal fibroblasts (data not shown). Two mock transfectants and two β5-transfectants were selected from each of four normal fibroblast strains, and all of the following experiments were performed using these eight mock transfectants and eight β5-transfectants. To prove the functionally active state of the newly expressed αvβ5 in β5-transfectants, we assayed the attachment of either transfectant to vitronectin and type I collagen. β5-Transfectants strongly attached to vitronectin compared with mock transfectants, whereas the attachment of either transfectant to type I collagen was at a similar level (Figure 2A). The specificity of adhesion was confirmed by the application of anti-αvβ5 antibody (Figure 2B). Thus, the newly expressed αvβ5 was functionally active.
Figure 1.

Confirmation of the establishment of β5-transfectants. Total RNAs were extracted from confluent quiescent cells, and the levels of β5 mRNA (A) or αv mRNA (C) were determined by Northern blotting. Cell surface proteins were labeled with biotin, and whole-cell lysates were subjected to immunoprecipitation using goat anti-β5 antibody (B, left panel), goat anti-β1 antibody (D), or goat anti-β3 antibody (E). Precipitated proteins were subjected to SDS-PAGE, and Western blots were prepared. The blots were probed with streptavidin coupled to horseradish peroxidase. A parallel immunoprecipitation performed with preimmune goat IgG failed to show integrin immunoreactivities (B, right panel). The bands in immunoprecipitation were confirmed to be the indicated subunits by a reprobing analysis using specific antibodies to αv-, β1-, β3-, or β5-subunits (data not shown), whereas the identity of α1, α2, α3, and α5 was determined by the size on a gel. Experiments were repeated five times with similar results.
Figure 2.

Assay of mock and β5-transfectant attachment to vitronectin or type I collagen. A: Cells were incubated at 37°C for 1 hour in the wells coated with vitronectin or type I collagen. After centrifugation, attached cells were stained, and the absorbance of 595 nm was measured. B: Cells were preincubated at 37°C for 1 hour with anti-αvβ5 antibody or preimmune IgG, and cell attachment was determined. The mean from five separate experiments is shown.
β5-Transfectants Display the Myofibroblastic Differentiation
To assess the phenotypical alteration of β5-transfectants, we compared the expression of the COL1A2 gene between mock and β5-transfectants. The levels of COL1A2 mRNA and the activity of the −353COL1A2 promoter were significantly elevated in β5-transfectants (Figure 3, A and B). Furthermore, the level of α-smooth muscle actin (α-SMA) protein, a specific marker of myofibroblasts, was also markedly elevated in β5-transfectants (Figure 3C), indicating the myofibroblastic differentiation of β5-transfectants. This finding was confirmed by immunofluorescence of β5-transfectants, which showed the morphological changes of cellular hypertrophy and well-formed α-SMA fibers, specific features of myofibroblasts previously described31 (Figure 3D).
Figure 3.

Myofibroblastic differentiation of β5-transfectants. A: mRNA levels of α2(I) collagen and GAPDH genes were analyzed by Northern blotting. DNA probes against α2(I) collagen hybridized to cellular transcripts of 5.2 and 5.7 kb. One representative of five independent experiments is shown. B: The −353COL1A2 promoter activities were determined by CAT assays. Values represent the CAT activity relative to that of mock transfectants (100 arbitrary units [AU]). The means ± SD of five independent experiments are shown. *P < 0.05 versus mock transfectants. The expression of α-SMA protein was determined by immunoblotting (C) and by immunofluorescence (D). Experiments were repeated five times with similar results.
Constitutive Activation of TGF-β Signaling by Endogenous TGF-β Stimulation in β5-Transfectants
We next investigated the effects of anti-TGF-β antibody or TGF-β1 antisense oligonucleotide (5′-GAGGGCGGCATGGGGAGG-3′, AS-TGF-β1), which has been found to be sufficient to block TGF-β1 transcription in vitro32 and in vivo,33 in relation to the COL1A2 promoter activity. Mock transfectants treated with anti-TGF-β antibody showed little reduction in −353COL1A2 promoter activity, whereas treated β5-transfectants showed a marked, dose-dependent reduction (Figure 4A). The treatment with AS-TGF-β1 showed similar results to those obtained by anti-TGF-β antibody (Figure 4B). These results indicate that constitutive activation of β5-transfectants depends on stimulation by autocrine TGF-β. The TGF-β isoform that was primarily responsible for the activation of β5-transfectants was TGF-β1 because AS-TGF-β1 decreased the promoter activity to a similar extent as that achieved with anti-TGF-β antibody. This notion was confirmed by immunoprecipitation and DNA affinity precipitation, which showed constitutive phosphorylation of Smad3 and increased DNA binding ability of Smad3 in β5-transfectants, respectively (Figure 4, C and D).
Figure 4.

Constitutive activation of TGF-β/Smad signaling by endogenous TGF-β stimulation in β5-transfectants. Cells were transfected with 2 μg of −353COL1A2 promoter in the presence of anti-TGF-β antibody (anti-TGF-β) or preimmune IgG (A) or in the presence of TGF-β1 antisense oligonucleotide (AS-TGF-β1) or TGF-β1 sense oligonucleotide (S-TGF-β1) (B). Values represent the CAT activity relative to that of mock transfectants treated with preimmune IgG (A) or treated with S-TGF-β1 (B) (100AU). The means ± SD of five independent experiments are shown. *P < 0.05 versus β5-transfectants treated with preimmune IgG (A) or S-TGF-β1 (B). Whole-cell lysates derived from multiple clones of each transfectant were subjected to immunoprecipitation using anti-Smad3 antibody, and phospho-Smad3 was detected by immunoblotting using anti-phospho-serine antibody (C). The same membrane was stripped and reprobed with anti-Smad2/3 antibody (left panel). A parallel immunoprecipitation performed with preimmune mouse IgG failed to show Smad3 immunoreactivity (right panel). D: Nuclear extracts derived from multiple clones of each transfectant were incubated with biotin-labeled oligonucleotides. Proteins bound to these nucleotides were isolated with streptavidin-agarose beads, and Smad3 was detected by immunoblotting. All blots show one representative of five independent experiments.
Myofibroblastic Differentiation of β5-Transfectants Requires Cell Adhesion Signals and Stimulation by Autocrine TGF-β
As described above, myofibroblastic differentiation of β5-transfectants may be attributed to the stimulation by autocrine TGF-β. Because a previous report demonstrated that myofibroblastic differentiation is dependent on cell adhesion signaling, such as integrin/FAK, as well as TGF-β stimulation,34 we investigated the activation state of focal adhesion kinase (FAK). As shown in Figure 5A, the phosphorylation levels of FAK on Tyr-397 were elevated in β5-transfectants compared with mock transfectants. Treatment with PP-2, which is a pharmacological inhibitor of FAK/Src, or transient overexpression of a kinase-deficient FAK mutant, mutated by substituting Tyr-397 with Phe, markedly decreased the expression levels of α-SMA in β5-transfectants (Figure 5, B and C, respectively). Furthermore, the treatment of AS-TGF-β1 suppressed the expression of α-SMA and the phosphorylation of FAK on Tyr-397 (Figure 5, D and E, respectively). Cumulatively, these results support the previous finding that myofibroblastic differentiation by TGF-β stimulation is dependent on cell adhesion signaling.
Figure 5.

Myofibroblastic differentiation of β5-transfectants is dependent on both TGF-β signaling and integrin/FAK signaling. A: Whole-cell lysates derived from multiple clones of each transfectant were subjected to immunoblotting using anti-397Y-phospho-FAK antibody. The same membrane was stripped and reprobed with anti-FAK antibody. B: Quiescent fibroblasts were treated with the indicated reagent for 48 hours. Whole-cell lysates were subjected to immunoblotting using anti-α-SMA antibody and anti-β-actin antibody. C: Cells were transfected with 2 μg of expression vector of Y397F-FAK or corresponding empty vector. After a 48-hour incubation, whole-cell lysates were subjected to immunoblotting using anti-α-SMA antibody and anti-β-actin antibody. D: Quiescent cells were treated with 10 μmol/L TGF-β1 antisense oligonucleotide (AS-TGF-B1) or TGF-β1 sense oligonucleotide (S-TGF-β1) for 48 hours. Whole-cell lysates were subjected to immunoblotting using anti-α-SMA antibody and anti-β-actin antibody. E: Quiescent cells were treated with 10 μmol/L TGF-β1 antisense oligonucleotide (AS-TGF-B1) or TGF-β1 sense oligonucleotide (S-TGF-β1) for 48 hours. Whole-cell lysates were subjected to immunoblotting using anti-397Y-phospho-FAK antibody. The same membrane was stripped and reprobed with anti-FAK antibody. All experiments were performed using multiple clones of each transfectants, and one representative is shown. In right panels of B through D, the protein levels quantitated by scanning densitometry are shown relative to those in mock transfectants treated with dimethylsulfoxide (DMSO), empty vector, or S-TGF-β1, respectively (1 arbitrary unit [AU]). Data are expressed as the means ± SD of five independent experiments. *P < 0.05 versus mock transfectants under the same condition. **P < 0.05 versus β5-transfectants treated with DMSO, empty vector, or S-TGF-β1.
Latent TGF-β1 Is Recruited on the Cell Surface by the Interaction of LAP-β1 with αvβ5 in β5-Transfectants
We next compared the expression levels of TGF-β1 between mock and β5-transfectants. There was no significant difference in mRNA levels (Figure 6A). However, the levels of total TGF-β1 proteins (active + latent) in conditioned media were significantly decreased in β5-transfectants (Table 1, 0.284 ± 0.075 ng/ml versus 0.497 ± 0.105 ng/ml, P < 0.05), but there was no significant difference in the levels of active TGF-β1. Preincubation with anti-αvβ5 antibody restored the levels of total TGF-β1 proteins in β5-transfectants (Table 1), whereas the same treatment had no effect on the levels of TGF-β1 mRNA (data not shown). Because LAP-β1 binds αvβ5,22 these results suggest that SLC is recruited on the cell surface of β5-transfectants through the interaction of LAP-β1 with αvβ5. To confirm this hypothesis, we compared binding to SLC between mock and β5-transfectants (Figure 6B). SLC binding ability was significantly elevated in β5-transfectants (5.0-fold increase, P < 0.05). By the addition of anti-αvβ5 antibody, the increased SLC binding in β5-transfectants was significantly reduced to a level similar to that observed in mock transfectants. Because previous reports demonstrated that LAP-β1 binds to αv-containing integrins, such as αvβ6 and αvβ8, through its RGD motif,14,21 we further investigated whether LAP-β1 interacts with αvβ5 through its RGD motif. To this end, we generated mutant SLC, in which the RGD motif was changed to RGE, and performed binding assay. As expected, there was no significant difference in the binding of mutant SLC between mock and β5-transfectants. We further performed binding assays in the presence of RGD peptide or RGE peptide and demonstrated that RGD peptide, but not RGE peptide, significantly decreased SLC binding in β5-transfectants, whereas both peptides showed no effect in mock transfectants. Cumulatively, these results indicate that SLC is recruited on the cell surface of β5-transfectants through the interaction of RGD motif of LAP-β1 with αvβ5.
Figure 6.

SLC is recruited and activated by its interaction with αvβ5 in β5-transfectants. A: Northern blotting was performed with DNA probes for TGF-β1 and GAPDH genes. Experiments were repeated five times with similar results. B: Confluent quiescent cells were incubated at 4°C for 1 hour with 35S-labeled SLC or mutated SLC (mSLC) in the presence or absence of cold SLC. Cell surface-bound SLC was removed by extraction buffer and quantitated by scintillation counting. In some experiments, assays were performed in the presence of RGD peptide (10 μg/ml), RGE peptide (10 μg/ml), anti-αvβ5 antibody (anti-αvβ5, 10 μg/ml), or preimmune mouse IgG (IgG, 10 μg/ml). Values represent the scintillation count relative to that of mock transfectants treated with SLC (100AU). *P < 0.05 versus mock transfectants under the same condition. C: Cells were cultured in MEM containing 10 μmol/L AS-TGF-β1 for 48 hours. Then, the medium was changed to serum free-medium containing 10 μmol/L AS-TGF-β1, and cells were transfected with 2 μg of -353COL1A2 promoter. Cells were stimulated with the indicated concentration of TGF-β1 or SLC for the last 24 hours, and the CAT activities were determined. In some experiments, SLC stimulation was performed in the presence of RGD peptide (10 μg/ml), RGE peptide (10 μg/ml), anti-αvβ5 antibody (anti-αvβ5, 10 μg/ml), or preimmune mouse IgG (IgG, 10 μg/ml). Values represent the CAT activity relative to that of mock transfectants treated with S-TGF-β1 (100AU). The means ± SD of five independent experiments are shown. *P < 0.05 versus β5-transfectants treated with AS-TGF-β1. #P < 0.05 versus β5-transfectants treated with AS-TGF-β1, SLC, and RGE peptide. $P < 0.05 versus β5-transfectants treated with AS-TGF-β1, SLC, and preimmune mouse IgG.
Table 1.
Levels of Total (Active + Latent) and Active TGF-β1 Secreted by Either Transfectant into Culture Supernatants
| Total TGF-β1 (ng/ml) | Active TGF-β1 (ng/ml) | |
|---|---|---|
| Mock transfectants without any treatments | 0.497 ± 0.105 | 0.071 ± 0.015 |
| Mock transfectants treated with anti-αvβ5 antibody | 0.525 ± 0.112 | 0.080 ± 0.019 |
| β5-Transfectants without any treatments | 0.284 ± 0.075* | 0.063 ± 0.015 |
| β5-Transfectants treated with anti-αvβ5 antibody | 0.501 ± 0.106 | 0.070 ± 0.017 |
Values are means ± SD.
P < 0.05.
The Inducibility of the COL1A2 Promoter Activity against Exogenous SLC Stimulation Is Increased in β5-Transfectants
We next investigated the effect of exogenous SLC stimulation on −353COL1A2 promoter activity in mock and β5-transfectants (Figure 6C). To suppress the endogenous TGF-β1 production, cells were pretreated with 10 μmol/L AS-TGF-β1 for 48 hours before transfection. AS-TGF-β1 almost completely diminished the increased promoter activity in β5-transfectants, whereas it showed little reduction in mock transfectants. Exogenous TGF-β1 stimulation showed significant increase in the promoter activity in both transfectants pretreated with AS-TGF-β1. Exogenous SLC stimulation also showed significant increase in the promoter activity in β5-transfectants in a dose-dependent manner, whereas the same treatment revealed no significant effect in mock transfectants. In addition, RGD peptide, but not RGE peptide, significantly decreased SLC-induced promoter activity in β5-transfectants, whereas both peptides showed no effect on the promoter activity in mock transfectants treated with SLC. Furthermore, in the presence of anti-αvβ5 antibody, the effect of SLC in β5-transfectants was completely diminished. These results indicate that the inducibility of the COL1A2 promoter activity against exogenous SLC stimulation is increased in β5-transfectants by the αvβ5-dependent manner.
Endogenous Active TGF-β Is Localized on the Cell Surface of β5-Transfectants
To investigate the localization of active TGF-β, we co-cultured either transfectant with TMLC cells, a mink lung epithelial reporter cell line stably expressing a portion of the plasminogen activator inhibitor 1 promoter35 (Figure 7A). The luciferase activity was significantly elevated in TMLC cells co-cultured with β5-transfectants compared with those co-cultured with mock transfectants (about 10-fold increase, P < 0.05). This increase was almost completely abolished by antibodies against TGF-β or αvβ5, whereas preimmune mouse or rabbit IgG showed no effect. We also performed co-culture assays with inserts to separate TMLC cells and either transfectant while allowing soluble molecules to pass. In the absence of contact, β5-transfectants showed no significant induction of luciferase activity. These results indicate that activated endogenous TGF-β is localized on the cell surface of β5-transfectants. We next tested whether the activation of SLC in β5-transfectants was occurring through previously described mechanisms.20,22,36,37 Plasmin inhibitor (aprotinin), mannose-6-phosphate, metalloprotease inhibitor (GM6001), aspartic protease inhibitor (leupeptin, pepstatin A), cysteine protease inhibitor (leupeptin and E64), serine protease inhibitor (phenylmethylsulfonyl fluoride, aprotinin, and leupeptin), and functional blocking antibody against TSP-1 (monoclonal Ab 133) did not affect the COL1A2 promoter activity in β5-transfectants (Figure 7B). These results indicate that the activation of SLC in β5-transfectants does not require other known activators of SLC.
Figure 7.

SLC is activated on the cell surface of β5-transfectants. A: Transfectant and TMLC cells were mixed at a ratio of 1:1 and co-cultured in confluence. In experiments without cell-cell contact, either transfectant or TMLC cells were co-cultured separately by using inserts. After a 24-hour incubation, the luciferase activities were determined. In some experiments, assays were performed in the presence of anti-αvβ5 antibody (anti-αvβ5, 10 μg/ml), preimmune mouse IgG (M-IgG, 10 μg/ml), anti-TGF-β antibody (anti-TGF-β, 10 μg/ml), or preimmune rabbit IgG (R-IgG, 10 μg/ml). Values represent the luciferase activity relative to that of TMLC cells co-cultured with mock transfectants in the absence of contact (100AU). *P < 0.05 versus mock transfectants under the same condition. B: Mock or β5-transfectants were transfected with 2 μg of -353COL1A2 promoter in the presence of appropriate reagents. Values represent the CAT activity relative to that of β5-transfectants treated with DMSO (100AU). The means ± SD of five independent experiments are shown.
Interaction between αvβ5 and TGF-β Receptors
Previous reports demonstrated that TGF-β receptors and β5-subunit were located in clathrin-coated membranes.28,38,39 Furthermore, a recent report proposed a novel model of TGF-β signaling regulation in which clathrin-dependent internalization promotes TGF-β signaling by sequestering the complex of TGF-β and its receptors from the Smad7-Smurf-mediated degradation pathway, which is mediated by the caveolin-dependent internalization.40 Taken together, these results suggest that αvβ5 promotes the interaction of endogenous TGF-β1 with its receptors in clathrin-coated membranes, subsequently resulting in efficient activation of TGF-β signaling through the acceleration of the clathrin-dependent internalization. To confirm this point, we performed immunoprecipitation. Consistent with previous reports, anti-clathrin antibody precipitated detectable levels of αv-subunit, β5-subunit, TβRI, and TβRII in both transfectants (Figure 8A). Furthermore, as shown in immunoprecipitation assays using anti-clathrin antibody or anti-caveolin antibody, which effectively precipitated these proteins (Figure 8B, right panels), β5-subunits predominantly interacted with clathrin rather than with caveolin (Figure 8B, left panels). Moreover, the interaction of β5-subunits with TGF-β receptors was markedly elevated in β5-transfectants, whereas such interaction was marginal in mock transfectants (Figure 8C), suggesting that the levels of interaction between αvβ5 and TGF-β receptors depends on the levels of αvβ5. These results support the hypothesis described above.
Figure 8.

Interaction between αvβ5 and TGF-β receptors. A: Whole-cell lysates were subjected to immunoprecipitation using anti-clathrin antibody, and αv, β5, TβRI, TβRII, or clathrin were detected by immunoblotting. B: Whole-cell lysates were subjected to immunoprecipitation using anti-clathrin antibody (CL) or anti-caveolin antibody (CV), and β5, clathrin, or caveolin levels in precipitated proteins were determined by immunoblotting (left panels). Residual (unprecipitated) proteins were also subjected to immunoblotting using anti-clathrin antibody or anti-caveolin antibody to confirm the efficiency of immunoprecipitation (right panels). C: Immunoprecipitation was performed using anti-β5 antibody, and β5, TβRI, or TβRII was detected by immunoblotting. All blots show one representative of five independent experiments.
The Treatment of Anti-αvβ5 Antibody Reversed the Myofibroblastic Phenotype in Scleroderma Fibroblasts
Finally, we investigated whether αvβ5 was involved in the self-activation system in scleroderma fibroblasts, which might be activated by the stimulation of autocrine TGF-β. To this end, we examined the effect of anti-αvβ5 antibody on the expression levels of α-SMA in scleroderma fibroblasts (Figure 9). The expression levels of α-SMA were significantly elevated in scleroderma fibroblasts compared with normal fibroblasts. However, the treatment of anti-αvβ5 antibody significantly reduced the expression levels of α-SMA in scleroderma fibroblasts, whereas the treatment of preimmune IgG showed no effect. In contrast, neither anti-αvβ5 antibody nor preimmune IgG affected the expression levels of α-SMA in normal fibroblasts. These results indicate that the up-regulated expression of αvβ5 contributes to the establishment of myofibroblastic phenotype in scleroderma fibroblasts.
Figure 9.

The treatment of anti-αvβ5 antibody reverses the myofibroblastic phenotype in scleroderma fibroblasts. A: Whole-cell lysates prepared from quiescent normal and scleroderma fibroblasts were subjected to immunoblotting using anti-α-SMA antibody and anti-β-actin antibody. B: Quiescent normal and scleroderma fibroblasts were treated with anti-αvβ5 antibody (10 μg/ml) or preimmune mouse IgG (10 μg/ml) for 48 hours. Whole-cell lysates were subjected to immunoblotting using anti-α-SMA antibody and anti-β-actin antibody. Experiments were repeated five times with similar results.
Discussion
This study was undertaken to clarify the mechanism underlying the αvβ5-mediated myofibroblast differentiation of dermal fibroblasts. First, we demonstrated that stable transfection of β5-subunit into dermal fibroblasts resulted in the myofibroblast differentiation through the establishment of autocrine TGF-β signaling. This whole process was dependent on FAK activation, consistent with the notion that stable expression of the myofibroblast phenotype requires both TGF-β1 and integrin/FAK signals. Next, we showed that β5-transfectants recruited SLC on the cell surface through the interaction of αvβ5 with RGD motif in LAP and that endogenous active TGF-β was localized on the cell surface of β5-transfectants. Furthermore, we revealed that αvβ5 interacted with TGF-β receptors and that the complex of αvβ5 with TGF-β receptors was markedly elevated in β5-transfectants. Collectively, αvβ5 may promote a ternary complex formation of endogenous TGF-β and TGF-β receptors, leading to the establishment of autocrine TGF-β signaling. Finally, we elucidated that the blockade of αvβ5 reversed the myofibroblastic phenotype in scleroderma fibroblasts. To our knowledge, this is the first report to demonstrate the detailed mechanism underlying the αvβ5-mediated myofibroblast differentiation of dermal fibroblasts.
In general, the biological effect of cytokines is mainly determined by the occurrence of cytokine-receptor interaction, which is modulated by the concentration and the activity of cytokines and/or their receptors. For example, in scleroderma fibroblasts, the up-regulated expression of TGF-β receptors contributes to the increased biological effect of autocrine TGF-β, resulting in excessive ECM deposit.12 The present observations that αvβ5 recruits SLC on cell surface and complexes with TGF-β receptors indicate that αvβ5 is a novel molecule that enhances the occurrence of the interaction between endogenous TGF-β and its receptors. In support of this concept, αvβ5 is markedly elevated in scleroderma fibroblasts,24 and the blockade of αvβ5 reverses the myofibroblastic phenotype of scleroderma fibroblasts.
Because TGF-β is secreted in its latent form, normal TGF-β function is thought to be largely controlled by its activation from the latent state. So far, two mechanisms have been reported in the activation of SLC. One is the proteolysis of LAP, which results in the release of active TGF-β from SLC. Proteases such as plasmin, metalloproteases, aspartic proteases, cysteine proteases, and serine proteases have been reported to be involved in this process.20,22,37 In addition, αvβ8 activates SLC via a membrane type 1-matrix metalloproteinase-dependent proteolytic pathway.21 The other mechanism is the conformational change of LAP leading to the activation of SLC. This nonproteolytic process is thought to be dependent on an intrinsic ability of LAP to adopt different conformations.41 TSP-1 and αvβ6 have been demonstrated to be involved in this process.14,17,37 SLC binds to TSP-1 through the N-terminus of LAP and such interaction induces a conformational change and subsequent activation of SLC, although the active TGF-β molecules remain bound to TSP-1. SLC also interacts with αvβ6 through the C-terminus of LAP, but such interaction is not sufficient for its activation. After binding, αvβ6 requires interaction with actin cytoskeleton to activate bound SLC. In the present study, we demonstrated that the up-regulated expression of αvβ5 induced myofibroblast differentiation by the establishment of autocrine TGF-β signaling, suggesting that αvβ5 activates SLC. Because β5 has a cytoplasmic domain highly homologous to that of β6-subunit,42 we made a hypothesis that αvβ5 activates SLC by the nonproteolytic pathway. To clarify this point, we performed TGF-β bioassay. β5-Transfectants significantly increased the luciferase activity of TMLC cells in the presence of contact, suggesting that endogenous active TGF-β is localized on the cell surface of β5-transfectants. By contrast, β5-transfectants showed no significant effect on the luciferase activity of TMLC cells in the absence of cell contact. Consistently, there was no significant difference in the levels of active TGF-β1 in cultured media between mock and β5-transfectants. Furthermore, any known activators of SLC did not inhibit the luciferase activity of TMLC cells co-cultured with β5-transfectants. These observations suggest that SLC is activated on the cell surface of β5-transfectants via a nonproteolytic pathway. However, Munger et al14 previously demonstrated that TMLC cells co-cultured with β6-transfectants showed significantly increased luciferase activity compared with those co-cultured with mock transfectants even in the absence of cell contact, which is not consistent with the present observation in β5-transfectants. There are two possible explanations against this discrepancy. The first is that αvβ5 does not activate SLC but promotes formation of a ternary complex between endogenous TGF-β and the TGF-β receptors, which would result in the activation of downstream pathways in a TGF-β-dependent fashion. The second is that SLC is activated by αvβ5 on the cell surface and is rapidly exposed to and consumed by TGF-β receptors in β5-transfectants, which would lead to the low levels of freely diffusible TGF-β in cultured media. Although we cannot determine which hypothesis is likely, the interaction of αvβ5 with TGF-β receptors may play a central role in the establishment of autocrine TGF-β signaling in β5-transfectants.
Numerous previous findings indicate that integrin-mediated adhesion regulates transmission of growth factor receptor signals.43 Regarding TGF-β signaling, Thannickal et al34 demonstrated that stable expression of the myofibroblast phenotype requires both TGF-β1 and adhesion-dependent signals, whereas TGF-β receptor(s)-mediated signaling of Smad2 phosphorylation does not appear to require simultaneous integrin activation. In the present study, we demonstrated that β5-transfectants differentiate into myofibroblasts, and pharmacological inhibition of FAK or expression of kinase-deficient FAK reduced the expression of α-SMA in those cells. The treatment of AS-TGF-β1 reduced the phosphorylation levels of 397-FAK and the expression levels of α-SMA in β5-transfectants, suggesting that activation of integrin/FAK signaling as well as stimulation of autocrine TGF-β is also required in myofibroblastic differentiation of β5-transfectants. Alternatively, adhesion-dependent signals may induce the expression of other proteins modifying the TGF-β/Smad pathway. Further studies regarding the detailed mechanism of the interaction between TGF-β signaling and integrin-dependent adhesion signaling may give valuable clues for the treatment of fibrotic diseases characterized by persistent myofibroblast activation.
The novel finding in the present study is the interaction of αvβ5 with TGF-β receptors. To date, the interaction of integrins with receptor tyrosine kinases, such as epidermal growth factor receptor, platelet-derived growth factor receptor β, insulin-like growth factor receptor, and vascular endothelial growth factor receptor-2, have been well studied. The epidermal growth factor receptor forms a complex with β1 integrins and antibodies against the β1 integrin subunit cause autophosphorylation of this receptor, even in the absence of the ligand stimulation.44 αvβ3 has been shown to associate with receptor tyrosine kinases such as platelet-derived growth factor receptor β,45–47 insulin-like growth factor receptor,46 and vascular endothelial growth factor receptor-247 in the presence of ligand stimulation, and such interactions result in a synergistic signaling effect. In contrast, the interaction of integrins with nonreceptor tyrosine kinases, such as those for TGF-β, has been poorly understood. However, Scaffidi et al48 recently demonstrated that αvβ3 interacts with TβRII, and such interaction potentiates the proliferative effects of TGF-β1 in human lung fibroblasts. In the present study, TGF-β1 antisense oligonucleotide or anti-TGF-β1 antibody almost completely diminished the increased COL1A2 promoter activity in β5-transfectants. This observation excludes the possibility that the interaction of αvβ5 with TGF-β receptors causes autophosphorylation of these receptors even in the absence of ligand but strongly supports the concept that such interaction potentiates the fibrogenic effects of TGF-β1 on human dermal fibroblasts. Collectively, the interaction of integrins with growth factor receptors may be a major form of cross-talk as well as the sharing of intracellular signaling molecules. The interaction of integrins with TGF-β receptors may give new insight into the cooperation of these two signaling pathways to enhance cellular responses.
A previous report demonstrated that epithelium-restricted β6−/− mice exhibited exaggerated lung inflammation and protection from pulmonary fibrosis by the treatment of bleomycin.14 This observation is explained by the loss of paracrine effect of SLC, which is recruited and activated on the cell surface of lung and bronchial epithelium by αvβ6. The current data that the up-regulated expression of αvβ5 enhances the COL1A2 gene expression in normal dermal fibroblasts indicate that autocrine effect is biologically critical in the αvβ5-dependent activation of TGF-β signaling in dermal fibroblasts. This notion is strongly supported by the present result that the up-regulated αvβ5 interacts with TGF-β receptors in dermal fibroblasts. Considering that clathrin-dependent internalization sequesters activated TGF-β receptor complex from Smad7-Smurf-mediated degradation,40 the predominant interaction of αvβ5 with clathrin rather than with caveolin may also contribute to the establishment of autocrine TGF-β signaling through the acceleration of clathrin-dependent internalization. Because the cell surface expression of αvβ5 is controlled by the levels of β5-subunit and a major cytokine that increases the expression of β5-subunit is TGF-β,30,49 it is postulated that αvβ5 is one of candidates that play important roles in positive feedback of TGF-β signaling.
A previous study demonstrated that mice lacking integrin β5-subunit develop, grow, and reproduce normally. There was also no abnormality in wound healing and adenovirus infection, which are major biological processes in which αvβ5 participates.50 These results suggest that most roles of αvβ5 can be compensated for by other αvβ5-independent pathways. This functional redundancy in αvβ5 makes the pharmacological interference of αvβ5 functions a promising approach to the treatment of fibrotic diseases. It would be of interest to determine whether there is a defect of TGF-β activation in these mice. Alternatively, to our knowledge, investigations of TGF-β activity in animal models treated with αvβ5 blocking antibody have not been performed.
In summary, we demonstrated that the up-regulated expression of αvβ5 induces myofibroblastic differentiation of dermal fibroblasts. The interaction of αvβ5 with TGF-β receptors is a novel finding that can explain the establishment of autocrine TGF-β signaling in β5-transfectants. This evidence suggests the possibility of regulating fibrotic disorders, especially scleroderma, by targeting this integrin.
Acknowledgments
We thank Dr. Joseph C. Loftus (Mayo Clinic, Scottsdale, AZ), Dr. Daniel B. Rifkin (New York University School of Medicine, New York), and Dr. Joanne E. Murphy-Ullrich (University of Alabama, Birmingham, AL) for providing us with αv cDNA, TMLC cells, and anti-TSP1 antibody, respectively.
Footnotes
Address reprint requests to Hironobu Ihn, M.D., Ph.D., Department of Dermatology & Plastic and Reconstructive Surgery, Faculty of Medical and Pharmaceutical Sciences, Kumamoto University, 1-1-1 Honjo, Kumamoto-city, Kumamoto 860-8556, Japan. E-mail: ihn-der@kaiju.medic.kumamoto-u.ac.jp.
Supported in part by a grant for scientific research from the Japanese Ministry of Education (10770391) and by project research for progressive systemic sclerosis from the Japanese Ministry of Health and Welfare.
References
- Heldin CH, Miyazono K, ten Dijke P. TGF-β signaling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Jelaska A, Arakawa M, Broketa G, Korn JH. Heterogeneity of collagen synthesis in normal and systemic sclerosis skin fibroblasts: increased proportion of high collagen-producing cells in systemic sclerosis fibroblasts. Arthritis Rheum. 1996;39:1338–1346. doi: 10.1002/art.1780390811. [DOI] [PubMed] [Google Scholar]
- LeRoy EC. Increased collagen synthesis by SSc skin fibroblasts in vitro: a possible defect in the regulation or activation of the scleroderma fibroblast. J Clin Invest. 1974;54:880–889. doi: 10.1172/JCI107827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham RB, Prince RK, Rodnan GP, Taylor F. Increased collagen accumulation in dermal fibroblast cultures from patients with progressive systemic sclerosis (scleroderma). J Lab Clin Med. 1978;92:5–21. [PubMed] [Google Scholar]
- Peltonen J, Kahari L, Uitto J, Jimenez SA. Increased expression of type VI collagen genes in systemic sclerosis. Arthritis Rheum. 1990;33:1829–1835. doi: 10.1002/art.1780331211. [DOI] [PubMed] [Google Scholar]
- Rudnicka L, Varga J, Christiano AM, Iozzo RV, Jimenez SA, Uitto J. Elevated expression of type VII collagen in the skin of patients with systemic sclerosis: regulation by transforming growth factor-β. J Clin Invest. 1994;93:1709–1715. doi: 10.1172/JCI117154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WD, LeRoy EC, Smith EA. Fibronectin release by systemic sclerosis and normal dermal fibroblasts in response to TGF-β. J Rheumatol. 1991;18:241–246. [PubMed] [Google Scholar]
- Falanga V, Tiegs SL, Alstadt SP, Roberts AB, Sporn MB. Transforming growth factor-β: selective increase in glycosaminoglycan synthesis by cultures of fibroblasts from patients with progressive systemic sclerosis. J Invest Dermatol. 1987;89:100–104. doi: 10.1111/1523-1747.ep12580445. [DOI] [PubMed] [Google Scholar]
- Massague J. Transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB. Transforming growth factor-β: recent progress and new challenges. J Cell Biol. 1992;119:1017–1021. doi: 10.1083/jcb.119.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Border WA, Ruoslahti E. Transforming growth factor-β in disease: the dark side of tissue repair. J Clin Invest. 1992;90:1–7. doi: 10.1172/JCI115821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihn H, Yamane K, Kubo M, Tamaki K. Blockade of endogenous transforming growth factor-β signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor-β receptors. Arthritis Rheum. 2001;44:474–480. doi: 10.1002/1529-0131(200102)44:2<474::AID-ANR67>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGF-β activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, Ribeiro S, Gentry L, Murphy-Ullrich JE. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-β in a chemically defined system. J Biol Chem. 1994;269:26775–26782. [PubMed] [Google Scholar]
- Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Tada H, Isogai S. The fibronectin production is increased by thrombospondin via activation of TGF-β in cultured human mesangial cells. Nephron. 1998;79:38–44. doi: 10.1159/000044989. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-β by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor-β1 by plasmin. J Cell Biol. 1990;110:1361–1367. doi: 10.1083/jcb.110.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-β1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol Biol Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludbrook SB, Barry ST, Delves CJ, Horgan CMT. The integrin αvβ3 is a receptor for the latency-associated peptides of transforming growth factors β1 and β3. Biochem J. 2003;369:311–318. doi: 10.1042/BJ20020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Increased expression of integrin αvβ5 in scleroderma fibroblasts. Am J Pathol. 2004;164:1275–1292. doi: 10.1016/s0002-9440(10)63215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K, Furuhashi M, Aoki H, Goto D, Kuwano H, Sugamura K, Miyazono K, Kato M. c-myc is a downstream target of the Smad pathway. J Biol Chem. 2002;277:854–861. doi: 10.1074/jbc.M104170200. [DOI] [PubMed] [Google Scholar]
- Ihn H, Ohnishi K, Tamaki T, LeRoy EC, Trojanowska M. Transcriptional regulation of the human α2(I) collagen gene: combined action of upstream stimulatory and inhibitory cis-acting elements. J Biol Chem. 1996;271:26717–26723. doi: 10.1074/jbc.271.43.26717. [DOI] [PubMed] [Google Scholar]
- Danielpour D. Improved sandwich enzyme-linked immunosorbent assays for transforming growth factor-β1. J Immunol Meth. 1993;158:17–25. doi: 10.1016/0022-1759(93)90254-5. [DOI] [PubMed] [Google Scholar]
- Anders RA, Dore JJ, Jr, Arline SL, Garamszegi N, Loef EB. Differential requirement for type I and type II transforming growth factor-β receptor kinase activity in ligand-mediated receptor endocytosis. J Biol Chem. 1998;273:23118–23125. doi: 10.1074/jbc.273.36.23118. [DOI] [PubMed] [Google Scholar]
- Zwaagstra JC, El-Alfy M, O’Connor-McCourt MD. Transforming growth factor (TGF)-β1 internalization: modulation by ligand interaction with TGF-β receptors types I and II and a mechanism that is distinct from clathrin-mediated endocytosis. J Biol Chem. 2001;276:27237–27245. doi: 10.1074/jbc.M100033200. [DOI] [PubMed] [Google Scholar]
- Sheppard D, Cohen DS, Wang A, Busk M. Transforming growth factor-β differentially regulates expression of integrin subunits in guinea pig airway epithelial cells. J Biol Chem. 1992;267:17409–17414. [PubMed] [Google Scholar]
- Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- Falanga V, Greenberg AS, Ochoa SM, Roberts AB, Falabella A, Yamaguchi Y. Stimulation of collagen synthesis by anabolic steroid stanozol. J Invest Dermatol. 1998;111:1193–1197. doi: 10.1046/j.1523-1747.1998.00431.x. [DOI] [PubMed] [Google Scholar]
- Brunet CL, Sharpe PM, Ferguson MWJ. Inhibition of TGF-β3 (but not TGF-β1 or TGF-β2) activity prevents normal and mouse embryonic palate fusion. Int J Dev Biol. 1995;39:345–355. [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- Dennis PA, Rifkin DB. Cellular activation of latent transforming growth factor-β requires binding to the cation-independent mannose-6-phosphate/insulin-like growth factor type II receptor. Proc Natl Acad Sci USA. 1991;88:580–584. doi: 10.1073/pnas.88.2.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-β. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- Anders RA, Arline SL, Dore JJ, Leof EB. Distinct endocytic responses of heteromeric and homomeric transforming growth factor-β receptors. Mol Biol Cell. 1997;8:2133–2143. doi: 10.1091/mbc.8.11.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Deyne PG, O’Neill A, Resneck WG, Dmytrenko GM, Pumplin DW, Block RJ. The vitronectin receptor associates with clathrin-coated membrane domains via the cytoplasmic domain of its β5 subunit. J Cell Sci. 1998;111:2729–2740. doi: 10.1242/jcs.111.18.2729. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, LeRoy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- Mc Mahon GA, Dignam JD, Gentry LE. Structural characterization of the latent complex between transforming growth factor-β1 and β1-latency-associated peptide. Biochem J. 1996;313:343–351. doi: 10.1042/bj3130343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylänne J. Conserved functions of the cytoplasmic domains of integrin β subunits. Front Biosci. 1998;3:877–886. doi: 10.2741/a329. [DOI] [PubMed] [Google Scholar]
- Schwartz MA. Integrins, oncogenes, and anchorage independence. J Cell Biol. 1997;139:575–578. doi: 10.1083/jcb.139.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro L, Venturino M, Bozzo C, Silengo L, Altruda F, Beguinot L, Tarone G, Defilippi P. Integrins induce activation of EGF receptor: role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998;17:6622–6632. doi: 10.1093/emboj/17.22.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard AS, Garcia-Cardena G, Leong M, Madri JA, Sessa WC, Languino LR. The synergistic activity of alphavbeta3 integrin and PDGF receptor increases cell migration. J Cell Sci. 1998;111:469–478. doi: 10.1242/jcs.111.4.469. [DOI] [PubMed] [Google Scholar]
- Schneller M, Vuori K, Ruoslahti E. αvβ3 integrin associates with activated insulin and PDGFβ receptors and potentiates the biological activity of PDGF. EMBO J. 1997;16:5600–5607. doi: 10.1093/emboj/16.18.5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges E, Jan Y, Ruoslahti E. Platelet-derived growth factor receptor β and vascular endothelial growth factor receptor 2 bind to the β3 integrin through its extracellular domain. J Biol Chem. 2000;275:39867–39873. doi: 10.1074/jbc.M007040200. [DOI] [PubMed] [Google Scholar]
- Scaffidi AK, Petrovic N, Moodley YP, Fogel-Petrovic M, Kroeger KM, Seeber RM, Eidne KA, Thompson PJ, Knight DA. αvβ3 Integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–37733. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- Zambruno G, Marchisio PC, Marconi A, Vaschieri C, Melchiori A, Giannetti A, De Lucall M. Transforming growth factor-βl modulates β1 and β5 integrin receptors and induces the de novo expression of the αvβ6 heterodimer in normal human keratinocytes: implications for wound healing. J Cell Biol. 1995;129:853–865. doi: 10.1083/jcb.129.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Griffiths M, Wu J, Farese RV, Jr, Sheppard D. Normal development, wound healing, and adenovirus susceptibility in β5-deficient mice. Mol Cell Biol. 2000;20:755–759. doi: 10.1128/mcb.20.3.755-759.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]