

Abstract
Ameliorating the function of the glomerular barrier to circulating proteins by blocking angiotensin II (Ang II) translates into less risk of progression toward end-stage renal failure in diabetic and nondiabetic nephropathies. However, the mechanisms underlying this barrier protection are not clear. Specialized contacts between adjacent podocytes are major candidate targets, and the actin cytoskeleton is emerging as a regulatory element. Here, we present data demonstrating that Ang II induced reorganization of F-actin fibers and redistribution of zonula occludens-1 (ZO-1) that is physically associated with actin in murine podocytes. These effects were paralleled by increased albumin permeability across podocyte monolayers. The F-actin stabilizer jasplakinolide prevented both ZO-1 redistribution and albumin leakage, suggesting that actin cytoskeleton rearrangement is instrumental to podocyte permselective dysfunction induced by Ang II. Changes in both F-actin and ZO-1 patterns were confirmed in glomeruli of rat isolated perfused kidneys on short infusion of Ang II, leading to increased protein excretion. Podocyte dysfunction was mediated by Ang II type 1 receptor and was partly dependent on Src kinase-phospholipase C activation. These data demonstrate that strategies aimed at stabilizing podocyte-podocyte contacts and targeting the relevant intracellular signal transduction are crucial to renoprotection.
The population of patients with end-stage renal failure is expanding worldwide at an alarming rate, partly because of steady rises in the prevalence of conditions such as diabetic nephropathy, HIV nephropathy, obesity, and chronic allograft rejection. The relevance of investigating the dysfunction of the glomerular barrier to macromolecules relies on the evidence that proteinuria is both a strong predictor and a contributing factor of progression of nephropathy.1,2 Mechanisms underlying the loss of the barrier function and the associated pathology have recently emerged on the wave of the identification of specific molecules of the foot process in the podocyte.3–5 Besides location at or near the intercellular contact, these molecules share interactions with each other and the actin cytoskeleton.6–8 The latter is increasingly thought to play a central role in maintaining podocyte structure and function3,9,10 and to contribute to reorganization of the foot process in proteinuric nephropathies.11,12
One pivotal mediator of progressive renal injury, angiotensin II (Ang II), may play a direct role in the pathogenesis of proteinuria.13 Infusion of Ang II in rat isolated kidneys caused impairment of the glomerular barrier leading to enhanced filtration of molecules of radii ≥34 Å and increased protein excretion.14 Evidence for roles of Ang II in progressive disruption of glomerular permselectivity rests on the observation that angiotensin-converting enzyme inhibitors (ACE-I) preserve the size-selective restriction to macromolecular probes in animals and in humans.15,16 The case for Ang II as a candidate promoter of progressive podocyte dysfunction in vivo was suggested by findings that ACE-I prevented proteinuria and the associated glomerular redistribution of the foot process protein zonula occludens-1 (ZO-1) in a rat model of glomerulosclerosis.17 Molecular mechanisms whereby Ang II could produce such change are unknown. The perijunctional actin cytoskeleton may be crucial to the integrity of the tight junction and to the regulation of paracellular permeability.18 No studies have yet comparatively investigated the effects of Ang II on actin cytoskeleton and functional integrity of podocyte layer and the relevant signal transduction.
In epithelial cells, ZO-1 at the cytoplasmic surface of the tight junction couples transmembrane proteins to actin microfilaments and other associated molecules.19,20 In podocytes, ZO-1 is similarly located at the cytoplasmic side of the slit diaphragm21 and is linked directly or indirectly through catenin intermediates to the transmembrane proteins, Neph122 and P-cadherin.23 This analogy, together with the evidence that Ang II induces cytoskeletal changes in rat podocytes,24 prompted us to investigate whether perturbation of cytoskeletal organization by Ang II might be instrumental in inducing changes in ZO-1 expression and permselective dysfunction. To test this hypothesis and to study the intracellular signaling pathways involved in Ang II effects, we examined differentiated mouse podocytes. The significance of the in vitro data in respect to events occurring in the glomerulus was established directly by studying the effects of Ang II infusion on ZO-1 expression and protein flux in the rat isolated perfused kidney.
Materials and Methods
Cell Culture and Incubation
Immortalized mouse podocytes (kind gift from P. Mundel) were grown on rat tail collagen type I (BD Bioscience, Bedford, MA) in permissive conditions at 33°C in RPMI 1640 medium (Invitrogen, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 10 U/ml mouse recombinant γ-interferon (Sigma-Aldrich, St. Louis, MO), and 100 U/ml penicillin plus 0.1 mg/ml streptomycin (Sigma-Aldrich).23 To induce differentiation, podocytes were maintained at 37°C in nonpermissive conditions in RPMI 1640 medium plus 10% FBS and antibiotics without γ-interferon for at least 2 weeks.23 Selected experiments (immunocytochemistry of ZO-1 and nephrin as well as permeability studies) were performed in another murine podocyte cell line (kindly provided by K. Endlich). In this case, differentiation of podocytes was achieved by maintaining cells at 38°C on type IV collagen (BD Bioscience) in the absence of γ-interferon for the same time as above.25 Both cell lines were identified as adult podocytes by high synaptopodin expression. Differentiated podocytes were routinely maintained for 24 hours in RPMI 1640 plus 1% FBS (test medium) before all of the experiments. For immunofluorescence studies, podocytes were seeded on type I or IV collagen-coated glass coverslips for Mundel’s and Endlich’s podocytes, respectively, and then exposed to Ang II 10−7 mol/L (Sigma-Aldrich) or to test medium for 1 hour. All of the experiments were performed at 37 or at 38°C for Mundel or Endlich’s podocytes, respectively.
To investigate roles of Ang II type 1 (AT1) receptor, actin cytoskeleton, and Src family tyrosine kinases, we incubated podocytes (by Mundel) for 1 hour with the highly potent and selective AT1 receptor antagonist L-158,80926 (10 nmol/L; kind gift from Merck Sharp Dohme Research Laboratories, Rahway, NJ), jasplakinolide (JASP; 100 nmol/L; Molecular Probes Inc., Eugene, OR), which is a cell-permeable monocyclic peptide that binds and stabilizes F- actin,27 or the highly potent selective inhibitor of Src kinases PP128 (1 μmol/L; BIOMOL Research Laboratories Inc., Plymouth Meeting, PA), respectively, followed by Ang II (10−7 mol/L) for 1 hour in the continued presence of drugs. JASP alone had no effects on the distribution of podocyte molecules (data not shown). The involvement of phospholipase C (PLC) was studied by incubating the podocytes with the PLC inhibitor U-7312229 (1 μmol/L; Sigma-Aldrich) for 30 minutes before and during 1 hour of exposure to Ang II 10−7 mol/L.
Fluorescence Microscopy
At the end of incubation, cells were fixed with 2% paraformaldehyde plus 4% sucrose for 10 minutes at 37 or 38°C as appropriate, washed once with phosphate buffered saline (PBS), and permeabilized with 0.3% Triton X-100 (Sigma-Aldrich) in PBS for 4 minutes at room temperature.23 Nonspecific binding sites were blocked with blocking solution (2% FBS, 2% bovine serum albumin, and 0.2% bovine gelatin in PBS) for 30 minutes at room temperature. Cells were then incubated (overnight, 4°C) with polyclonal rabbit anti-ZO-1 (10 μg/ml; Zymed Laboratories, Inc., South San Francisco, CA), anti-α-catenin (10 μg/ml; Santa Cruz Biotechnology Inc., Santa Cruz, CA), or anti-α-actinin (10 μg/ml; Santa Cruz Biotechnology) antibodies (Abs) or with a polyclonal guinea pig anti-nephrin Ab (1:100; Progen, Heidelberg, Germany) or with an anti-β-catenin monoclonal Ab (10 μg/ml, Santa Cruz Biotechnology) followed by fluorescein isothiocyanate (FITC)-conjugated secondary Abs (Jackson Immunoresearch Laboratories, West Grove, PA) for 1 hour at room temperature. Negative control experiments with FITC-conjugated secondary Ab alone resulted in complete prevention of staining. For double labeling of ZO-1 and F-actin, cells were preincubated with anti-ZO-1 Ab followed by rhodamine phalloidin (20 U/ml; Molecular Probes) for 45 minutes at room temperature. At the end of incubation, coverslips were washed and mounted in 1% N-propyl-gallate in 50% glycerol and 0.1 mol/L Tris-HCl (pH 8). In Mundel’s podocytes, fluorescence was analyzed by an observer blinded to the nature of experimental samples using an inverted light microscope (IX70; Olympus Optical Co., Tokyo, Japan) equipped with epifluorescence followed by computer-based image analysis (Image Pro-Plus 4.5). Separate images from the same field were acquired using appropriate filters, imported into Adobe Photoshop 2.0, and digitally merged. Immunofluorescence staining for ZO-1 and nephrin in Endlich’s podocytes was analyzed by an inverted confocal laser scanning microscope (LS 510 Meta; Zeiss, Jena, Germany). In additional experiments, immunostaining for ZO-1 was further analyzed by three-dimensional (3-D) reconstruction of the podocyte signal. To this purpose, resting or Ang II-stimulated Endlich’s podocytes were double labeled for ZO-1, as described above, and the nuclear cell marker 4,6-diamidine-2′-phenylindole dihydrocloride (DAPI; Boehringer, Mannheim, Germany) for 30 minutes at 37°C. Ten confocal images for each experimental setting were randomly acquired with a z-axis scan of 21 to 36 images at 0.32 μm distance (LS 510 Meta; Zeiss). Three-dimensional reconstructions of the image series were performed to obtain 3-D models of ZO-1-stained regions and of DAPI staining using the “marching cubes” technique implemented in the Visualization Tool Kit libraries (http://public.kitware.com/).
Morphometry
The pattern of ZO-1 distribution in unstimulated and Ang II-treated podocytes in the absence or presence of the inhibitors was evaluated by morphometry. Briefly, about 20 fields for each sample showing ZO-1-labeled podocytes were randomly digitized, in a blinded manner, from the fluorescence microscope and stored on digital images. The image was binarized using a threshold to segment areas of ZO-1 staining. For each fluorescent segment, the perimeter in pixels was automatically calculated using the image analysis software Image J1.32 (http://rsb.info.nih.gov). Then, the length of each segment was calculated as the semiperimeter, and the sum of all segment lengths per field was computed. Exact enlargement in micrometers per pixel of digital images was calculated from images of a reference grid digitized at the same resolution. Finally, the average number of segments per field, the average of total length per field, and the mean length per field (the ratio between the total length and the number of segments per field) for each experimental setting (from 70 to 130 fields in each group, podocytes incubated with medium alone, Ang II, or Ang II plus inhibitor) were calculated and expressed in micrometers per field.
Permeability Studies
Permeability was determined by measuring the transepithelial passage of FITC-bovine serum albumin (BSA) from apical to basolateral compartment of transwell bicameral chambers (0.4-μm pore; Corning Costar Corporation, Cambridge, MA) as described previously30 with slight modifications. Confluent differentiated podocytes were exposed to test medium or Ang II 10−7 mol/L for 1 hour. FITC-BSA (100 μg/ml; Sigma-Aldrich) was loaded into apical compartment for 1 hour. L-158,809 (10 nmol/L), JASP (100 nmol/L), or PP1 (1 μmol/L) was added to wells 1 hour before Ang II. The role of PLC was studied by preincubation with U-73122 30 minutes before Ang II. At the end of the incubation, fluorescence in the basolateral compartment was measured using fluorescence spectroscopy (excitation = 490; emission = 525 nm).
To quantify the trans-membrane flux of FITC-BSA in micrograms per hour, we calculated the BSA concentrations in the lower chambers using fluorescent arbitrary units of the albumin solution added to the apical compartment, taking into account the volume of the basolateral compartment. The permeability assays were performed at 37°C (Mundel’s podocytes) or at 38°C (Endlich’s podocytes).
Western Blot Analysis
Podocytes were incubated in a radioimmunoprecipitation assay buffer (0.1% sodium dodecyl sulfate [SDS], 0.5% deoxycholate, 1% Triton X-100, 20 mmol/L HEPES (pH 7.5), and 150 mmol/L NaCl) in the presence of protease inhibitor cocktail (Sigma-Aldrich) and 1 mmol/L EDTA for 30 minutes on ice, lysed by sonication, and centrifuged (15,000 × g for 15 minutes at 4°C) to remove detergent-insoluble materials. Protein concentration was determined by bicinchoninic acid assay (Pierce Chemical Co., Rockford, IL). Equal amounts of proteins were separated on 5% SDS-PAGE under reducing conditions and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA). After blocking with 5% skim milk in Tris-buffered saline supplemented with 0.1% Tween-20, membranes were incubated overnight at 4°C with anti-ZO-1 Ab (0.5 μg/ml) followed by horseradish peroxidase-conjugated anti-rabbit IgG (room temperature, 1 hour). Bound Abs were detected by diaminobenzidine (Merck, Darmstadt, Germany).
For differential detergent extraction, podocytes were lysed with 1% Triton X-100 (in 20 mmol/L HEPES [pH 7.5] and 150 mmol/L NaCl plus protease inhibitors) at 4°C (1 hour, gentle agitation). After centrifugation (15,000 × g, 4°C for 30 minutes), pellets were resuspended to identical volume as supernatants in Laemmli buffer and solubilized by sonication. Proteins were analyzed by 8% SDS-PAGE and immunoblotted with polyclonal anti-ZO-1 or anti-actin (Sigma-Aldrich) Abs followed by horseradish peroxidase-conjugated goat anti-rabbit IgG. Bound Abs were detected by chemiluminescence (Pierce). Bands were quantified by densitometry with Microscan 1.0.5 and NIH Image 1.61/ppc software.
Isolated Perfused Kidney
Rat kidneys were isolated and perfused in a recirculating system with an artificial cell-free medium.14 After a 20-minute equilibration period, a 10-minute baseline urine collection and a perfusate sample were obtained at the end of the clearance period. Then Ang II (16 ng/minute) or vehicle (n = 3 for each group) was infused into the renal artery for 10 minute, and both urine and perfusate samples were collected at the end of the clearance for creatinine assay. Urinary protein concentration was also determined. Urine output, urinary protein excretion rate, and glomerular filtration rate (GFR) were evaluated. The infusion time was chosen on the basis of previous time-course experiments, showing that after 10 minutes of infusion of Ang II (16 ng/minute), urinary protein excretion rate was already significantly increased in respect to baseline values and preceded the fall of GFR. At the end of perfusion, kidney specimens were embedded in OCT medium and frozen in liquid nitrogen for immunohistochemistry. Small fragments of cortical kidney tissue were fixed overnight at 4°C in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4, for transmission electron microscopy (TEM).
For ZO-1 and F-actin staining, cryostat sections of kidney tissue (3 μm) were fixed (10 minutes at room temperature) in pre-chilled acetone and incubated (overnight, 4°C) with rabbit anti-ZO-1 Ab (10 μg/ml) followed by Cy3-conjugated goat anti-rabbit Ab (1 hour at room temperature) or with rhodamine phalloidin (5 U/ml for 45 minutes). In each section, the glomerular distribution of immunofluorescence staining for ZO-1 was evaluated, assigning a score to individual glomeruli as follows: 0, linear distribution along the glomerular capillary wall; 0.5, heterogeneous distribution of the protein along the glomerular membrane, with changes from a linear to a finely punctate pattern; and 1.0, markedly discontinuous distribution of ZO-1 with focal loss of staining. The final score (SZO-1) per section was then calculated as the weighted mean
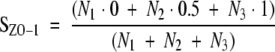 |
where Ni (i = 1 to 3) is the number of glomeruli in each category. Seventy glomeruli on average per section were evaluated by an observer blinded to the nature of the experimental groups. As negative control, incubation of the sections with the secondary antibody alone resulted in a complete prevention of staining at glomerular level.
For TEM, samples were washed repeatedly in cacodylate buffer, postfixed in 1% OsO4 for 1 hour, and then dehydrated through ascending grades of alcohol and embedded in Epon resin. Ultrathin sections were stained with uranyl acetate for examination using a Philips Morgagni transmission electron microscope.
Statistics
Results are expressed as means ± SEM. Morphometric values estimating ZO-1 fragmentation and data on trans-membrane flux of FITC-BSA were compared by analysis of variance and Bonferroni test. Data on urinary protein excretion rate and GFR in isolated perfused kidneys were analyzed by paired Student’s t-test. Score of glomerular distribution of ZO-1 in rat isolated perfused kidneys was analyzed by Mann-Whitney test. Statistical significance was defined as P < 0.05.
Results
Effects of Ang II on Actin Cytoskeleton and ZO-1 Expression in Murine Podocytes
Results of double immunolabeling for F-actin and ZO-1 are reported in Figure 1. Podocytes displayed actin stress fibers that were arranged in parallel (Figure 1a). On Ang II addition, the pattern changed into reorganization of F-actin to the cell periphery at the expense of transcytoplasmic microfilaments (Figure 1d). In untreated podocytes, the ZO-1 staining was peripherally distributed at contacts and process interdigitations of adjacent cells as fine segments (Figure 1b), while it appeared markedly fragmented, and its density decreased in response to Ang II (Figure 1e). Morphometric evaluation of ZO-1 labeling confirmed that the average number of ZO-1 segments per field as well as their total and mean length in podocytes exposed to Ang II were significantly lower than in unstimulated cells (Table 1). Double staining showed ZO-1 colocalization with peripheral F-actin subsets (Figure 1, c and f).
Figure 1.

Ang II induces ZO-1 redistribution in podocytes by a mechanism dependent on actin rearrangement. Staining for F-actin and ZO-1 in control (a–c) and Ang II-stimulated podocytes in the absence (d–f) or presence (g–i) of the F-actin stabilizer jasplakinolide. On Ang II exposure (10−7 mol/L for 1 hour), F-actin filaments stained predominantly at the periphery of differentiated podocytes rather than along the cell axis (d versus a). Cytoskeletal changes were associated with ZO-1 fragmentation at podocyte-podocyte contacts (e versus b). Merged images show colocalization of ZO-1 and F-actin at discrete sites of cell-cell contact (c and f). Actin stabilization (g versus d) prevented Ang II-induced ZO-1 fragmentation (h versus e).
Table 1.
Morphometric Analysis of ZO-1 Staining in Murine Podocytes
| NP/field | ΣLi (μm/field) | Lm (μm/field) | |
|---|---|---|---|
| Control | 187 ± 5 | 347 ± 8 | 1.88 ± 0.04 |
| Ang II | 170 ± 3* | 273 ± 6† | 1.61 ± 0.02† |
| Ang II + JASP | 195 ± 6‡ | 349 ± 14‡ | 1.77 ± 0.03‡ |
| Ang II + L-158,809 | 208 ± 5‡ | 343 ± 12‡ | 1.63 ± 0.03 |
| Ang II + PP1 | 189 ± 5§ | 330 ± 10‡ | 1.75 ± 0.04‡ |
| Ang II + U-73122 | 202 ± 5‡ | 346 ± 7‡ | 1.73 ± 0.03 |
P < 0.05 versus control.
P < 0.01 versus control.
P < 0.05 versus Ang II.
P < 0.01 versus Ang II.
Data are means ± SEM. NP, number of fluorescent segments per field; ΣLi, sum of segments per field; Lm, mean length per field.
To investigate whether changes in ZO-1 staining reflected changes in the amount of protein expressed in podocytes exposed to Ang II, we further analyzed ZO-1 expression by Western blot. As shown in Figure 2a, ZO-1 appeared as a band of 225 kd in control and in Ang II-treated podocyte. Comparable ZO-1 levels indicated no effect of Ang II on the amount of protein expressed.
Figure 2.

ZO-1 protein in Ang II-treated podocytes shows normal levels of expression and maintains physical association with actin. a: Western blot analysis of ZO-1 in differentiated podocytes either unstimulated (control, lanes 1 to 3) or exposed to Ang II (10−7 mol/L for 1 hour) (lanes 4 to 6). b: Differential detergent experiments showing distribution of ZO-1 and actin in detergent-soluble (S) and -insoluble (P) fractions of control (lanes 1 and 2) and Ang II-stimulated podocytes (lanes 3 and 4). Numbers on the left indicate molecular values (× 10−3) of protein standards. In a and b, blots are representative of three and four experiments, respectively.
After differential detergent extraction, the majority of ZO-1 co-sedimented with actin in the insoluble fraction in untreated podocytes. ZO-1 remained physically associated with actin in response to Ang II exposure (Triton X-100-insoluble fraction: control, 79.3 ± 2.6% of total ZO-1; Ang II, 81.4 ± 2.2%; n = 9 for each experimental setting; Figure 2b). These findings are consistent with the possibility that Ang II-induced actin cytoskeleton rearrangement might be responsible for reorganization of the peripheral membrane architecture of podocytes, as reflected by altered ZO-1, and ultimately for barrier dysfunction of podocyte-podocyte contact. Evaluation of transepithelial passage of fluorescent albumin to the basolateral compartment of bicameral chambers disclosed significant increases in albumin permeability across the monolayer in response to Ang II (Figure 3). Ang II exposure had no apparent effect on podocyte attachment, because the cell density was not affected by Ang II (data not shown). Finally, stabilization of F-actin by JASP (Figure 1, g and i) prevented Ang II-induced ZO-1 redistribution as documented by immunofluorescent pattern (Figure 1, h and i) and estimation of the density and length of ZO-1 segments per field (Table 1), which were similar to control one. Preservation of ZO-1 distribution in podocytes by JASP was paralleled by a significant reduction of albumin leakage across the podocyte monolayer (Figure 3).
Figure 3.

Ang II increases albumin permeability across podocyte monolayers. Transepithelial albumin permeability in control (open bar) and Ang II-stimulated podocytes in the absence (gray bar) or presence (hatched bar) of the F-actin stabilizer JASP. JASP significantly prevented podocyte permselective dysfunction. Data are means ± SEM of three independent experiments, *P < 0.01 versus control; °P < 0.01 versus Ang II.
Ang II likewise altered podocyte expression of α-catenin, (which binds both to actin and ZO-1), β-catenin (which binds both to α-catenin and to P-cadherin at the slit diaphragm), and α-actinin (linker of the cadherin/catenin complex to actin via α-catenin31 ) (Figure 4). Immunofluorescent patterns of both catenins on Ang II exposure showed punctate distribution of the proteins and a loss of peripheral staining in respect to unstimulated podocytes (Figure 4, b versus a and d versus c). Moreover, α-actinin was densely present in the perinuclear area instead of being localized on microfilament bundles (Figure 4, f versus e). These findings suggest that actin rearrangement may play a major role on the redistribution of closely interacting proteins other than ZO-1 in response to Ang II.
Figure 4.

Ang II alters catenins and α-actinin distribution in podocytes. Immunostaining of α- and β-catenin and α-actinin in control (a, c, and e) and Ang II-stimulated podocytes (b, d, and f). On Ang II exposure (10−7 mol/L for 1 hour), catenins showed punctate patterns, whereas α-actinin was concentrated at perinuclear sites.
Role of AT1 Receptors in ZO-1 Redistribution and Podocyte Dysfunction
Ang II is known to modulate several podocyte functions through AT1 receptors.32 Thus, we next investigated whether the observed Ang II effects could be related to activation of AT1 receptors, which are expressed in the murine podocyte line.33 Pretreatment of podocytes with the specific AT1 receptor antagonist L-158,809 prevented both the redistribution of ZO-1 (Figure 5, c versus b; Table 1) and the increase in albumin permeability by Ang II (Figure 5d).
Figure 5.

AT1 receptor mediates Ang II-induced ZO-1 redistribution and permselective dysfunction. a and b: Micrographs show ZO-1 redistribution in podocytes exposed to Ang II (b) compared with control (a). c: L-158,809 prevented changes in ZO-1 staining in podocytes on Ang II exposure. d: Transepithelial albumin permeability in control (open bar) and Ang II-stimulated podocytes in the absence (gray bar) or presence (hatched bar) of AT1 receptor blocker L-158,809. Data are means ± SEM of six independent experiments, *P < 0.01 versus control; °P < 0.01 versus Ang II.
Intracellular Signals Downstream of the AT1 Receptor
It has been shown that tyrosine kinases serve as intracellular signals for Ang II-induced actin reorganization in human mesothelial and rat glomerular epithelial cells.34,35 To investigate whether tyrosine kinases may contribute to Ang II-induced ZO-1 redistribution, we focused on Src family tyrosine kinases, which mediate AT1 receptor signal events for cell adhesion and spreading.36 Pretreatment of podocytes with a selective inhibitor of Src family kinases, PP1, prevented ZO-1 redistribution (Figure 6, a–c; Table 1) and significantly improved podocyte permselectivity (trans-membrane albumin flux: Ang II + PP1, 18.55 ± 1.31 versus 35.78 ± 3.02 μg/hour; P < 0.01; n = 3 independent experiments). One major target of Src, PLC γ1, is activated by Ang II.37 Thus, we evaluated the role of PLC in actin reorganization and ZO-1 redistribution. Double immunostaining showed that the PLC inhibitor U-73122 prevented both actin reorganization and ZO-1 redistribution in Ang II-stimulated cells (Figure 6, d–f; Table 1). These effects resulted in complete prevention of albumin filtration across the monolayer (trans-membrane albumin flux: Ang II + U-73122, 14.17 ± 0.26 μg/hour versus Ang II, 41.92 ± 7.48 μg/hour; P < 0.01; n = 3 independent experiments).
Figure 6.

Src kinase activation of PLC plays role in the podocyte response to Ang II. a–c: Immunostaining for ZO-1 in control (a) and Ang II-stimulated podocytes in absence (b) or presence (c) of the Src kinase inhibitor PP1. d–f: Double immunofluorescent staining of ZO-1 and actin in control (d) and Ang II-stimulated podocytes in absence (e) or presence (f) of the PLC inhibitor U-73122.
The effects of Ang II on ZO-1 distribution and albumin permeability were further evaluated in a newly developed murine podocyte cell line (by K. Endlich) that stably expresses nephrin and forms a continuous epithelial layer in the differentiated state,25 thus possessing features of in situ podocyte cell-cell contacts. In the unstimulated podocytes, ZO-1 staining was visible by immunofluorescence as a continuous belt outlining the cell borders (Figure 7a). On Ang II exposure, ZO-1 was still peripheral but interrupted (Figure 7b). 3-D reconstruction of podocytes double stained with ZO-1 and DAPI showed that disruption and fragmentation of ZO-1 in response to Ang II was independent of the focal plane of observa-tion, as shown by supplemental material (online at http://ajp.amjpathol.org).
Figure 7.

Ang II induces ZO-1 redistribution and permselective dysfunction in a novel podocyte cell line. a: Confocal images of confluent differentiated podocytes exposed to medium alone displayed immunoreactivity for ZO-1 that appeared as continuous bands along the periphery of cells. b: In Ang II-treated cells, ZO-1 stained as peripheral interrupted lines. c: Transepithelial albumin permeability in control (open bar) and Ang II-stimulated podocytes in the absence (gray bar) or presence (hatched bars) of AT1 receptor blocker L-158,809, Src family kinase inhibitor PP1, and PLC inhibitor U-73122. Data are means ± SEM of four experiments, *P < 0.05 versus control; °P < 0.05 versus Ang II.
In our experimental settings, podocytes expressed nephrin at the perinuclear region and at cell-cell contacts (Figure 8a). This pattern of immunoreactivity is similar to that previously described by Schiwek et al.25 In response to Ang II, nephrin staining was weaker around the nucleus and even absent at podocyte-podocyte contact (Figure 8b). These changes were paralleled by an increase in the albumin permeability across the podocyte monolayer, an event that was significantly prevented by Ang II receptor blockade and by inhibitors of Src family kinases and PLC, confirming the data described above (Figure 7).
Figure 8.

Ang II alters the distribution pattern of nephrin in a novel podocyte cell line. a: Confocal images of confluent differentiated podocytes exposed to medium alone show nephrin staining at the perinuclear region and at the cell-cell contacts (arrows). b: No or much fainter peripheral signal was observed in response to Ang II.
Effect of Ang II on ZO-1 Distribution in the Isolated Perfused Kidney
To investigate whether changes in ZO-1 on Ang II exposure may also occur in the glomerulus and may possibly reflect alterations responsible for proteinuria in vivo, we infused Ang II in rat isolated perfused kidneys, as a tool to study directly the effects of Ang II on glomerular capillary wall function.14 Ten minutes of Ang II infusion significantly increased urinary protein excretion rate in comparison with baseline values without affecting GFR (Table 2). In glomeruli of vehicle-infused kidneys, immunohistochemical analysis of ZO-1 showed a predominant linear pattern along the peripheral capillary wall (Figure 9a). After infusion of Ang II, the staining was markedly altered in more than 50% of glomeruli examined, exhibiting more heterogeneous staining with a punctate pattern or loss of protein at focal areas within glomeruli (Figure 9b). The score averaged 0.40 ± 0.04 in this group, a value significantly higher (P < 0.05) than that computed in vehicle-infused isolated kidneys (0.19 ± 0.04). Moreover, altered ZO-1 distribution was associated with F-actin reorganization. In isolated kidneys exposed to vehicle, the capillary wall as well as the mesangium of nearly all glomeruli examined (89 ± 3%) were strongly positive for F-actin (Figure 9c). This percentage was significantly reduced to 58 ± 6% after infusion with Ang II because of the presence of glomeruli showing F-actin mainly concentrated in mesangial areas rather than in the peripheral capillary loops (Figure 9d). These changes were not associated with ultrastructural evidence of podocyte damage or apoptotic loss (Figure 10).
Table 2.
Effect of Short Time Infusion of Ang II on Urinary Protein Excretion Rate and GFR in Rat Isolated Perfused Kidneys
| Vehicle
|
Ang II (16 ng/minute)
|
|||
|---|---|---|---|---|
| 10 minutes* | 20 minutes | 10 minutes* | 20 minutes | |
| Proteinuria (μg/minute) | 13.8 ± 2.6 | 14.3 ± 2.4 | 10.7 ± 1.6 | 30.7 ± 5.2† |
| GFR (ml/minute) | 0.89 ± 0.14 | 0.82 ± 0.11 | 0.90 ± 0.09 | 0.89 ± 0.03 |
Baseline pre-infusion values.
P < 0.05 versus basal.
Data are mean ± SEM (n = 3 for each group).
Figure 9.

Short infusion of Ang II alters glomerular ZO-1 and F-actin distribution in rat isolated perfused kidney. Immunofluorescence staining for ZO-1 (a and b) and F-actin (c and d) in isolated kidneys infused with vehicle or Ang II (16 ng/minute) for 10 minutes. a: Glomerulus of vehicle-infused kidney shows immunoreactivity for ZO-1 along the peripheral capillary wall. b: Glomerulus of Ang II-infused kidney showing heterogeneous/discontinuous distribution of ZO-1 staining. c and d: Compared with vehicle (c), short infusion of Ang II induced also a decrease in F-actin staining in the peripheral capillary wall reflecting sites of podocyte foot processes (d, arrowheads).
Figure 10.

Electron micrographs of glomerular capillary walls of isolated kidney preparations. Samples were infused with either vehicle (a) or Ang II (b). No significant podocyte damage can be observed. Magnification, ×8000.
Discussion
The present report documents that Ang II induces actin cytoskeleton reorganization, leading to redistribution of a functionally important molecule of the foot process, ZO-1, in mouse podocytes. The effects of increasing protein permeability both in podocyte monolayers and in the rat isolated perfused kidney in concomitance with ZO-1 redistribution disclose a direct action of Ang II in perturbing the glomerular sieving function.
Several studies attest to the importance of the actin cytoskeleton in regulating the tight junction paracellular barrier.18,20 Our data on differential detergent extraction of podocyte lysates show physical association between ZO-1 and actin, suggesting a framework whereby the cytoskeleton affects podocyte-podocyte contacts. The direct interaction between actin and ZO-1 has been suggested to enhance the retention and stabilization of ZO-1 at the cell junction for assembly and regulation of transmembrane proteins of the barrier.19 ZO-1 is also concentrated at cadherin-based cell-cell contacts in nonepithelial cells, serving as a cross-linker between cadherin/catenin complex and actin38 to confer strong cell-cell adhesion.39 Consistently, the disorganization of actin cytoskeleton by cytochalasin D promotes a shift from strong to weak adhesion.39 In murine podocytes generated by Mundel and colleagues,23 ZO-1 is co-expressed with catenins and P-cadherin at the slit diaphragm. In this cell line, we have shown that actin-associated ZO-1 is redistributed in response to Ang II, as are α- and β-catenin and α-actinin, which colocalize with ZO-1 in the foot process.10,23 Paracellular permeability assays performed in a murine podocyte line by using anionic dextran (3 to 70 kd) have recently documented that podocytes establish a size-selective barrier dependent on differentiation.40 One key finding here is that the altered ZO-1 distribution in differentiated mouse podocytes exposed to Ang II was paralleled by increased albumin permeability across the monolayer. We have obtained similar results using the podocyte line recently developed by Endlich et al25 that displays in situ podocyte-podocyte contacts. Our findings that JASP prevented Ang II-induced ZO-1 redistribution and albumin permeability through stabilization of F-actin demonstrate that actin rearrangements underlie podocyte dysfunction. Thus, actin cytoskeleton changes and increased permeability to albumin confers, as shown for the first time, functional significance to cytoskeleton reorganization relevant to the specialized function of the podocyte in vivo. Our findings of peripheral loss of nephrin on Ang II exposure are in support of this interpretation. In line with our data, exposure of cultured human glomerular epithelial cells to Ang II was also reported to result in actin rearrangement and physical dislocation of nephrin.41
In the glomerulus, the capillary wall permeability to macromolecules is controlled at the filtration slit in such a way that plasma proteins leak if the podocyte-podocyte contact is altered. It is plausible that changes in actin cytoskeleton induced by Ang II alter both ZO-1 distribution and foot process-foot process interaction, resulting in increased transglomerular flux of plasma proteins. In support of such interpretation, our data in rat isolated perfused kidneys show that short infusions of Ang II induced proteinuria without modifying GFR. The increased glomerular permeability was associated with abnormal patterns of F-actin staining in glomerular epithelial areas at peripheral capillary walls. In the light of our findings in cultured podocytes, the loss of barrier function was ultimately attributable, at least partly, to the concomitant ZO-1 redistribution in podocyte foot processes.
Overall, these data indicate that Ang II directly promotes perturbation of the podocyte barrier and that the ensuing redistribution of ZO-1 closely reproduces that found at slit diaphragms in disease. Major insight can be derived regarding the molecular mechanism underlying proteinuria in rats with spontaneous glomerulosclerosis showing ZO-1 redistribution and permselective dysfunction before any morphological lesion can be detected.17 In Munich Wistar Frömter rats, glomerular redistribution of ZO-1 occurred without changes in the quantity of protein expressed.17 Here, Western blot findings are in agreement with the possibility that an altered interaction of ZO-1 with other associated proteins may disrupt the barrier. Preliminary evidence in db/db mice and in podocytes exposed to high glucose suggests that altered expression of ZO-1 also contributes to the pathogenesis of albuminuria in diabetes.42
Ang II binding to its receptors elicits many intracellular events that are likewise associated with receptor activation by growth factors.43 Such events had not been investigated previously in relation to specialized podocyte function. That AT1 receptor signaling in podocytes may lead to protein leakage in vivo has been suggested by findings of albuminuria and podocyte injury in transgenic rats with targeted overexpression of human AT1 receptor in podocytes.44 The inhibition of both ZO-1 redistribution and permeability change of cell monolayer by L-158,809, as shown here, reveals activation of the receptor on Ang II exposure. Despite no intrinsic kinase activity, AT1 receptor interacts with nonreceptor tyrosine kinases of peculiar interest, the Src family kinases, having a pivotal role in AT1 receptor-mediated cell growth and vascular remodeling.36,45 Our data of specific inhibition of Ang II-induced ZO-1 redistribution and permeability change using PP1 extend the role of Src kinase activation to the induction of cell-cell contact change and permselective dysfunction in podocytes. Src kinase activation was previously found to mediate epithelial contact disassembly in response to migratory growth factors or v-Src.46 Thus, the intracellular response to Ang II in podocytes seems to conform to a more generalized signaling for control of cell-cell contact.
Effects of U-73122 in preventing actin change, ZO-1 redistribution, and high permeability are in support of the role of PLC, after activation by the Src kinase, in mediating the response of podocytes to Ang II. Because Ang II through Src kinase promotes tyrosine-phosphorylation of PLC-γ1,37 which induces reorganization of cytoskeleton in other cell types,47 the phosphorylation of PLC-γ1 conceivably represents an early signal for cytoskeleton alterations including those that lead to ZO-1 redistribution in podocytes. Tyrosine phosphorylation of PLC-γ1 by hydrolyzing activity leads to decreased cellular content of α-actinin- and vinculin-bound phosphatidylinositol 4,5-bisphosphate (PIP2), thus contributing to impaired actin bundle formation.48 Of note, we have documented that in the presence of Ang II, the loss of transcytoplasmic F-actin filaments was associated with changes in α-actinin distribution. Our findings that the PLC inhibitor U-73122 prevents podocyte dysfunction, by blocking PIP2 hydrolysis, suggest that the control of PIP2 concentration might regulate the dynamic function of the filtration slit in vivo. Further studies should address this important aspect of the response of podocyte to Ang II.
In summary, our findings offer basic insight into the glomerular dysfunction that leads to abnormal efflux of proteins across the glomerular membrane and to renal parenchymal injury. Independently of antihypertensive properties, ACE-I and AT1 receptor blockers exert specific protection by preserving podocyte integrity, namely, regulatory functions of actin cytoskeleton on podocyte-podocyte contact. This provides specific rationale to their use in practice. Investigation should be focused on strategies that modulate transduction pathways and foot process function to most effectively prevent renal disease progression in patients resistant to currently available treatments.
Supplementary Material
Acknowledgments
We thank Fabio Sangalli, Nadia Stucchi, and Emmanuel Centemeri for technical help. Sara Conti provided technical assistance for TEM. We are indebted to Dr. Luca Antiga for assistance in image processing for 3-D reconstructions of ZO-1 staining. We also acknowledge Dr. Marina Noris, Norberto Perico, and Ariela Benigni for helpful advice and discussions.
Footnotes
Address reprint requests to Daniela Macconi, “Mario Negri” Institute for Pharmacological Research, Via Gavazzeni, 11, 24125 Bergamo, Italy. E-mail: macconi@marionegri.it.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Ruggenenti P, Perna A, Mosconi L, Matalone M, Pisoni R, Gaspari F, Remuzzi G. Proteinuria predicts end-stage renal failure in non-diabetic chronic nephropathies. Kidney Int. 1997;52(Suppl 63):S54–S57. [PubMed] [Google Scholar]
- Remuzzi G. Nephropathic nature of proteinuria. Curr Opin Nephrol Hypertens. 1999;8:655–663. doi: 10.1097/00041552-199911000-00001. [DOI] [PubMed] [Google Scholar]
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003;83:253–307. doi: 10.1152/physrev.00020.2002. [DOI] [PubMed] [Google Scholar]
- Chugh SS, Kaw B, Kanwar YS. Molecular structure-function relationship in the slit diaphragm. Semin Nephrol. 2003;23:544–555. doi: 10.1053/s0270-9295(03)00137-2. [DOI] [PubMed] [Google Scholar]
- Ly J, Alexander M, Quaggin SE. A podocentric view of nephrology. Curr Opin Nephrol Hypertens. 2004;13:299–305. doi: 10.1097/00041552-200405000-00006. [DOI] [PubMed] [Google Scholar]
- Saleem MA, Ni L, Witherden I, Tryggvason K, Ruotsalainen V, Mundel P, Mathieson PW. Co-localization of nephrin, podocin, and the actin cytoskeleton: evidence for a role in podocyte foot process formation. Am J Pathol. 2002;161:1459–1466. doi: 10.1016/S0002-9440(10)64421-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Takeuchi E, Salant DJ. Podocyte slit-diaphragm protein nephrin is linked to the actin cytoskeleton. Am J Physiol Renal Physiol. 2002;282:F585–F591. doi: 10.1152/ajprenal.00290.2001. [DOI] [PubMed] [Google Scholar]
- Welsch T, Endlich N, Kriz W, Endlich K. CD2AP and p130Cas localize to different F-actin structures in podocytes. Am J Physiol Renal Physiol. 2001;281:F769–F777. doi: 10.1152/ajprenal.2001.281.4.F769. [DOI] [PubMed] [Google Scholar]
- Drenckhahn D, Franke R-P. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59:673–682. [PubMed] [Google Scholar]
- Ichimura K, Kurihara H, Sakai T. Actin filament organization of foot processes in rat podocytes. J Histochem Cytochem. 2003;51:1589–1600. doi: 10.1177/002215540305101203. [DOI] [PubMed] [Google Scholar]
- Whiteside CI, Cameron R, Munk S, Levy J. Podocytic cytoskeletal disaggregation and basement-membrane detachment in puromycin aminonucleoside nephrosis. Am J Pathol. 1993;142:1641–1653. [PMC free article] [PubMed] [Google Scholar]
- Shirato I, Sakai T, Kimura K, Tomino Y, Kriz W. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am J Pathol. 1996;148:1283–1296. [PMC free article] [PubMed] [Google Scholar]
- Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 2000;57:1803–1817. doi: 10.1046/j.1523-1755.2000.00031.x. [DOI] [PubMed] [Google Scholar]
- Lapinski R, Perico N, Remuzzi A, Sangalli F, Benigni A, Remuzzi G. Angiotensin II modulates glomerular capillary permselectivity in rat isolated perfused kidney. J Am Soc Nephrol. 1996;7:653–660. doi: 10.1681/ASN.V75653. [DOI] [PubMed] [Google Scholar]
- Remuzzi A, Puntorieri S, Battaglia C, Bertani T, Remuzzi G. Angiotensin converting enzyme inhibition ameliorates glomerular filtration of macromolecules and water and lessens glomerular injury in the rat. J Clin Invest. 1990;85:541–549. doi: 10.1172/JCI114470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisoni R, Ruggenenti P, Sangalli F, Lepre MS, Remuzzi A, Remuzzi G. Effect of high dose ramipril with or without indomethacin on glomerular selectivity. Kidney Int. 2002;62:1010–1019. doi: 10.1046/j.1523-1755.2002.00535.x. [DOI] [PubMed] [Google Scholar]
- Macconi D, Ghilardi M, Bonassi ME, Mohamed EI, Abbate M, Colombi F, Remuzzi G, Remuzzi A. Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J Am Soc Nephrol. 2000;11:477–489. doi: 10.1681/ASN.V113477. [DOI] [PubMed] [Google Scholar]
- Turner JR. ‘Putting the squeeze’ on the tight junction: understanding cytoskeletal regulation. Semin Cell Dev Biol. 2000;11:301–308. doi: 10.1006/scdb.2000.0180. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J. 2002;16:1835–1837. doi: 10.1096/fj.02-0121fje. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Mitic LL, Anderson JM. Transmembrane proteins in the tight junction barrier. J Am Soc Nephrol. 1999;10:1337–1345. doi: 10.1681/ASN.V1061337. [DOI] [PubMed] [Google Scholar]
- Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol. 1990;111:1255–1263. doi: 10.1083/jcb.111.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TB, Schmidts M, Gerke P, Schermer B, Zahn A, Hartleben B, Sellin L, Walz G, Benzing T. The carboxyl terminus of neph family members binds to the PDZ domain protein zonula occludens-1. J Biol Chem. 2003;278:13417–13421. doi: 10.1074/jbc.C200678200. [DOI] [PubMed] [Google Scholar]
- Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- Sharma R, Lovell HB, Wiegmann TB, Savin VJ. Vasoactive substances induce cytoskeletal changes in cultured rat glomerular epithelial cells. J Am Soc Nephrol. 1992;3:1131–1138. doi: 10.1681/ASN.V351131. [DOI] [PubMed] [Google Scholar]
- Schiwek D, Endlich N, Holzman L, Holthofer H, Kriz W, Endlich K. Stable expression of nephrin and localization to cell-cell contacts in novel murine podocyte cell lines. Kidney Int. 2004;66:91–101. doi: 10.1111/j.1523-1755.2004.00711.x. [DOI] [PubMed] [Google Scholar]
- Chang RSL, Siegl PKS, Clineschmidt BV, Mantlo NB, Chakravarty PK, Greenlee WJ, Patchett AA, Lotti VJ. In vitro pharmacology of L-158,809, a new highly potent and selective angiotensin II receptor antagonist. J Pharmacol Exp Ther. 1992;262:133–138. [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AMJ, Sausville EA, Duncan KLK, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and src family-selective tyrosine kinase inhibitor: study of lck- and fyn-T dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Lea JP, Jin SG, Roberts BR, Shuler MS, Marrero MB, Tumlin JA. Angiotensin II stimulates calcineurin activity in proximal tubule epithelia through AT-1 receptor-mediated tyrosine phosphorylation of the PLC-γ1 isoform. J Am Soc Nephrol. 2002;13:1750–1756. doi: 10.1097/01.asn.0000022029.50356.2c. [DOI] [PubMed] [Google Scholar]
- Oshima T, Laroux FS, Coe LL, Morise Z, Kawachi S, Bauer P, Grisham MB, Specian RD, Carter P, Jennings S, Granger DN, Joh T, Alexander JS. Interferon-γ and interleukin-10 reciprocally regulate endothelial junction integrity and barrier function. Microvasc Res. 2001;61:130–143. doi: 10.1006/mvre.2000.2288. [DOI] [PubMed] [Google Scholar]
- Knudsen KA, Soler AP, Johnson KR, Wheelock MJ. Interaction of α-actinin with the cadherin/catenin cell-cell adhesion complex via α-catenin. J Cell Biol. 1995;130:67–77. doi: 10.1083/jcb.130.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavenstadt H. Roles of the podocyte in glomerular function. Am J Physiol Renal Physiol. 2000;278:F173–F179. doi: 10.1152/ajprenal.2000.278.2.F173. [DOI] [PubMed] [Google Scholar]
- Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- Kuwahara M, Kuwahara M. Involvement of Rho and tyrosine kinase in angiotensin II-induced actin reorganization in mesothelial cells. Eur J Pharmacol. 2002;436:15–21. doi: 10.1016/s0014-2999(01)01591-6. [DOI] [PubMed] [Google Scholar]
- McCarthy ET, Sharma M, Sharma R, Savin VJ. Angiotensin II induced reorganization of cytoskeleton in glomerular epithelial cells is blocked by inhibitors of protein tyrosine kinase. J Am Soc Nephrol. 2000;11:424A. [Google Scholar]
- Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest. 1999;103:789–797. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrero MB, Schieffer B, Paxton WG, Schieffer E, Bernstein KE. Electroporation of pp60c-src antibodies inhibits the angiotensin II activation of phospholipase C-γ1 in rat aortic smooth muscle cells. J Biol Chem. 1995;270:15734–15738. doi: 10.1074/jbc.270.26.15734. [DOI] [PubMed] [Google Scholar]
- Itoh M, Nagafuchi A, Moroi S, Tsukita S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to a catenin and actin filaments. J Cell Biol. 1997;138:181–192. doi: 10.1083/jcb.138.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura Y, Itoh M, Maeno Y, Tsukita S, Nagafuchi A. Functional domains of α-catenin required for the strong state of cadherin-based cell adhesion. J Cell Biol. 1999;144:1311–1322. doi: 10.1083/jcb.144.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JL, Pollak MR, Denker BM. Cultured podocytes establish a size-selective barrier regulated by specific signaling pathways and demonstrate synchronized barrier assembly in a calcium switch model of junction formation. J Am Soc Nephrol. 2005;16:1593–1602. doi: 10.1681/ASN.2004080679. [DOI] [PubMed] [Google Scholar]
- Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy. Evidence for a distinctive role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- Rincon-Choles H, Vasylyeva TL, Bhandari K, Barnes JL, Bhandari B, Simon M, Pergola PE, Gorin Y, Abboud HE. Regulation of ZO-1 expression and phosphorylation in experimental diabetes. J Am Soc Nephrol. 2003;14:47A. doi: 10.2337/diabetes.55.04.06.db05-0355. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Schieffer B, Ma H, Bernstein KE, Ling BN. ANG II-induced tyrosine phosphorylation stimulates phospholipase C- gamma 1 and Cl-channels in mesangial cells. Am J Physiol. 1996;270:C1834–C1842. doi: 10.1152/ajpcell.1996.270.6.C1834. [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Podlich D, Hahnel B, Kriz W, Gretz N. Angiotensin II type 1 receptor overexpression in podocytes induces glomerulosclerosis in transgenic rats. J Am Soc Nephrol. 2004;15:1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- Schieffer B, Paxton WG, Chai Q, Marrero MB, Bernstein KE. Angiotensin II controls p21ras activity via pp60c-src. J Biol Chem. 1996;271:10329–10333. doi: 10.1074/jbc.271.17.10329. [DOI] [PubMed] [Google Scholar]
- Owens DW, McLean GW, Wyke AW, Paraskeva C, Parkinson EK, Frame MC, Brunton VG. The catalytic activity of the src family kinases is required to disrupt cadherin-dependent cell-cell contacts. Mol Biol Cell. 2000;11:51–64. doi: 10.1091/mbc.11.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Fukami K, Itoh T, Takenawa T. Phosphorylation of phospholipase C γ1 on tyrosine residue 783 by platelet-derived growth factor regulates reorganization of the cytoskeleton. Exp Cell Res. 1998;243:113–122. doi: 10.1006/excr.1998.4132. [DOI] [PubMed] [Google Scholar]
- Fukami K, Endo T, Imamura M, Takenawa T. α-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J Biol Chem. 1994;269:1518–1522. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}