

Abstract
Background
Angioedema without major urticarial flares (hives) is poorly understood. Its causes are diverse, and little is known about its pathogenic mechanisms. We report on our 11 years of experience with this condition and propose a classification of patients affected by angioedema unaccompanied by urticaria.
Methods
From 1993 through 2003 at our outpatient clinic, 929 consecutive patients were examined for recurrent angioedema unaccompanied by urticaria. A detailed history was taken to identify known causes of angioedema, with special attention to external allergenic agents. Each patient underwent a complete physical examination, routine laboratory tests (blood cell count, protein electrophoresis, erythrosedimentation rate, examination of stool for ova and parasites, pharyngeal and urine cultures, sinus and dental radiography, and measurements of antitissue autoantibodies and rheumatoid factor in plasma), and complement parameters (C1 inhibitor, C4 and C1q). Further testing was done when pertinent, based on clinical findings. When all results were negative, response to H1-antihistamine was considered.
Results
Angioedema could not be classified in 153 patients who were lost to follow-up (16.4%). Among the 776 cases with adequate data, these types of angioedema were identified: 124 (16%) related to external agents such as a drug, insect bite or foodstuff; 85 (11%) related to treatment with angiotensin-converting enzyme inhibitor; 55 (7%) associated with an autoimmune disease or infection; and 197 (25%) caused by C1 inhibitor deficiency. In the other 315 cases (41%), the etiology was undiscovered: 254 cases (33% of the study sample) were idiopathic histaminergic; 40 (5%) were idiopathic nonhistaminergic; and 21 (3%) had other causes of peripheral or generalyzed edema.
Interpretation
Our data indicated that angioedema without urticaria could be classified according to specific clinical and pathogenic characteristics, and we have suggested a work-up for patients experiencing this condition.
Angioedema — a self-limited, localized swelling that involves subcutaneous tissues or mucosa — can involve the face and other areas. Skin colour is usually unchanged, although it may be heralded by erythema marginatum. There is no burning sensation and itchiness is generally absent. Lesions tend to be large and without clear demarcation; most resolve within 2 or 3 days.1
Frequently the result of the release of histamine, angioedema is often associated with urticarial flares (hives; the association is often called urticaria–angioedema syndrome) and responds to antihistamines and corticosteroids.2 Occasionally, angioedema occurs without urticaria, does not respond to drugs against allergies and tends to last 5–7 days — for example, when it is caused by a deficiency in plasma of C1 esterase inhibitor (C1Inh).
To investigate and form classifications for different forms of angioedema unaccompanied by urticaria, we analyzed 929 consecutive patients referred to our university hospital over an 11-year period (January 1993 through December 2003).
Methods
Our outpatient clinic for angioedema is a tertiary-level centre where patients are referred mostly by specialists. All clinic patients with a history of recurrent angioedema without major urticaria between January 1993 and December 2003 were considered for the study.
From each patient, we obtained a clinical history with information about allergies and their relation to potential causative agents (e.g., foods, drugs, chemicals) and carried out a complete physical examination. We obtained sinus and dental radiographs as well as test results for blood cell count, serum protein electrophoresis, erythrocyte sedimentation rate, C-reactive protein, hepatic enzymes, renal function, thyroid function and anti-tissue antibodies (antinuclear antibodies and antibodies to extractable nuclear antigens, to double-stranded DNA, to thyroglobulin and to thyroperoxidase). Stool examinations (for ova and parasites) and urinanalyses were carried out, and pharyngeal and urine samples were cultured. Further specific analyses were performed if deemed necessary.
If all test results were negative, response to long-term H1-antihistamine therapy was considered: patients were given cetirizine (20 mg) or desloratadine (10 mg) daily for at least a month. In the first 27 patients who did not respond to cetirizine, we administered higher doses of sedating antihistamine (25 mg of hydroxyzine, 3 times daily). Since none improved and the side effects were considerable, we stopped using sedating agents.
Plasma samples were collected and stored at –80°C. C1Inh function was measured with a commercially available chromogenic assay (Technochrome C1-INH, Technoclone GmbH, Vienna, Austria). C1Inh, C4, C3 and C1q antigen levels were measured with radial immunodiffusion plates (NOR-Partigen and [for C1q] LC-Partigen, Behring, Marburg, Germany). Autoantibodies to C1Inh in serum were measured with ELISA (enzyme-linked immunosorbent assay).3
Types of angioedema unaccompanied by urticaria were categorized as follows. Angiotensin-converting-enzyme inhibitor (ACEI)– related angioedema was diagnosed when angioedema recurred during ACEI therapy and disappeared upon withdrawal of the treatment.4
Hereditary angioedema (HAE) was diagnosed according to the published criteria,5 that is, when angioedema was subcutaneous, noninflammatory, self-limiting, without major urticaria, (often) recurrent and lasting more than 12 hours, with serum levels of C1Inh antigen or C1Inh function below 50% of normal. Type 1 HAE was diagnosed when both antigenic and functional levels of C1Inh were low; type 2, when functional levels of C1Inh were low and antigenic levels normal or higher.
Diagnosis of acquired C1Inh deficiency was based on the presence of angioedema identical to HAE but with an onset during or after the fourth decade of life, absence of a family history, and measurements of serum C1Inh antigen or C1Inh function below 50% of normal.6
Results
From 1993 through 2003, 929 patients were referred to our clinic for angioedema without major urticaria; 565 (61%) were female and 364 (39%), male. Median age at first visit was 41 years (range 1–87 yr). We did not consider 153 patients who dropped out before completing the work up. The remaining 776 patients were classified according to the cause of the angioedema (Table 1).
Table 1
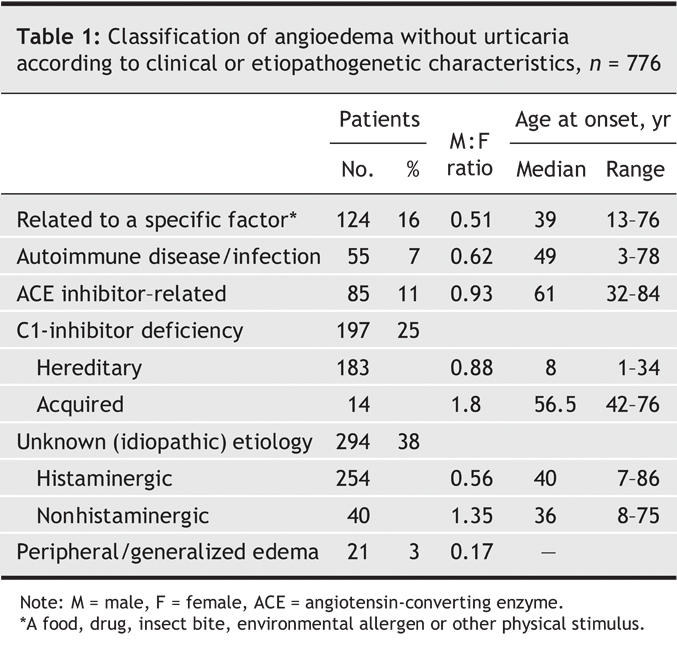
In 124 patients (16%), the recurrence of symptoms was clearly related to an exogenous stimulus, with a consistent cause– effect relation. Stimuli were identified as a medication in 56 cases (45%), a foodstuff in 45 (36%), both in 10 (8%), an insect bite in 5 (4%), another environmental allergen in 4 (3%) and a physical irritation or other stimulus in 4.
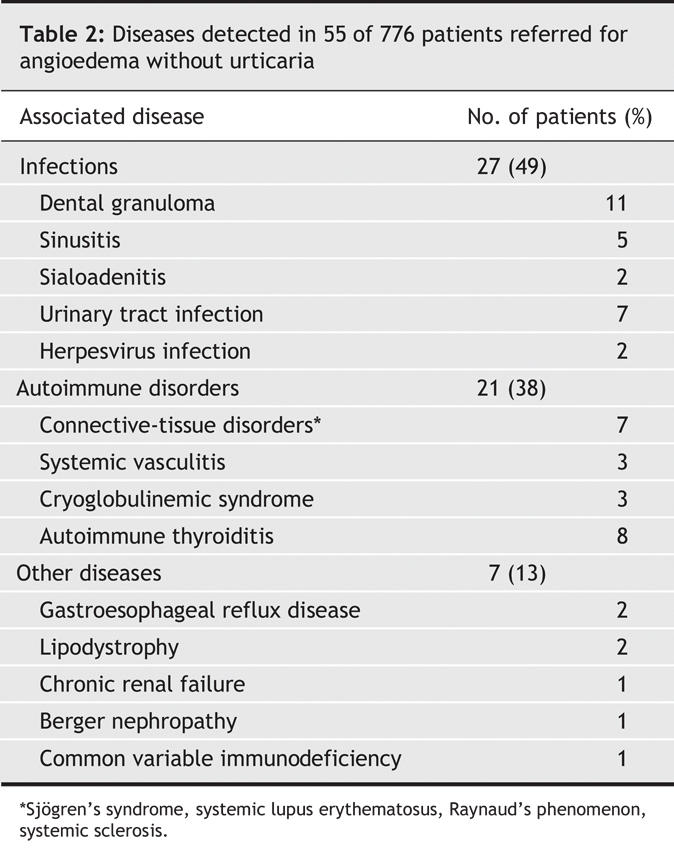
In 55 patients (7%), a concomitant disease was identified (Table 2). In 27 (49%) of these cases, it was an infection. Appropriate treatment of the infection markedly improved the angioedema in all patients with dental granuloma; 3 out of 5 patients with sinusitis; and 5 out of 7 patients with urinary tract infection. Neither of 2 patients with sialoadenitis showed improvement with treatment. In 1 woman, true angioedema of the lips relapsed upon recurrences of herpes simplex type 1; in another patient, angioedema had been present during a herpes zoster (shingles) outbreak. Of 2 patients infected with Helicobacter pylori who experienced gastroesophageal reflux, both the angioedema and reflux improved after proper eradication therapy in 1 case only.
Table 2
In 85 cases (11%), angioedema developed during treatment with ACEI for hypertension (82 patients) or heart failure (3 patients). The median duration of ACEI therapy before symptom onset of was 12 months (range 1 day to 13 years); angioedema appeared during the first month of therapy in 16 patients. Median time between the first attack and the withdrawal of therapy was 12 months (range 1 day to 10 years). The site of edema was the face in 69 patients (81%); the tongue and oral cavity in 38 (45%); other cutaneous sites in 15 (17%); upper airways (i.e., the oral, pharyngeal and laryngeal cavities) in 17 (20%); and the bowel in 3 (3%). ACEI was stopped in all cases. Ten patients (12%) were lost to follow-up; of the remaining 75, 67 (89%) had a clear improvement: 58 had no further recurrence of angioedema, and 9 had just minor sporadic episodes. The angioedema in 8 patients (11%) did not improve.
HAE was diagnosed in 197 patients (25%), who belonged to 120 unrelated families. Type 2 angioedema was present in 28 people (15%) from 14 families. Complement parameters are summarized in Table 3. Median age at diagnosis was 29 years (range 1–89 yr). Subcutaneous edema was present in 178 cases (97%); edema of the bowel, in 143 (78%); and laryngeal edema, in 68 (37%).
Table 3
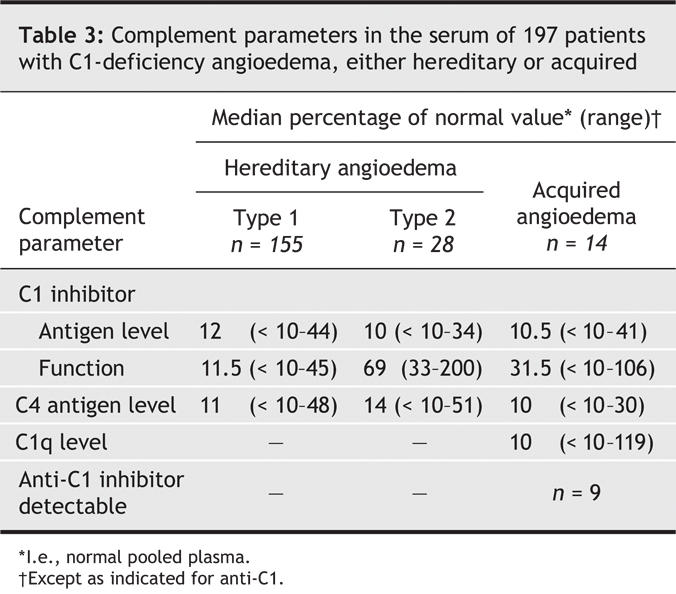
Acquired C1Inh deficiency was diagnosed in 14 patients, whose median age at diagnosis was 57.5 (range 42–76) years. All patients had cutaneous edema; 7 patients (50%), recurrent edema of the bowel; and 9 (64%), edema of the upper airways. A concomitant disease was found in 9 patients (64%): 7 cases of monoclonal gammopathy of uncertain significance (MGUS) and 2 of non-Hodgkins lymphoma.
In 294 patients (38%), physical examination and all laboratory test results were normal, and the cause of the angioedema or associated condition was inevident. All these patients began long-term antihistamine therapy for a month at minimum. In 254 (86%) of them, their angioedema disappeared or was drastically reduced; in the other 40 cases, it did not improve. Of the 254 whose condition improved, 7 had reported involvement of an upper airway; none needed resuscitative manoeuvres.
Of the 40 who were not helped by the antihistamines, angioedema had occurred in the upper airways of 14 (35%), including 1 patient whose laryngeal edema had been severe enough to have required endotracheal intubation. Eleven patients (27%) had experienced recurrent abdominal pain. Eight patients (20%) had 1 or more affected relatives. Tranexamic acid for acute treatment was effective in 8 patients and partially effective in 3. Twenty-two patients (55%) who were having more than 1 episode of severe angioedema monthly were administered continuous prophylaxis with tranexamic acid: in 16 patients (40%), symptoms disappeared completely, and in 6 (15%) their frequency and severity was reduced. The median duration of tranexamic therapy was 46 months (range 1–94 months).
The remaining 21 patients (2.7%) had other conditions that caused peripheral or generalized edema: Melkersson– Rosenthal syndrome (4 cases), lymphedema (3), capillary leak syndrome (3) and idiopathic edema (11).
Discussion
To date, no clinical survey of angioedema without urticaria has been reported; diagnostically, this condition remains confined within the general problem of the urticarial syndrome. The commonly applied label of “allergic” is frequently wrong, and standard anti-allergic therapeutic approaches can be totally ineffective. The growing relevance of this condition has been highlighted, moreover, by a recent study7 that showed that angioedema is the most frequent cause of hospital admission of all acute allergic, nonasthmatic diseases.
A comprehensive personal and familial history followed by specific tests allowed us to relate the presence of angioedema to exposure to external agents in 124 patients (16%): drugs (ASA, other nonsteroidal anti-inflammatory drugs, antibiotics), insect bites, foods and environmental allergens. These agents lead to episodes of angioedema, always in a close cause– effect relation. Patients who come to a physician to confirm their suspicion often identify the causative agent themselves. These reactions are very common; the diagnosis does not present notable problems, and patients are therefore seldom referred to specialized centres — which is the most likely explanation for the low percentage (16%) of such direct causations in our series. The mechanism can be an immediate classical, type 1, immunoglobulin E–mediated hypersensitivity reaction or a nonallergic hypersensitivity.8–10 Further investigations in the patient should therefore be tailored to the individual findings. IgE-mediated reaction to environmental allergens can be confirmed by results from skin-prick testing or the radioallergosorbent test (RAST).2 Provocation tests can confirm a diagnosis of nonallergic hypersensitivity.
At least 0.2% of patients taking ACEI develop angioedema, a well-documented but still frequently unrecognized side-effect of these drugs.11 As well as acting on angiotensin, ACE inactivates bradykinin,12 and ACEIs increase the bioavailability of that peptide.13 Patients who develope angioedema during treatment with ACEI likely have an altered metabolism in which bradykinin episodically accumulates in the plasma.14–17 A clinical history is crucial to the diagnosis. Symptoms may appear several years after beginning ACEI therapy, which makes the diagnosis more difficult. Clinical manifestations are angioedema, typically of the face and oral cavity, although other cutaneous sites, upper-airway mucosa and the bowel can be involved. It is well known that if ACEIs are not withdrawn, the symptoms tend to worsen,18 and there are reports of death from laryngeal edema.19 If angioedema does not disappear despite stopping of ACEI, additional analysis becomes necessary. In our case list, further investigation allowed different a classification of angioedema in 8 patients, as reported previously.4
Angioedema due to C1Inh deficiency is also dependent on the generation of bradykinin.20,21 The prevalence of HAE in the population is set at 1:50 000 — a number without solid epidemiological basis, but nevertheless probably fairly accurate.22 The frequency of HAE among patients with angioedema without urticaria is also unknown; in our series, it was present in 183 patients (24%), although our being a reference centre for diagnosis and treatment of HAE for more than 25 years probably biases this percentage. The possibility of a deficiency in C1Inh is strongly suggested by clinical clues such as widespread, purely noninflammatory angioedema of the extremities; frequent abdominal symptoms; and a family history. In patients whose HAE is type 1 (85%), the diagnosis is confirmed by measurements of plasma levels of C1Inh and C4 antigen, which most laboratories can perform. Despite easy clinical and laboratory tools, diagnosis of C1Inh deficiency is still astonishingly delayed in most patients. When these patients are only superficially evaluated, the misdiagnosis of “allergy” is a common feature.
It is important to identify a deficiency of C1Inh because its management is completely different:23,24 antihistamine, corticosteroids and adrenaline are ineffective, both as acute and chronic treatment. Severe acute attacks should be treated with replacement therapy: C1Inh concentrate. In countries such as the United States where C1Inh concentrate is unavailable, patients who develop a laryngeal attack must be relocated as soon as possible to where resuscitation facilities are immediately available. Prophylaxis with attenuated androgens (danazol, stanazolol, oxandrolone) is effective in preventing symptoms when their frequency and severity is so high as to affect the quality of life. In children, tranexamic acid can be used for prophylaxis of attacks.
Acquired C1Inh deficiency, an apparently rare condition,6 was diagnosed in 14 patients (2% of the total number). Its clinical symptoms are almost the same as those for HAE. The acquired form differs in an absence of family history, late symptom onset and a different response to treatment. It also is frequently associated with B-lymphocyte disorders ranging from MGUS to B-cell malignancies. The treatment of acute attacks, as in HAE, is based on C1Inh concentrate; however, markedly higher doses may be needed in some patients.3 Antifibrinolytic agents tend to be more effective than androgen derivatives for angioedema prophylaxis.6
In a small proportion of our patients, H1-receptor blockers were ineffective; C1Inh measurements were normal; no cause for angioedema could be identified; test results for antithyroid autoantibody were negative; clinical symptoms were almost identical to those of HAE; and, in a few cases, other family members were affected. As previously reported,25 these patients were treated with tranexamic acid, which effectively prevents angioedema symptoms. Identified as idiopathic nonhistaminergic angioedema, this form could have a pathogenetic mechanism similar to that of C1Inh deficiency and bradykinin as mediators of symptoms.26 Despite some analogies, this condition does not seem to be the same as the recently described HAE type 3, which affects only women, is frequently estrogen-dependent, has a clear hereditary pattern and does not respond to tranexamic acid.27–29
As expected, a small proportion of patients (7%) had an associated disease. Defining a cause–effect relation between such disease and angioedema is frequently difficult. We found a sustained rate of accordance between the cure of localized infections and the disappearance of the angioedema. This finding reinforces the hypothesis of a connection between the 2 conditions and the importance of screening for infectious foci when no obvious cause of angioedema is detectable. Whether or not Helicobacter pylori infection can cause angioedema is still controversial,2 and we cannot provide additional evidence.
We found 21 patients (2%) with an autoimmune disorder, a percentage slightly higher than that found in the normal population.30 The association of urticaria and autoimmune thyroid disease,31 connective-tissue disorder or systemic vasculitis is well known;32 in some cases, chronic urticaria itself can be autoimmune.33,34 Hence, angioedema can be a major feature of the so-called hypocomplementemic urticaria vasculitis syndrome (HUVS).35 Although we have seen some patients so diagnosed, the angioedema in each case coexisted with clinically significant urticaria; the patients were therefore not included in our study.
We found a single patient with common variable immunodeficiency and angioedema. Such an association has been already described,36 but its significance is unclear.36
Twenty-one patients referred with suspected angioedema had other diseases that may be masqueraders of true angioedema. Melkersson–Rosenthal syndrome, which was diagnosed in 4 patients, is a rare idiopathic noncaseating granulomatous condition characterized by lip swelling (or other oro facial form of edema), facial nerve palsy and stable lingua plicata.37 The symptoms differ from true angioedema in not being episodic: they tend to last up to several months. Three patients had primary lymphedema, but this condition can easily be distinguished from angioedema by its peculiar clinical characteristics.38
Idiopathic systemic capillary-leak syndrome (ISCLS) or idiopathic edema was diagnosed in 14 patients. These are totally different diseases, but share the characteristic of recurrent diffuse subcutaneous edema mainly of the face, hips and hands. ISCLS is a rare acute, lethal condition of recurrent generalized increase in vascular permeability. A similar condition has been described in patients treated with recombinant interleukin-2, but the etiopathogenetic mechanism of this syndrome remains unknown.39 Two of the 3 patients we observed died within 5 years of diagnosis; the third did not return for follow-up.
Eleven women had the characteristic of idiopathic edema, a poorly understood syndrome characterized by periodic episodes of peripheral edema in women who have weight changes unrelated to their menstrual cycle.38 Symptoms are described as edema of hands, legs and face, which may be real or perceived by the patient.
To conclude this survey, we propose an algorithm (Fig. 1) to guide clinicians in investigating cases of angioedema unaccompanied by urticaria.

Fig. 1: Diagnostic flowchart for various types of angioedema (AE) unaccompanied by uticaria. C1Inh = C1 esterase inhibitor, Ag = antigen.
@ See related article page 1083
Footnotes
This article has been peer reviewed.
Contributors: Lorenza Zingale conceived and designed the study and, along with Laura Beltrami, Lorena Maggioni, Emanuela Pappalardo and Benedetta Cicardi, contributed to the acquisition and interpretation of the data and the drafting of the article. Andrea Zanichelli contributed to the acquisition and interpretation of the data. Marco Cicardi substantially contributed to the conception and design of the study. All authors participated in the critical revision of the article and approved the version to be published.
Competing interests: Lorenza Zingale, Marco Cicardi and Andrea Zanichelli have consultancy agreements with companies that are developing drugs for C1 inhibitor–deficiency angioedema (Pharming, Jerini and Dyax); Benedetta Cicardi, with Pharming and Jerini. Lorena Maggioni and Emanuela Pappalardo have been paid by Jerini for performing laboratory tests as part of a clinical trial on a new drug for hereditary angioedema. Marco Cicardi and Laura Beltrami are involved in a research program funded by Novartis on angioedema related to use of an angiotensin-converting-enzyme inhibitor.
Correspondence to: Dr. Lorenza Zingale, Università degli Studi di Milano, Dipartimento di Medicina Interna, Ospedale San Giuseppe, Via San Vittore 12, 20123 Milano, Italy; fax 39 02 85994165; lorenza.zingale@unimi.it
REFERENCES
- 1.Friedmann PS. Assessment of urticaria and angio-oedema. Clin Exp Allergy 1999; 29 (Suppl 3):109-12 [discussion 113-5]. [DOI] [PubMed]
- 2.Grattan C, Powell S, Humphreys F. Management and diagnostic guidelines for urticaria and angio-oedema. Br J Dermatol 2001;144:708-14. [DOI] [PubMed]
- 3.Cicardi M, Bisiani G, Cugno M, et al. Autoimmune C1 inhibitor deficiency: report of eight patients. Am J Med 1993;95:169-75. [DOI] [PubMed]
- 4.Cicardi M, Zingale LC, Bergamaschini L, et al. Angioedema associated with angiotensin-converting enzyme inhibitor use: outcome after switching to a different treatment. Arch Intern Med 2004;164:910-3. [DOI] [PubMed]
- 5.Agostoni A, Aygoren-Pursun E, Binkley KE, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol 2004;114(3 Suppl):S51-131. [DOI] [PMC free article] [PubMed]
- 6.Cicardi M, Zingale LC, Pappalardo E, et al. Autoantibodies and lymphoproliferative diseases in acquired C1-inhibitor deficiencies. Medicine (Baltimore) 2003;82:274-81. [DOI] [PubMed]
- 7.Lin RY, Cannon AG, Teitel AD. Pattern of hospitalizations for angioedema in New York between 1990 and 2003. Ann Allergy Asthma Immunol 2005;95:159-66. [DOI] [PubMed]
- 8.Sampson HA. Update on food allergy. J Allergy Clin Immunol 2004;113:805-19 (quiz 820). [DOI] [PubMed]
- 9.Gruchalla RS. 10. Drug allergy. J Allergy Clin Immunol 2003;111(2 Suppl):S548-59. [DOI] [PubMed]
- 10.Bircher AJ. Drug-induced urticaria and angioedema caused by non-IgE mediated pathomechanisms. Eur J Dermatol 1999;9:657-63; quiz 663. [PubMed]
- 11.Vleeming W, van Amsterdam JG, Stricker BH, et al. ACE inhibitor-induced angioedema: incidence, prevention and management. Drug Saf 1998;18:171-88. [DOI] [PubMed]
- 12.Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol 2002;109:195-209. [DOI] [PubMed]
- 13.Nussberger J, Cugno M, Amstutz C, et al. Plasma bradykinin in angio-oedema. Lancet 1998;351:1693-7. [DOI] [PubMed]
- 14.Molinaro G, Cugno M, Perez M, et al. Angiotensin-converting enzyme inhibitor– associated angioedema is characterized by a slower degradation of des-arginine (9)- bradykinin. J Pharmacol Exp Ther 2002;303:232-7. [DOI] [PubMed]
- 15.Adam A, Cugno M, Molinaro G, et al. Aminopeptidase P in individuals with a history of angio-oedema on ACE inhibitors. Lancet 2002;359:2088-9. [DOI] [PubMed]
- 16.Duan QL, Nikpoor B, Dube MP, et al. A variant in XPNPEP2 is associated with angioedema induced by angiotensin I–converting enzyme inhibitors. Am J Hum Genet 2005;77:617-26. [DOI] [PMC free article] [PubMed]
- 17.Cugno M, Nussberger J, Cicardi M, et al. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol 2003;3:311-7. [DOI] [PubMed]
- 18.Brown NJ, Snowden M, Griffin MR. Recurrent angiotensin-converting enzyme inhibitor–associated angioedema. JAMA 1997;278:232-3. [DOI] [PubMed]
- 19.Dean DE, Schultz DL, Powers RH. Asphyxia due to angiotensin converting enzyme (ACE) inhibitor mediated angioedema of the tongue during the treatment of hypertensive heart disease. J Forensic Sci 2001;46:1239-43. [PubMed]
- 20.Davis AE III. The pathophysiology of hereditary angioedema. Clin Immunol 2005;114:3-9. [DOI] [PubMed]
- 21.Fields T, Ghebrehiwet B, Kaplan AP. Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykinin. J Allergy Clin Immunol 1983;72:54-60. [DOI] [PubMed]
- 22.Cicardi M, Agostoni A. Hereditary angioedema. N Engl J Med 1996;334:1666-7. [DOI] [PubMed]
- 23.Bowen T, Cicardi M, Farkas H, et al. Canadian 2003 international consensus algorithm for the diagnosis, therapy, and management of hereditary angioedema. J Allergy Clin Immunol 2004;114:629-37. [DOI] [PubMed]
- 24.Gompels MM, Lock RJ, Abinun M, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol 2005;139:379-94. [DOI] [PMC free article] [PubMed]
- 25.Cicardi M, Bergamaschini L, Zingale LC, et al. Idiopathic nonhistaminergic angioedema. Am J Med 1999;106:650-4. [DOI] [PubMed]
- 26.Nussberger J, Cugno M, Cicardi M. Bradykinin-mediated angioedema. N Engl J Med 2002;347:621-2. [DOI] [PubMed]
- 27.Binkley KE, Davis AE III. Estrogen-dependent inherited angioedema [review]. Transfus Apheresis Sci 2003;29:215-9. [DOI] [PubMed]
- 28.Binkley KE, Davis A III. Clinical, biochemical, and genetic characterization of a novel estrogen-dependent inherited form of angioedema. J Allergy Clin Immunol 2000;106:546-50. [DOI] [PubMed]
- 29.Bork K, Barnstedt SE, Koch P, et al. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 2000;356:213-7. [DOI] [PubMed]
- 30.Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmun Rev 2003;2:119-25. [DOI] [PubMed]
- 31.Leznoff A, Sussman GL. Syndrome of idiopathic chronic urticaria and angioedema with thyroid autoimmunity: a study of 90 patients. J Allergy Clin Immunol 1989;84:66-71. [DOI] [PubMed]
- 32.Kaplan AP. Clinical practice. Chronic urticaria and angioedema. N Engl J Med 2002;346:175-9. [DOI] [PubMed]
- 33.Kaplan AP. Chronic urticaria: pathogenesis and treatment. J Allergy Clin Immunol 2004;114:465-74 (quiz 475). [DOI] [PubMed]
- 34.Greaves M. Chronic urticaria. J Allergy Clin Immunol 2000;105:664-72. [DOI] [PubMed]
- 35.Wisnieski JJ, Baer AN, Christensen J, et al. Hypocomplementemic urticarial vasculitis syndrome. Clincal and serologic findings in 18 patients. Medicine (Baltimore) 1995;74:24-41. [DOI] [PubMed]
- 36.Altschul A, Cunningham-Rundles C. Chronic urticaria and angioedema as the first presentations of common variable immunodeficiency. J Allergy Clin Immunol 2002;110:664-5. [DOI] [PubMed]
- 37.Ang KL, Jones NS. Melkersson–Rosenthal syndrome. J Laryngol Otol 2002;116:386-8. [DOI] [PubMed]
- 38.Cho S, Atwood JE. Peripheral edema. Am J Med 2002;113:580-6. [DOI] [PubMed]
- 39.Cicardi M, Gardinali M, Bisiani G, et al. The systemic capillary leak syndrome: appearance of interleukin-2-receptor–positive cells during attacks. Ann Intern Med 1990;113:475-7. [DOI] [PubMed]