

Abstract
Gap junctions play a critical role in hearing and mutations in connexin genes cause a high incidence of human deafness. Pathogenesis mainly occurs in the cochlea, where gap junctions form extensive networks between non-sensory cells that can be divided into two independent gap junction systems, the epithelial cell gap junction system and the connective tissue cell gap junction system. At least four different connexins have been reported to be present in the mammalian inner ear, and gap junctions are thought to provide a route for recycling potassium ions that pass through the sensory cells during the mechanosensory transduction process back to the endolymph. Here we review the cochlear gap junction networks and their hypothesized role in potassium ion recycling mechanism, pharmacological and physiological gating of cochlear connexins, animal models harboring connexin mutations and functional studies of mutant channels that cause human deafness. These studies elucidate gap junction functions in the cochlea and also provide insight for understanding the pathogenesis of this common hereditary deafness induced by connexin mutations.
Keywords: Cochlea, Supporting cell, Gap junction, Connexin, Potassium, Deafness
Introduction
The mammalian cochlea is a highly organized sensory organ for hearing composed of auditory hair cells and supporting cells. Cochlear supporting cells include pillar cells, Deiters cells, Hensen cells, and Claudius cells, and these supporting cells are electrically and metabolically coupled by gap junction channels. In contrast, gap junctions are not found between the sensory cells (inner and outer hair cells), or between sensory and non-sensory cells in mammals (Kikuchi et al., 1995; Lautermann et al., 1998; Zhao & Santos-Sacchi, 1999, 2000; Zhao 2000). Additional gap junctional coupling exists in the stria vascularis, spiral limbus, and other cochlear structures (Jahnke, 1975; Gulley & Reese, 1976; Iurato et al., 1976; Hama & Saito, 1977; Santos-Sacchi & Dallos, 1983; Santos-Sacchi, 1987; Zwislocki et al., 1992; Kikuchi et al., 1995, 2003; Zhao & Santos-Sacchi, 1998, 2000; Forge et al., 1999). Thus, two distinct gap junctional networks have been described in the cochlea: an epithelial cell gap junction network between the non-sensory epithelial cells, and a connective tissue gap junction network between the connective tissue cells (Kikuchi et al., 1995). These gap junction networks are believed to be responsible for cochlear ionic homeostasis (Santos-Sacchi, 1985; Oesterle & Dallos, 1990, Kikuchi et al., 2000a; Santos-Sacchi, 2000).
The cochlea is organized in a manner that segregates the ionic environment presented to the sensory cells within this organ. It contains two major fluid-filled compartments, which hold the perilymph and endolymph (Fig. 1a). The perilymphatic space contains a high Na+ and low K+ solution, similar to other extracellular fluids. In contrast, the apices of the cochlear epithelial cells face the endolymph, which has an opposite cationic composition of high K+ and low Na+. The endolymph is further characterized by the presence of a positive endocochlear potential of 80 to 100 mV. Acoustically evoked receptor potentials are generated by the influx of potassium ions from the endolymph into the sensory hair cells. These K+ ions are then released into the basolateral extracellular spaces of the perilymph prior to being taken up by supporting cells and recycled back into the endolymph. The supporting cell gap junction networks are thought to play a critical role in the recirculation of cochlear K+ ions by providing an intercellular route of transport (Spicer & Schulte, 1996; Kikuchi et al., 1995, 2000b).
Gap junctions are membrane specializations containing intercellular channels for the passage of a wide variety of small molecules, including ions, nucleotides, siRNAs, and inositol phosphates (Veenstra, 1996; Harris, 2001; Goldberg et al., 2004; Valiunas et al., 2005). Each gap junction is recognized in transmission electron micrographs as a segment where the plasma membranes of two adjacent cells are separated by a uniform narrow gap of about 2 nm (Fig. 1b). Freeze-fracture electron micrographs demonstrate that each gap junction is composed of closely aggregated intramembranous channel particles (Fig. 1c), which in turn are hexameric assemblies of connexin proteins. Several different connexin (Cx) subunits have been reported to be expressed in the mammalian inner ear. These include Cx26, Cx30, Cx31 and Cx43 (Kikuchi et al., 1995, 2000a; Lautermann et al., 1998; Forge et al., 1999, 2003; Xia et al., 2000; Ahmad et al. 2003; Suzuki et al., 2003; Zhao, 2005). However, the precise cellular localization of Cx43 in the cochlea has been contested (Cohen-Salmon et al., 2004). It is widely accepted that Cx26 and Cx30 are the predominant isoforms expressed in cochlear supporting cells. Cx26 is present in the epithelial cell gap junction system and the connective tissue cell gap junction system, and Cx30 has a comparable distribution pattern. Additional connexins may also be present within the cochlea (Buniello et al., 2004).
Because intercellular channels span two neighboring cells, it is possible for each cell to contribute different mixtures of connexin proteins, thereby increasing the functional diversity of gating and permeability (White & Bruzzone, 1996). Gap junction channels can be gated by transjunctional voltage (Vj), membrane potential (Vm), pH, Ca2+, or membrane tension (Bennett et al., 1991; Harris, 2001). Each connexin has unique voltage-gating sensitivities, and hybrid channel configurations add further complexity. A homotypic channel generally has symmetric voltage-gating properties on either side of its symmetrical structure. A heterotypic channel can have asymmetrically rectified voltage-gating properties owing to each hemichannel having its own unique gating (Barrio et al., 1991; Rubin et al., 1992; Verselis et al., 1994; White et al., 1994). Cx26 and Cx30 can form heterotypic channels in vitro that show asymmetrically rectified voltage gating (Dahl et al., 1996). Cx26 and Cx30 can also co-assemble to form heteromeric hemichannels in liposomes, allowing for further structural variety (Locke et al., 2004). Thus, the precise nature of the communication between adjacent supporting cells could be profoundly influenced by the available complement of expressed connexin subunits.
Mutations in human Cx26, Cx30, or Cx31 (GJB2, GJB6, and GJB3) have been linked to both syndromic and nonsyndromic forms of deafness (Kelsell et al., 1997; Zelante et al., 1997; Richard et al., 1998; Xia et al., 1998; Grifa et al., 1999). In addition, genetically engineered ablation or mutation of Cx26 or Cx30 has produced deafness in mice (Cohen-Salmon et al., 2002; Teubner et al., 2003; Kudo et al., 2003). These findings strongly demonstrate the importance of gap junctional communication in the normal functioning of the inner ear, yet they do not fully explain why loss of a single gap junction subunit within the cochlea (i.e., by genetic mutation) would not be compensated for by the remaining connexin isoforms. Because there are connexin-dependent differences in permeation and gating, one must also consider that loss of one subunit type would not only change the macroscopic levels and gating of ionic communication, but could also significantly alter the range of molecules being exchanged between the coupled supporting cells. In this article, the detailed organization of the cochlear gap junction networks and the hypothesized potassium ion recycling mechanism are described, and related to what is known about pharmacological and physiological gating of Cx26 and Cx30. Studies of the functional properties of mutant channels that cause human deafness and animal models harboring connexin mutations are also discussed.
Gap Junction Organization in the Mammalian Cochlea
The presence of two independent gap junction networks, the epithelial cell system and the connective tissue cell system (Fig. 2) was first described by Kikuchi et al. (1995), and both Cx26 and Cx30 were shown to be widely distributed throughout these systems (Kikuchi et al., 1995; Lautermann et al., 1998). The epithelial cell gap junction system consists of interdental cells in the spiral limbus, all of the cochlear supporting cells, and root cells within the lower part of the spiral ligament. No direct communication via gap junctions exists between the hair cells and the neighboring supporting cells within the organ of Corti. The connective tissue cell gap junction system is composed of various types of fibrocytes in the spiral ligament and suprastrial zone, basal and intermediate cells in the stria vascularis, mesenchymal cells that line the perilymphatic space of the scala vestibuli, the supralimbal mesenchymal dark cells and the fibrocytes in the spiral limbus. The constituent cells of the epithelial cell gap junction system are lined by a basement membrane, and have no direct contact with the underlying connective tissue cells.
Potassium Ion Recycling Mechanism via Gap Junctions in the Cochlea
The formation and maintenance of the unique ion composition of the endolymph and the resulting endocochlear potential are essential for the auditory function. Activation of cochlear sensory cells by acoustic stimuli induces the influx of K+ ions from the endolymph to the hair cells. These K+ ions are then released basolaterally to the extracellular spaces of the organ of Corti, from which they can be picked up by the cochlear supporting cells (Boettger et al., 2002; Boettger et al., 2003). Once inside the supporting cells they move via the epithelial cell gap junction system laterally to the lower part of the spiral ligament. The K+ ions are released into the extracellular space within the spiral ligament by root cells, which are the outermost elements of the epithelial cell gap junction system, and are taken up by the Na, K-ATPase and Na-K-Cl cotransporter (NKCC1) in the plasma membrane of type II fibrocytes. This uptake incorporates the K+ into the connective tissue gap junction system. Within this system the K+ ions pass through the tight junctional barrier at the basal cell layer of the stria vascularis and are released to the extracellular space within the stria vascularis. The marginal cells of the stria vascularis also take up K+ ions via the combined activities of Na, K-ATPase and NKCC1, and return the K+ to the endolymphatic space, where it can be used again in mechanosensory transduction (Schulte & Adams, 1989; Crouch et al., 1997). An alternative model for potassium cycling involving inositol triphosphate-induced calcium waves has also been described (Bruzzone & Cohen-Salmon, 2005).
Voltage Gating of Cx26 and Cx30 in Cochlear Cell Pairs and in vitro Expression Systems
In cochlear supporting cells, multiple examples of asymmetrical (rectifying) gap junctional voltage gating have been documented, with marked variation between different cell pairs. Analysis of coupling in either Hensen or Deiters cells revealed a large heterogeneity in the voltage dependence of the gap junction coupling in individual cell pairs (Zhao & Santos-Sacchi, 1998, 2000; Zhao, 2000; Todt et al., 2001). In some pairs, a nearly symmetrical, but less pronounced dependence of Gj on Vj (Gj/Vj) was observed with Gj decreasing by 15% when Vjs ranged from 0 mV to ± 120 mV. In contrast, other cell pairs displayed a rectifying Gj/Vj behavior with a near constant Gj value at negative Vjs and Gj decreasing by about 50% at positive voltages. A less pronounced rectification of opposite polarity was also observed in other cases, with Gj constant at positive Vjs and decreased by 15% at negative voltages. Finally, a linear decrease of Gj by about 60% was also observed when Vj was varied from)120 mV to 120 mV. In addition to the asymmetric responses to the polarities of Vj, the channels were also sensitive to the membrane resting potential, and this Vm gating was independent of the Vj gating (Zhao & Santos-Sacchi, 2000). This combination of the asymmetry in transjunctional voltage gating with the dependence of coupling on membrane potential could produce asymmetric current flow between cells in the intact cochlea, and in turn influence K+ passage through the supporting cell gap junction network.
Gap junction channels between cochlear supporting cells are primarily composed of Cx26 and Cx30 (Kikuchi et al., 1995; Lautermann et al., 1998; Ahmad et al. 2003; Forge et al., 2003), and the voltage-gating properties of these connexins has been well documented in vitro. However, the Gj/Vj relationships obtained from homotypic gap junction channels in functional expression systems were not directly comparable to the results obtained in freshly isolated pairs of cochlear supporting cells. Cx26 expressed in Xenopus oocytes formed gap junction channels with slightly asymmetric voltage dependence (Barrio et al. 1991; Rubin et al., 1992; Oh et al., 1999). The instantaneous conductance increased linearly by about 10% as the transjunctional voltage Vj changed from)60 mV to 60 mV, while transjunctional potentials larger than ± 60 mV symmetrically reduced the steady-state conductance (Barrio et al. 1991; Verselis et al., 1994; Oh et al. 1999). When Cx30 was expressed in Xenopus oocytes or HeLa cells, it formed homotypic gap junction channels, which were characterized by a voltage dependency with a bell shape. Gj was rapidly reduced at Vjs greater than ±30 mV to a residual conductance of about 25% the initial value (Dahl et al., 1996; Valiunas et al., 1999; Manthey et al., 2000; Valiunas & Weingart, 2000; Essenfelder et al., 2004). Thus, simple homotypic Cx26 or Cx30 channels could not explain the gap junctional voltage gating seen in cochlear supporting cell pairs.
Since the Gj/Vj relationships observed in pairs of isolated cochlear supporting cells were not easily explained by the Gj/Vj relationships of homotypic gap junctions formed by Cx26 or Cx30 in vitro, it was proposed that supporting cells form heterotypic or heteromeric gap junction channels with variable stoichiometry and/or posttranslational modifications (Zhao & Santos-Sacchi, 2000). The asymmetrical voltage gating observed in cochlear supporting cells could play an important role in the cochlea by inducing a directional transjunctional current between coupled supporting cells (Zhao, 2000, 2005). If voltage-gating variability resulted from a positional related differentiation of cells within the population, directional currents would facilitate a directed funneling of ions (e.g., K+ ions) away from the sensory cells after acoustic stimulation (Todt et al., 2001). To fully understand the importance of the variability of the voltage-dependent modulation of the gap junction coupling in cochlear supporting cells, it would be desirable to analyze gap junction coupling in an intact cochlea preparation.
Further support for the formation of mixed Cx26 and Cx30 channels in the cochlea has been provided by co-immunoprecipitation experiments and immunohistological studies. Cx26 and Cx30 have been co-immunoprecipitated from a cochlear homogenate that included the organ of Corti, as well as vestibular, bone and neural tissues. (Ahmad et al. 2003; Forge et al., 2003; Sun et al., 2005). Forge et al. (2003) also reported that Cx26 and Cx30 could be co-localized in the same gap junctional plaque in the cochlear tissue. Although these studies do not provide conclusive proof that Cx26 and Cx30 indeed assemble into heteromeric or heterotypic channel configurations, they are consistent with the in situ electrophysiological data suggesting that Cx26 and Cx30 form hybrid channels in supporting cells. Hybrid Cx26/Cx30 channels suggest a possible mechanism of connexin mutation-induced hearing loss (Zhao & Santos-Sacchi, 2000). If hybrid Cx26/Cx30 channels are strictly required for K+ recycling and cochlear homeostasis, then mutation of either connexin would induce altered channel function even when the other partner remained normal. A need for hybrid channels would account for why continued expression and homotypic function of either Cx26 or Cx30 could not compensate for the mutations in the other gene.
Inner Ear Gap Junction Permeability
The diameter of the gap junction channel lumen is 1 to 1.5 nm for all connexins that have been structurally studied, consistent with the observed upper size limit for the minor diameter of molecules that can traverse the channel (Harris, 2001; Goldberg et al., 2004; Valiunas et al., 2005). Despite this shared pore diameter, channels formed by different connexin isoforms have distinct charge preferences and selectivity for molecules with diameters well below the upper limit (Elfgang et al., 1995; Veenstra, 1996; Bevans et al., 1998; Cao et al., 1998). In transfected cells, Cx26 channels show permeability to both cationic and anionic dyes (Elfgang et al., 1995; Nicholson et al., 2000; Beltramello et al., 2003), whereas Cx30 channels show a strong cationic preference and are impermeable to the anionic dyes Lucifer yellow and Alexa Fluor 488 (Manthey et al., 2001; Beltramello et al., 2003; Sun et al., 2005). In the cochlear sensory epithelium, gap junctional coupling also showed a strong charge-selectivity in permeability. Permeation of negatively charged dyes correlated with Cx26 expression, suggesting that Cx26 could be primarily responsible for anionic molecular permeability in the cochlear epithelial network (Zhao, 2005). Given most important cell signaling molecules, such as ATP, cAMP, cGMP, glutamine, and IP3, are negative anions, Cx26 channels may play an important role in intercellular signaling and nutrient sharing in the cochlea. Functional analysis of deafness causing mutations has provided strong support for this view (Beltramello et al. 2005; Zhang et al., 2005; see below).
Pharmacological Modulation of Gap Junction Coupling in Cochlear Supporting Cells
It has also been postulated that supporting cell gap junctional coupling is involved in cochlear pathologies such as drug-induced ototoxicity. In isolated Hensen cells, it was shown that the ototoxic antibiotic gentamicin reduced the conductance of the gap junctions (Todt et al., 1999). In line with this observation another ototoxic drug, cisplatin, was able to uncouple the gap junctions of fibroblasts (Zhao et al., 2004). Since the effects of gentamicin on gap junction coupling of Hensen cells could be counterbalanced by application of catalase, it was concluded that gentamicin first stimulates the production of H2O2, which! in turn induces the inhibition of gap junctional coupling (Todt et al., 1999). This conclusion was supported by experiments in which H2O2 was directly applied on Hensen cells, where it inhibited gap junction coupling in a concentration-dependent manner (Todt et al., 2001). Furthermore, it was shown that the effect of gentamicin was related to presence of Fe2+, since the Fe2+ chelator deferoxamine was able to block the gentamicin-induced gap junction uncoupling of Hensen cells (Ngezahayo et al., unpublished data). Thus, gentamicin stimulated production of H2O2, which was transformed by a Fenton reaction into free radicals like hydroxyl, which in turn inhibited the gap junction coupling.
The mechanism by which free radicals induce closure of gap junction channels of Hensen cells is still a matter of speculation. Lin and Takemoto (2005) have shown that accumulation of reactive oxygen species (ROS) in lens epithelial cells stimulated PKC-c, which in turn phosphorylated Cx43, thereby reducing the conductance of the gap junction channels (Moreno, 2005). A similar mechanism can be postulated in supporting cells of the organ of Corti. Although Cx26 cannot be phosphorylated (Traub et al., 1986), it can be speculated that free radicals stimulate PKCin Hensen cells, which could then phosphorylate Cx30 and cause closure of gap junction channels containing Cx30. Further studies are needed to determine the pathways by which free radicals inhibit gap junction coupling in the cochlear supporting cells.
Nitric oxide (NO) has also been identified as another pharmacological modulator of gap junctional coupling in cochlear supporting cells (Blasits et al., 2000). It was shown that the gap junction coupling between Deiters cells could be blocked by application of the NO donor sodium nitroprusside (SNP). The effect of SNP on gap junction coupling could be suppressed by intracellular application of the Ca2+-chelator BAPTA and was mimicked by application of the membrane-permeable 8-bromo-cGMP. Thus, NO affected the gap junction coupling of Deiters cells by increasing the intracellular free Ca2+ concentration ([Ca2+]i) and production of cGMP, which stimulated a cGMP-dependent cascade of reactions to reduce the gap junction coupling of the Deiters cells. The origin of the Ca2+ (Ca2+-entry versus Ca2+-release from intracellular stores) as well as the relationship between the rise in [Ca2+]i and the production of cGMP have not yet been studied. However, NO was demonstrated to induce a rise in [Ca2+]i as well as a production of cGMP, and both messengers were required for gap junction uncoupling (Blasits, et al., 2000). Calmodulin has been postulated as an additional potential mediator of Ca2+-dependent gap junctional gating (Peracchia et al., 1983; Perrachia et al., 2000). In isolated Hensen cells, Blödow et al. (2003) have shown that calmodulin inhibitors like W7, trifluoperazine (TFP), or MLCK suppressed gap junction coupling. A calmodulin binding motif has been identified by sequence analysis in the C-termini of both Cx26 and Cx30, suggesting that calmodulin could directly interact with the connexins to modulate gap junction coupling between Hensen cells. Additional modulators of gap junction coupling in cochlear supporting cells, such as temperature, membrane tension, and pH, have been described, although their detailed mechanisms of action are still unclear. (Santos-Sacchi, 1986; Sato & Santos-Sacchi, 1994; Sato et al., 1998; Zhao & Santos-Sacchi, 1998).
Mouse Models of Cochlear Connexin Loss of Function
Data supporting a critical role for gap junctional coupling in cochlear homeostasis have emerged through the generation of mice with mutated or deleted Cx26 and Cx30 genes. Complete knockout of Cx26 in mice results in neonatal lethality, preventing analysis of its function in hearing (Gabriel et al., 1998). Cohen-Salmon et al. (2002) successfully performed targeted ablation of Cx26 specifically in the epithelial gap junctional network in the cochlea. These animals displayed normal patterns of cochlear development, but following the onset of hearing on postnatal day 14, showed an increase in postnatal cell death within the cochlea along with significant hearing loss. The initiation of cell death was found to occur near the inner hair cells, and coincided with decreases in the endolymphatic potential and endolymphatic potassium concentration. It was hypothesized that loss of Cx26 prevented recycling of K+ ions after sound stimulation and that elevated K+ in the extracellular perilymph inhibited uptake of the neurotransmitter glutamate, which ultimately resulted in cell death within the hair cell population. These data demonstrated that the epithelial gap junction network plays a critical role in cochlear function, but delineating the precise role of Cx26 becomes more complex since the tissue-specific deletion of the Cx26 gene did not alter the expression pattern of Cx30 in these mice (Cohen-Salmon et al., 2002). Cx30 is highly permeable to K+ ions (Valiunas et al., 1999), yet it alone could not prevent hearing loss in the absence of Cx26, and the presence of a single type of connexin with similar ionic selectivity was unable to rescue the phenotype observed in these mice. Further investigation of these conditional knockout animals will be invaluable toward further elucidation of the role of gap junctional communication in the recycling of K+ ions in the etiology of deafness.
A different mouse model examined the pathology of dominant mutations by transgenically expressing the dominant-negative Cx26 mutant R75W in the inner ear (Kudo et al., 2003). This mutation caused syndromic deafness associated with skin disease and its dominant-negative effects were verified by showing that R75W lacked channel activity when expressed alone and efficiently inhibited the activity of co-expressed wild-type Cx26 (Richard et al., 1998). Transgenic mice expressing the R75W mutation exhibited profound deafness that was evident as early as at two weeks of age. In addition, these mice developed significant histological abnormalities within the inner ear by two weeks of age, including deformities of the tunnel of Corti, supporting cells and hair cell degeneration (Kudo et al., 2003). However, the stria vascularis and spiral ligament, which are also rich in Cx26 expression, appeared normal. By seven weeks of age, hair cells and the tunnel of Corti had completely degenerated in R75W animals (Kudo et al., 2003). These findings suggested that R75W exerted its effects within the epithelial transport system of the inner ear by altering the circulation of K+ ions into the cortilymph, instead of the extracellular perilymph, as was reported in the conditional Cx26 model (Cohen-Salmon et al., 2002). These data further highlight that diverse cochlear pathologies may arise depending on the pattern of inheritance and/or the nature of the Cx26 mutation.
A Cx30 knockout mouse model has been successfully established as well (Teubner et al., 2003). Similar to Cx26 ablation in cochlear supporting cells, deletion of Cx30 in mice resulted in hearing loss but the cochlea and vestibular end organs developed normally (Teubner et al., 2003). Despite normal development, the Cx30 knockouts lacked the endocochlear potential that normally results from the asymmetric Na+ and K+ concentrations in the endolymph and perilymph. At postnatal day 18, shortly after the onset of hearing, Cx30 knockouts displayed increased apoptosis within the cochlear sensory epithelium. Deletion of Cx30 did not alter the cochlear expression of Cx26, again raising the question of why the continued expression of Cx26 was not able to compensate for the loss of Cx30 (Teubner et al., 2003). One possible explanation for these phenotypes is that gap junctions may have other roles in addition to recycling K+ ions. This idea is supported by data from functional studies showing that while the K+ conductance of Cx26 and Cx30 is similar, the channels display significant differences regarding the permeability of larger molecules (Valiunas et al., 1999; Manthey et al., 2001). Thus, specific loss of either Cx26 or Cx30 within cochlear epithelial cells would not simply reduce the intercellular passage of K+ ions, but could also significantly alter the exchange of larger solutes between the coupled cells.
Functional Properties of Mutant Channels
Mutations in Cx26 are associated with pathogenesis of most autosomal recessive and dominant hearing loss, as well as sporadic congenital deafness (Kelsell et al., 1997; Estivill et al., 1998; Rabionet et al., 2000; Kenneson et al., 2002). These mutations include nonsense or insertion/deletion, which results in the premature termination of protein translation, and missense mutations that alter a single amino acid. 35delG is the most frequently found mutation in Cx26 involved in 70-85% of Cx26-related deafness (Zelante et al., 1997). Deletion of this one base results in a frame shift introducing a premature stop codon and resulting in termination of protein translation. In addition to 35delG, numerous missense mutations have been identified throughout the Cx26 gene (http://davinci.crg.es/deafness/) and many of them have been analyzed for alteration of their functional properties (White, 2000). For example, Bruzzone et al. (2003) found that the V84L mutation was able to form functional channels as well as wild-type Cx26 and that the voltage-gating properties shared similar features. In another study by D’Andrea et al. (2002), two additional Cx26 mutations, L90P and R127H, induced low levels of Lucifer Yellow dye transfer between transfected HeLa cells. Thus, not all deafnesscausing mutations resulted in complete loss of activity and the functional mutations may provide unique insights into the specific role of Cx26 in hearing.
This view has been supported by other functional studies of connexin mutants. Thus far, most mutants of Cx26 cannot form functional gap junction channels in vitro (White et al., 1998; Martin et al., 1999; Choung et al., 2002; D’Andrea et al., 2002; Thonnissen et al., 2002; Bruzzone et al., 2003; Marziano et al., 2003; Wang et al., 2003; Meş e et al., 2004; Melchionda et al., 2005; Piazza et al., 2005). However, some deafness-associated mutants in the Cx26, for example T8M, V84L, A88S, V95M, E114G and N206S, were still able to form functional homotypic gap junction channels in vitro (Choung et al., 2002; Thonnissen et al., 2002; Bruzzone et al., 2003; Wang et al., 2003; Meş e et al., 2004; Zhang et al., 2005). Many of these functional mutants were shown to induce electrical coupling in experimental expression systems. Given that potassium is the dominant permeant ion of normal cytoplasm, it is very likely that these mutant channels retain high permeability to K+ ions. Thus, deafness associated with these missense mutations in the Cx26 gene may not solely depend on the loss of K+ coupling and K+ re-circulation. Instead, these mutations may interfere with the coupling of larger biochemical solutes in the inner ear. This concept has been validated by recent experiments showing that the V84L mutation had impaired permeability to the cell signaling molecule inositol 1,4,5-trisphosphate (IP3) compared to wild-type Cx26 (Beltramello et al., 2005). This study showed that potassium conductance and open probability of V84L were indistinguishable from those of Cx26, but V84L failed to propagate Ca2+ waves stimulated by direct intracellular injection of IP3. Beltramello et al. (2005) further documented that injection of IP3 into supporting cells of the rat organ of Corti produced a regenerative wave of Ca2+ throughout the tissue, although the precise role played by calcium waves in the process of sound transduction remains speculative (Bruzzone & Cohen-Salmon, 2005).
Additional evidence that gap junction-mediated exchange of larger biochemical molecules was required for normal cochlear function came from a second study examining IP3 and Ca2+ waves. Zhang et al. (2005) also showed that organotypic cochlear cultures mediated the propagation of intracellular Ca2+ waves by facilitating intercellular diffusion of injected IP. They then documented that a subset of Cx26 mutations, V84L, V95M, and A88S, specifically altered the intercellular exchange of larger biochemical solutes like propidium iodide and IP3, but retained permeability to simple ions, as measured by both electrical coupling and experiments where Na+ and Ca2+ were directly injected into one cell and then detected in a neighboring cell using fluorescent dyes (Zhang et al., 2005). These data suggested that inositol 1,4,5-trisphosphate was the critical permeant molecule for calcium wave propagation and confirmed that additional Cx26 deafness-linked mutations that retained ionic coupling were deficient in their permeability to larger biochemical molecules. The combined results of Beltramello et al. (2005) and Zhang et al. (2005) strongly suggest that potassium permeability is not the only function of supporting cell gap junctions, and that permeability to cytoplasmic signaling molecules and metabolites may also be required for normal hearing.
Fig. 1.
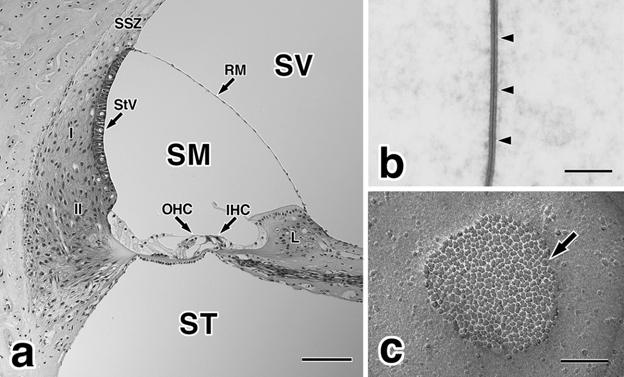
(a) A paraffin section of the guinea pig cochlea stained with hematoxylin and eosin. The perilymphatic space is composed of the scala vestibuli (SV) and scala tympani (ST), and contains high levels of sodium and low levels of potassium ions. Endolymph is present in the scala media (SM), and contains high levels of K+ and low levels of Na+. I, type I fibrocytes of the spiral ligament; II, type II fibrocytes of the spiral ligament; IHC, inner hair cells; L, spiral limbus; OHC, outer hair cells; RM, Reissner’s membrane; SSZ, suprastrial zone; StV, stria vascularis. Bar = 100 lm. (b) Thin section electron micrograph of a typical gap junction (arrowheads) between supporting cells in the guinea pig organ of Corti. Bar = 100 nm. (c) Freeze-fracture electron micrograph of a typical gap junction (arrow) between supporting cells of the guinea pig organ of Corti. Gap junctions are clusters of homogeneous intramembranous particles, connexons, in the protoplasmic fracture face of the plasma membrane. Bar = 100 nm.
Fig. 2.
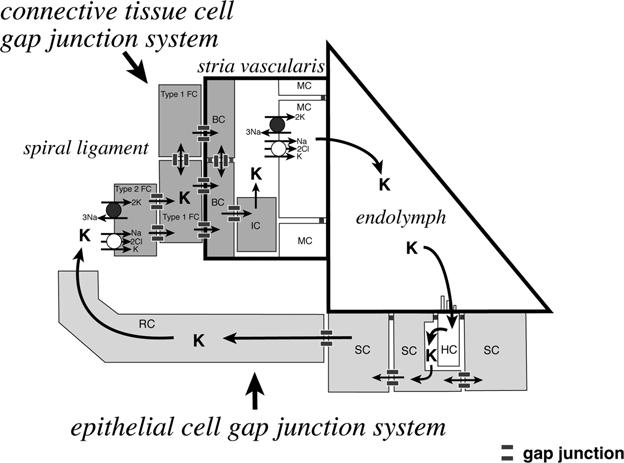
Schematic illustration of a potassium ion recycling mechanism in the mammalian cochlea. There are two independent gap junction systems, the epithelial cell gap junction system and the connective tissue cell gap junction system in the cochlea. Potassium ions, which play a pivotal role in the mechanoelectrical sound transduction process in the cochlea, are recycled via these two gap junction systems. BC, basal cells; HC, hair cells; IC, intermediate cells, MC, marginal cells; RC, root cells; SC, supporting cells; Type 1 FC, type I fibrocytes; Type II FC, type II fibrocytes. This illustration is modified with permission from Kikuchi et al. (2000a).
Acknowledgments
The authors gratefully acknowledge J.C. Adams (Harvard Medical School) and G. Meş e (SUNY Stony Brook) for their critical reading of this manuscript. Work in our laboratories is supported in part by NIH grants DC06652 (T.W.W.) and DC05989 (H.-B.Z.). We apologize to colleagues whose work could not be cited here due to space constraints.
Footnotes
All authors contributed equally to this article.
References
- Ahmad S, Chen S, Sun J, Lin X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem. Biophys. Res. Commun. 2003;307:362–368. doi: 10.1016/s0006-291x(03)01166-5. [DOI] [PubMed] [Google Scholar]
- Barrio LC, Suchyna T, Bargiello T, Xu LX, Roginski RS, Bennett M, Nicholson BJ. Gap junctions formed by connexins 26 and 32 alone and in combination are differently affected by applied voltage. Proc. Natl. Acad. Sci. USA. 1991;88:8410–8414. doi: 10.1073/pnas.88.19.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramello M, Bicego M, Piazza V, Ciubotaru CD, Mammano F, D’Andrea P. Permeability and gating properties of human connexins 26 and 30 expressed in HeLa cells. Biochem. Biophys. Res. Commun. 2003;305:1024–1033. doi: 10.1016/s0006-291x(03)00868-4. [DOI] [PubMed] [Google Scholar]
- Beltramello M, Piazza V, Bukauskas FF, Pozzan T, Mammano F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell. Biol. 2005;7:63–69. doi: 10.1038/ncb1205. [DOI] [PubMed] [Google Scholar]
- Bennett MVL, Barrio LC, Bargiello TA, Spray DC, Hertzberg E, Saez JC. Gap junctions: New tools, new answers, new questions. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- Bevans CG, Kordel M, Rhee SK, Harris AL. Isoform composition of connexin channels determines selectivity among second messengers and uncharged molecules. J. Biol. Chem. 1998;273:2808–2816. doi: 10.1074/jbc.273.5.2808. [DOI] [PubMed] [Google Scholar]
- Blasits S, Maune S, Santos-Sacchi J. Nitric oxide uncouples gap junctions of supporting Deiters cells from Corti’s organ. Pflügers Arch. 2000;440:710–712. doi: 10.1007/s004240000355. [DOI] [PubMed] [Google Scholar]
- Blödow A, Ngezahayo A, Ernst A, Kolb HA. Calmodulin antagonists suppress gap junction coupling in isolated Hensen cells of the guinea pig cochlea. Pflügers Arch. 2003;446:36–41. doi: 10.1007/s00424-002-1004-9. [DOI] [PubMed] [Google Scholar]
- Boettger T, Hubner CA, Maler H, Rust MB, Beck FX, Jentsch TJ. Deafness and renal tubular acidosis in mice lacking the K-Cl co-transporter Kcc4. Nature. 2002;416:874–878. doi: 10.1038/416874a. [DOI] [PubMed] [Google Scholar]
- Boettger T, Rust MB, Maier H, Seidenbecher T, Schweizer M, Keating DJ, Faulhaber J, Ehmke H, Pfeffer C, Scheel O, Lemcke B, Horst J, Leuwer R, Pape H-C, Volkl H, Hubner A, Jentsch TJ. Loss of K-Cl co-transporter KCC3 causes deafness, neurodegeneration and reduced seizure threshold. EMBO J. 2003;22:5422–5434. doi: 10.1093/emboj/cdg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone R, Cohen-Salmon M. Hearing the messenger: Ins(1,4,5) P3 and deafness. Nat. Cell Biol. 2005;7:14–16. doi: 10.1038/ncb0105-14. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D’Andrea P, White TW. Loss-offunction and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- Buniello A, Montanaro D, Volinia S, Gasparini P, Marigo V. An expression atlas of connexin genes in the mouse. Genomics. 2004;83:812–820. doi: 10.1016/j.ygeno.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Cao F, Eckert R, Elfgang C, Nitsche JM, Snyder SA, Hulser DF, Willecke K, Nicholson BJ. A quantitative analysis of connexin-specific permeability differences of gap junctions expressed in HeLa transfectants and Xenopus oocytes. J. Cell. Sci. 1998;111:31–43. doi: 10.1242/jcs.111.1.31. [DOI] [PubMed] [Google Scholar]
- Choung YH, Moon SK, Park HJ. Functional study of GJB2 in hereditary hearing loss. Laryngoscope. 2002;112:1667–1671. doi: 10.1097/00005537-200209000-00026. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, Maxeiner S, Kruger O, Theis M, Willecke K, Petit C. Expression of the connexin43- and connexin45-encoding genes in the developing and mature mouse inner ear. Cell Tissue Res. 2004;316:15–22. doi: 10.1007/s00441-004-0861-2. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch JJ, Sakaguchi N, Lytle C, Schulte BA. Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J. Histochem. Cytochem. 1997;45:773–778. doi: 10.1177/002215549704500601. [DOI] [PubMed] [Google Scholar]
- Dahl E, Manthey D, Chen Y, Schwarz HJ, Chang YS, Lalley PA, Nicholson BJ, Willecke K. Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J. Biol. Chem. 1996;271:17903–17910. doi: 10.1074/jbc.271.30.17903. [DOI] [PubMed] [Google Scholar]
- D’Andrea P, Veronesi V, Bicego M, Melchionda S, Zelante L, Di Iorio E, Bruzzone R, Gasparini P. Hearing loss: frequency and functional studies of the most common connex-in26 alleles. Biochem. Biophys. Res. Commun. 2002;296:685–691. doi: 10.1016/s0006-291x(02)00891-4. [DOI] [PubMed] [Google Scholar]
- Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA, Hulser DF, Willecke K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell. Biol. 1995;129:805–817. doi: 10.1083/jcb.129.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essenfelder GM, Bruzzone R, Lamartine J, Charollais A, Blanchet-Bardon C, Barbe MT, Meda P, Walksman G. Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activity. Hum. Mol. Genet. 2004;13:1703–1714. doi: 10.1093/hmg/ddh191. [DOI] [PubMed] [Google Scholar]
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D’Agruma L, Mansfield E, Rappaport E, Govea N, Mila M, Zelante L, Gasparini P. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998;351:394–398. doi: 10.1016/S0140-6736(97)11124-2. [DOI] [PubMed] [Google Scholar]
- Forge A, Becker D, Casalotti S, Edwards J, Evans WH, Lench N, Souter M. Gap junctions and connexin expression in the inner ear. Novartis Found. Symp. 1999;219:134–156. doi: 10.1002/9780470515587.ch9. [DOI] [PubMed] [Google Scholar]
- Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J. Comp. Neurol. 2003;467:207–231. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- Gabriel HD, Jung D, Butzler C, Temme A, Traub O, Winterhager E, Willecke K. Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 1998;140:1453–1461. doi: 10.1083/jcb.140.6.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg GS, Valiunas V, Brink PR. Selective permeability of gap junction channels. Biochim. Biophys. Acta. 2004;1662:96–101. doi: 10.1016/j.bbamem.2003.11.022. [DOI] [PubMed] [Google Scholar]
- Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 1999;23:16–18. doi: 10.1038/12612. [DOI] [PubMed] [Google Scholar]
- Gulley RS, Reese TS. Intercellular junctions in the reticular lamina of the organ of Corti. J. Neurocytol. 1976;5:479–507. doi: 10.1007/BF01181652. [DOI] [PubMed] [Google Scholar]
- Hama K, Saito K. Gap junctions between the supporting cells in some acousticovestibular receptors. J. Neurocytol. 1977;6:1–12. doi: 10.1007/BF01175410. [DOI] [PubMed] [Google Scholar]
- Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 2001;34:325–472. doi: 10.1017/s0033583501003705. [DOI] [PubMed] [Google Scholar]
- Iurato S, Franke K, Luciano L, Wermber G, Pannese E, Reale E. Intercellular junctions in the organ of Corti as revealed by freeze fracturing. Acta. Otolaryngol. 1976;82:57–69. doi: 10.3109/00016487609120863. [DOI] [PubMed] [Google Scholar]
- Jahnke K. The fine structure of freeze-fractured intercellular junctions in the guinea pig inner ear. Acta. Otolaryngol. 1975;336:1–40. [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Van Naarden Braun K, Van, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet. Med. 2002;4:258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Adams JC, Miyabe Y, So E, Kobayashi T. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness. Med. Electron Microsc. 2000a;33:51–56. doi: 10.1007/s007950070001. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 1995;191:101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Kimura RS, Paul DL, Takasaka T, Adams JC. Gap junction systems in the mammalian cochlea. Brain Res. Brain Res. Rev. 2000b;32:163–166. doi: 10.1016/s0165-0173(99)00076-4. [DOI] [PubMed] [Google Scholar]
- Kudo T, Kure S, Ikeda K, Xia AP, Katori Y, Suzuki M, Kojima K, Ichinohe A, Suzuki Y, Aoki Y, Kobayashi T, Matsubara Y. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum. Mol. Genet. 2003;12:995–1004. doi: 10.1093/hmg/ddg116. [DOI] [PubMed] [Google Scholar]
- Lautermann J, ten Cate WJF, Altenhoff P, Grümmer R, Traub O, Frank HG, Janhke K, Winterhager E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998;294:415–420. doi: 10.1007/s004410051192. [DOI] [PubMed] [Google Scholar]
- Locke D, Stein T, Davies C, Morris J, Harris AL, Evans WH, Monaghan P, Gusterson B. Altered permeability and modulatory character of connexin channels during mammary gland development. Exp. Cell Res. 2004;298:643–660. doi: 10.1016/j.yexcr.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Lin D, Takemoto DJ. Oxidative activation of protein kinase Cc through the C1 domain. Effects on gap junctions. J. Biol. Chem. 2005;280:13682–13693. doi: 10.1074/jbc.M407762200. [DOI] [PubMed] [Google Scholar]
- Manthey D, Banach K, Desplantez T, Lee CG, Kozak CA, Traub O, Weingart R, Willecke K. Intracellular domains of mouse connexin26 and -30 affect diffusional and electrical properties of gap junction channels. J. Membrane. Biol. 2001;181:137–148. doi: 10.1007/s00232-001-0017-1. [DOI] [PubMed] [Google Scholar]
- Martin PE, Coleman SL, Casalotti SO, Forge A, Evans WH. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum. Mol. Genet. 1999;8:2369–2376. doi: 10.1093/hmg/8.13.2369. [DOI] [PubMed] [Google Scholar]
- Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003;12:805–812. doi: 10.1093/hmg/ddg076. [DOI] [PubMed] [Google Scholar]
- Melchionda S, Bicego M, Marciano E, Franzè A, Morgutti M, Bortone G, Zelante L, Carella M, D’Andrea P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Comm. 2005;337:799–805. doi: 10.1016/j.bbrc.2005.09.116. [DOI] [PubMed] [Google Scholar]
- Meş e G, Londin E, Mui R, Brink PR, White TW. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum. Genet. 2004;115:191–199. doi: 10.1007/s00439-004-1142-6. [DOI] [PubMed] [Google Scholar]
- Moreno AP. Connexin phosphorylation as a regulatory event linked to channel gating. Biochim. Biophys. Acta. 2005;1711:172–182. doi: 10.1016/j.bbamem.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Nicholson BJ, Weber PA, Cao F, Chang H, Lampe P, Goldberg G. The molecular basis of selective permeability of connexins is complex and includes both size and charge. Braz. J. Med. Biol. Res. 2000;33:369–378. doi: 10.1590/s0100-879x2000000400002. [DOI] [PubMed] [Google Scholar]
- Oesterle EC, Dallos P. Intracellular recordings from supporting cells in the guinea pig cochlea: DCpotentials. J. Neurophysiol. 1990;64:617–636. doi: 10.1152/jn.1990.64.2.617. [DOI] [PubMed] [Google Scholar]
- Oh S, Rubin JB, Bennett MV, Verselis VK, Bargiello TA. Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J. Gen. Physiol. 1999;114:339–364. doi: 10.1085/jgp.114.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peracchia C, Bernadini G, Pernacchia LL. Is calmodulin involved in the regulation of gap junction permeability? Pfügers Arch. 1983;99:152–154. doi: 10.1007/BF00663912. [DOI] [PubMed] [Google Scholar]
- Peracchia C, Sotkis A, Wang XG, Pernacchia LL, Persechini A. Calmodulin directly gates gap junction channels. J. Biol. Chem. 2000;275:26220–26224. doi: 10.1074/jbc.M004007200. [DOI] [PubMed] [Google Scholar]
- Piazza V, Beltramello M, Menniti M, Colao E, Malatesta P, Argento R, Chiarella G, Gallo LV, Catalano M, Perrotti N, Mammano F, Cassandro E. Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss. Clin. Genet. 2005;68:161–166. doi: 10.1111/j.1399-0004.2005.00468.x. [DOI] [PubMed] [Google Scholar]
- Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum. Mutat. 2000;16:190–202. doi: 10.1002/1098-1004(200009)16:3<190::AID-HUMU2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 1998;103:393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Verselis VK, Bennett MVL, Bargiello TA. Molecular analysis of voltage dependence of heterotypic gap junctions formed by connexins 26 and 32. Biophys. J. 1992;62:183–195. doi: 10.1016/S0006-3495(92)81804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Sacchi J. The effects of cytoplamic acidification upon electrical coupling in the organ of corti. Hear. Res. 1985;19:207–215. doi: 10.1016/0378-5955(85)90140-6. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J. Temperature dependence of electrical coupling in organ of Corti. Hear. Res. 1986;21:205–211. doi: 10.1016/0378-5955(86)90219-4. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J. Cell coupling differs in the in vitro and in vivo organ of Corti. Hear. Res. 1987;25:227–232. doi: 10.1016/0378-5955(87)90094-3. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J. Cell coupling in the organ of Corti. Brain Res. Brain Res. Rev. 2000;32:167–171. doi: 10.1016/s0165-0173(99)00077-6. [DOI] [PubMed] [Google Scholar]
- Santos-Sacchi J, Dallos P. Intercellular communication in the supporting cells of the organ of Corti. Hear. Res. 1983;9:317–326. doi: 10.1016/0378-5955(83)90034-5. [DOI] [PubMed] [Google Scholar]
- Sato Y, Handa T, Matsumura M, Orita Y. Gap junction change in supporting cells of organ of Corti with ryanodine and caffeine. Acta. Otolaryngol. 1998;118:821–825. doi: 10.1080/00016489850182512. [DOI] [PubMed] [Google Scholar]
- Sato Y, Santos-Sacchi J. Cell coupling in the supporting cells of Corti’s organ: sensitivity to intracellular H+ and Ca2+ Hear. Res. 1994;80:21–24. doi: 10.1016/0378-5955(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Schulte BA, Adams JC. Distribution of immunoreactive Na+, K+-ATPase in the gerbil cochlea. J. Histochem. Cytochem. 1989;7:127–134. doi: 10.1177/37.2.2536055. [DOI] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA. The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear. Res. 1996;100:80–100. doi: 10.1016/0378-5955(96)00106-2. [DOI] [PubMed] [Google Scholar]
- Sun J, Ahmad S, Chen S, Tang W, Zhang Y, Chen P, Lin X. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am. J. Physiol. 2005;288:C613–C623. doi: 10.1152/ajpcell.00341.2004. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Takamatsu T, Oyamada M. Expression of gap junction protein connexin43 in the adult rat cochlea: comparison with connexin26. J. Histochem. Cytochem. 2003;51:903–912. doi: 10.1177/002215540305100705. [DOI] [PubMed] [Google Scholar]
- Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of en-docochlear potential. Hum. Mol. Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- Thonnissen E, Rabionet R, Arbones ML, Estivill X, Willecke K, Ott T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002;111:190–197. doi: 10.1007/s00439-002-0750-2. [DOI] [PubMed] [Google Scholar]
- Todt I, Ngezahayo A, Ernst A, Kolb HA. Inhibition of gap junctional coupling in cochlear supporting cells by gentamicin. Pflügers Arch. 1999;438:865–867. doi: 10.1007/s004249900109. [DOI] [PubMed] [Google Scholar]
- Todt I, Ngezahayo A, Ernst A, Kolb HA. Hydrogen peroxide inhibits gap junctional coupling and modulates intracellular free calcium in cochlear Hensen cells. J. Membrane. Biol. 2001;181:107–114. doi: 10.1007/s00232001-0014-4. [DOI] [PubMed] [Google Scholar]
- Traub O, Look J, Dermietzel R, Brummer F, Hulser D, Willecke K. Comparative characterization of the 21-kD and 26-kD gap junction proteins in murine liver and cultured hepatocytes. J. Cell Biol. 1986;108:1039–1051. doi: 10.1083/jcb.108.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V, Manthey D, Vogel R, Willecke K, Weingart R. Biophysical properties of mouse connexin30 gap junction channels studied in transfected human HeLa cells. J. Physiol. 1999;519:631–644. doi: 10.1111/j.1469-7793.1999.0631n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V, Polosina YY, Miller H, Potapova IA, Valiuniene L, Doronin S, Mathias RT, Robinson RB, Rosen MR, Cohen IS, Brink PR. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 2005;568:459–468. doi: 10.1113/jphysiol.2005.090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiunas V, Weingart R. Electrical properties of gap junction hemichannels identified in transfected HeLa cells. Pflügers Arch. 2000;440:366–379. doi: 10.1007/s004240000294. [DOI] [PubMed] [Google Scholar]
- Veenstra RD. Size and selectivity of gap junction channels formed from different connexins. J. Bioenerg. Biomembr. 1996;28:327–337. doi: 10.1007/BF02110109. [DOI] [PubMed] [Google Scholar]
- Verselis VK, Ginter CS, Bargiello TA. Opposite voltage gating polarities of two closely related connexins. Nature. 1994;368:348–351. doi: 10.1038/368348a0. [DOI] [PubMed] [Google Scholar]
- Wang HL, Chang WT, Li AH, Yeh TH, Wu CY, Chen MS, Huang PC. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 2003;84:735–742. doi: 10.1046/j.1471-4159.2003.01555.x. [DOI] [PubMed] [Google Scholar]
- White TW. Functional analysis of human Cx26 mutations associated with deafness. Brain Res. Brain Res. Rev. 2000;32:181–183. doi: 10.1016/s0165-0173(99)00079-x. [DOI] [PubMed] [Google Scholar]
- White TW, Bruzzone R. Multiple connexin proteins in single intercellular channels: connexin compatibility and functional consequences. J. Bioenerg. Biomembr. 1996;28:339–350. doi: 10.1007/BF02110110. [DOI] [PubMed] [Google Scholar]
- White TW, Bruzzone R, Goodenough DA, Paul DL. Voltage gating of connexins. Nature. 1994;371:208–209. doi: 10.1038/371208a0. [DOI] [PubMed] [Google Scholar]
- White TW, Deans MR, Kelsell DP, Paul DL. Connexin mutations in deafness. Nature. 1998;394:630–631. doi: 10.1038/29202. [DOI] [PubMed] [Google Scholar]
- Xia AP, Ikeda K, Katori Y, Oshima T, Kikuchi T, Takasaka T. Expression of connexin 31 in the developing mouse cochlea. Neuroreport. 2000;11:2449–2453. doi: 10.1097/00001756-200008030-00022. [DOI] [PubMed] [Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 1998;20:370–373. doi: 10.1038/3845. [DOI] [PubMed] [Google Scholar]
- Zelante L, Gasparini P, Estivill X, Melchionda S, D’Agruma L, Govea N, Mila M, Monica MD, Lutfi J, Shohat M, Mansfield E, Delgrosso K, Rappaport E, Surrey S, Fortina P. Connexin26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans. Hum. Mol. Genet. 1997;6:1605–1609. doi: 10.1093/hmg/6.9.1605. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tang W, Ahmad S, Sipp JA, Chen P, Lin X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc. Natl. Acad. Sci. USA. 2005;102:15201–15206. doi: 10.1073/pnas.0501859102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HB, Santos-Sacchi J. Effect of membrane tension on gap junctional conductance of supporting cells in Corti’s organ. J. Gen. Physiol. 1998;112:447–455. doi: 10.1085/jgp.112.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HB, Santos-Sacchi J. Auditory collusion and a coupled couple of outer hair cells. Nature. 1999;399:359–362. doi: 10.1038/20686. [DOI] [PubMed] [Google Scholar]
- Zhao HB, Santos-Sacchi J. Voltage gating of gap junctions in cochlear supporting cells: Evidence for nonhomotypic channels. J. Membrane. Biol. 2000;175:17–24. doi: 10.1007/s002320001051. [DOI] [PubMed] [Google Scholar]
- Zhao HB. Directional rectification of gap junctional voltage gating between Deiters cells in the inner ear of Guinea pig. Neurosci. Lett. 2000;296:105–108. doi: 10.1016/s0304-3940(00)01626-8. [DOI] [PubMed] [Google Scholar]
- Zhao HB. Connexin26 is responsible for anionic molecule permeability in the cochlea for intercellular signalling and metabolic communications. Eur. J. Neurosci. 2005;21:1859–1868. doi: 10.1111/j.1460-9568.2005.04031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Lin ZX, Zhang ZQ. Cisplatin-induced pre-mature senescence with concomitant reduction of gap junctions in human fibroblasts. Cell Res. 2004;14:60–66. doi: 10.1038/sj.cr.7290203. [DOI] [PubMed] [Google Scholar]
- Zwislocki JJ, Slepecky NB, Cefaratti LK, Smith RL. Ionic coupling among cells in the organ of Corti. Hear. Res. 1992;57:175–194. doi: 10.1016/0378-5955(92)90150-l. [DOI] [PubMed] [Google Scholar]