

Abstract
In principle, given the amino acid sequence of a protein, it is possible to compute the corresponding three-dimensional structure. Methods for modelling structure based on this premise have been under development for more than 40 years. For the past decade, a series of community wide experiments (termed Critical Assessment of Structure Prediction (CASP)) have assessed the state of the art, providing a detailed picture of what has been achieved in the field, where we are making progress, and what major problems remain. The rigorous evaluation procedures of CASP have been accompanied by substantial progress. Lessons from this area of computational biology suggest a set of principles for increasing rigor in the field as a whole.
Keywords: protein structure prediction, community wide experiment, critical assessment of structure prediction, computational biology
1. Introduction
In the 1950s, work by Anfinsen & colleagues conclusively showed that the information determining the three-dimensional structure of a protein molecule is contained in the amino acid sequence. Recognition of this relationship rapidly led to the development of methods for computing structure from sequence. There were many early encouraging reports of partial success, starting in the 1960s and continuing through the 1970s and 1980s. And yet, during this long period, there were very few reports of computed structures in any way competing with those obtained experimentally. The mismatch between apparent success and the lack of useful applications suggested that the traditional peer reviewed publication system is not sufficient to ensure rigor in this area of computational biology. The Critical Assessment of Structure Prediction (CASP) experiments were devised as a means of addressing the specific needs of methods evaluation in structure modelling. CASP is one of a number of ways in which this problem may be addressed. As discussed later, the fundamental differences between computational and experimental biology dictate that new procedures be adopted in the field as a whole.
2. CASP
CASP is a community wide experiment with the goal of assessing the effectiveness of methods for modelling protein structure. The aims are to provide detailed information about the strengths and weaknesses of current structure modelling methods, to identify where progress has been made, to show where there are serious bottlenecks to further progress, and to indicate how these may eventually be removed. Key features are:
The use of bona fide blind predictions, rather than the previous practice of reproducing already known structures.
Participants provide models for the same set of proteins, greatly facilitating comparison of performance.
Predictions are made on a reasonably large set of proteins, reducing the impact of case specific artefacts.
There are multiple independent approaches to evaluation, reducing bias.
All models and analysis results are freely available to all, allowing maximum use to be made of the data.
The experiment has been conducted every 2 years since 1994 (CASP1), with the most recent one taking place in 2004 (CASP6). Information about soon-to-be experimentally determined protein structures is collected, and passed on to registered predictors. More than 200 prediction teams from 24 countries participated in CASP6, providing over 30 000 predictions on 90 protein domains. Predictions are evaluated using a battery of numerical criteria (Zemla et al. 2001) and more importantly, are carefully examined by independent assessors. A conference is held to discuss the results, and a special issue of the journal Proteins is published, with articles by the assessors and by some of the more successful prediction teams. Details for the 5th experiment can be found in the most recent journal issue (Moult et al. 2003). In particular, articles by the three assessment groups (Aloy et al. 2003; Kinch et al. 2003; Tramontano & Morea 2003) provide a detailed overview of the state of the art at that time, and another article puts the results in the context of previous CASPs (Venclovas et al. 2003). The Proteins issue for the sixth experiment will appear in early 2006. All participant registration, target management, prediction collection and numerical analysis are handled by the Protein Structure Prediction Center (Zemla et al. 2001). The Center web site (predictioncenter.org) provides access to details of the experiment and all results. A second web site (www.forcasp.org) provides a discussion forum for the CASP community.
3. Classes of structure prediction difficulty
Early work in the structure modelling field focused on understanding the nature of the natural protein folding process, and on the development of physics based force fields to determine the relative free energy of any conformation of a polypeptide chain. These methods were much in evidence at the first CASP, but have largely been supplanted by more successful ‘knowledge based’ approaches, which use the large and growing set of experimentally determined structures and sequences, in a variety of ways. As a consequence, accuracy of models depends on similarity to already known structures, and the number of related sequences that are available. Based on this consideration, CASP considers three classes of modelling difficulty, discussed in the following sections.
4. Comparative modelling based on a clear sequence relationship
For cases where there is an easily detectable sequence relationship between a target protein and one or more of known structure (a highly statistically significant score from a BLAST search; Altschul et al. 1990), an accurate core model (typically 2–3 Å RMS error on Ca atoms) can be obtained by copying from the structural template or templates (Tramontano & Morea 2003). Copying is often non-trivial, requiring a correct alignment of the target and template sequences. Improvements over the CASPs have resulted in largely correct alignments in this modelling zone. A single template structure rarely provides a complete model. Alternative templates may provide some additional structural features, and short regions of chain (‘loops’) are sometimes modelled in an approximately correct manner. Generally, reliably building regions of the structure not present in a template remains a challenge. Side chain conformations are very tightly correlated with backbone conformation (Chung & Subbiah 1995), so not surprisingly, side chain accuracy in these approximate models is poor.
A typical CASP6 comparative model is shown in figure 1, for Target 266, an Aeropyrum pernix homologue of the Haemophilus influenzae proline tRNA editing enzyme (An & Musier-Forsyth 2004). For large regions of the structure the template provides an accurate guide, resulting in good overall quality. Two non-template loop regions (A and B) are successfully modelled. The largest differences between the template and the target are in two helices (H1 and H2) flanking the active site, suggesting different substrate specificities. The best models leave the helices in the template orientation, so it is not possible to analyse possible specificity differences. In general, although the structure around active sites is usually well conserved between proteins with the same specificity, it is often the least conserved when the specificities differ.
Figure 1.
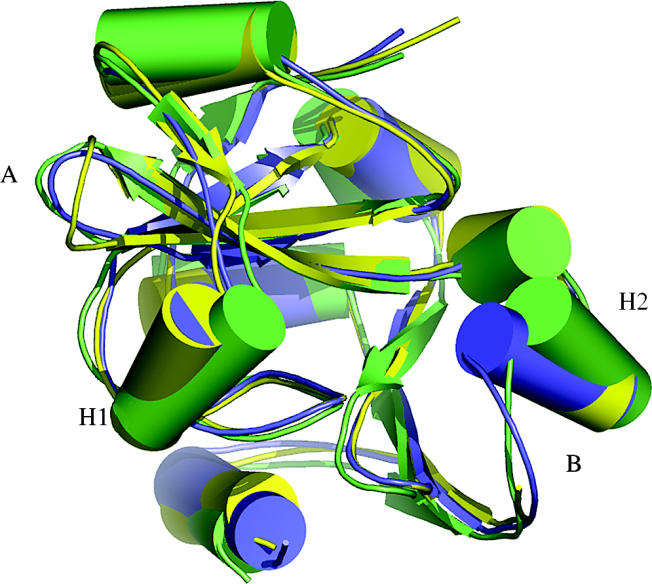
CASP6 Target 266, an example of a structure model based on a relatively close evolutionary relationship. The best model is blue, experimental structure (PDB entry 1wdv) is green, and the available template structure (28% sequence identity to target, 1dbu_A) is yellow. Where template and target are similar (yellow and green superpose), the model is accurate. Two loop regions not available in the template (A and B) are also reasonably correct. Helices H1 and H2 have different orientations in the template and the target, not corrected in the model. These structural features may be related to ligand specificity differences. Refinement of these models to rival experiment remains a central challenge, with signs of recent progress.
While large parts of this class of model are approximately correct, they require refinement to be competitive with experiment, and to reproduce key functional features. Refinement remains the principal bottleneck to progress, and is now receiving a large amount of attention. In spite of limitations, these models are very useful for a variety of purposes, often identifying which members of a protein family have the same detailed function, and which are different (DeWeese-Scott & Moult 2004).
5. Modelling based on more distant evolutionary relationships
A second class of model quality is provided by those cases where an evolutionary relationship can be detected with more sophisticated methods than just BLAST. The core of these methods is alignment of a set of sequences, so that the characteristics of protein families may be used to detect relationships (Altschul et al. 1997; Karplus et al. 1998; Karplus & Hu 2001; Marti-Renom et al. 2004; Kahsay et al. 2005). Structural information is also used in a number of ways to enhance the detection of homologues (Sippl 1993; Bates et al. 2001; Karplus et al. 2003; McGuffin & Jones 2003; Venclovas 2003; von Grotthuss et al. 2003; Przybylski & Rost 2004; Wrabl & Grishin 2004).
Models based on the detection of these more distant relationships are limited in accuracy by four factors: identifying suitable structural templates, accuracy of alignment of sequence onto a template, conformational differences between the core template and target structures, and the difficulty of modelling regions of the target not available from a template. Nevertheless, methodological improvements together with the increased size of the pool of known structures and sequences has resulted in a steady improvement in model quality over the course of the CASP experiments. Further progress will depend on two main factors: first, effective application of template free modelling methods to those regions not found in a template. As outlined below, improvements in that area make this possible. The second factor is accurate alignment. This will likely require refinement at an all-atom level, since the information needed to distinguish between alternative alignments is contained in the detailed atomic interactions.
Although these models are not highly accurate, they nevertheless are useful for providing an overall idea of what a structure is like, helping choose residues for mutagenesis experiments, for example. They also often establish evolutionary relationships to more studied proteins, and so provide valuable approximate information about molecular function. Figure 2 shows an example from CASP6.
Figure 2.
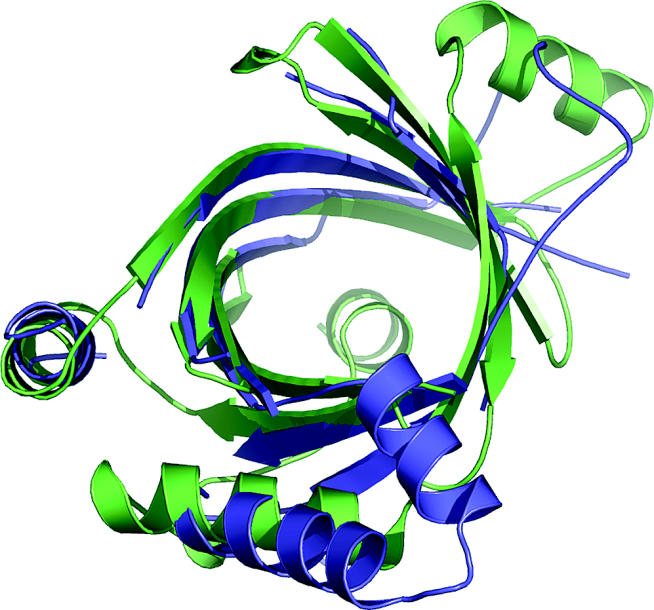
CASP6 Target 197, an example of comparative modelling based on a distant evolutionary relationship. The best model is blue, and the experimental structure (1xkc) is green. Accurately modelled regions of the beta barrel reflect available template information. Other regions, outside the beta barrel, have different conformations from the template, and are not accurately modelled. These structural features are likely related to detailed functional differences. In spite of limited accuracy, structure assisted recognition of these evolutionary relationships provides valuable information about function, in this case likely involvement in RNA editing.
6. Modelling of new folds
For proteins with folds that have not previously been found, and those where no relationship to a protein of known structure can be detected, a different set of methods are needed. Traditionally, this was the area where physics based approaches were used. These methods are still used by a few CASP participants, but have been largely displaced. Newer methods primarily utilize the fact that although we are far from observing all folds used in biology (Coulson & Moult 2002), we probably have seen nearly all substructures (Du et al. 2003). Methods make use of these partial structure relationships on a range of scales (Bystroff et al. 2004), from a few residues (Rohl et al. 2004), through secondary structure units, to super-secondary units (Jones & McGuffin 2003). Structure fragments are chosen on the basis of compatibility of the substructure with the local target sequence and compatibility of secondary structure propensity. Since the sequence/structure relationship is rarely strong enough to completely determine the structure of fragments (Bystroff et al. 1996), a range of possible conformations for each fragment are usually selected, and many possible combinations of sub-structures considered. Initial structures are assembled from fragments, and approximate potentials are used to guide a conformational search process, together with other information, such as prediction of residue contacts (Aloy et al. 2003). A large number of possible complete structures (1000–100 000) are usually generated. The most successful package using this strategy is Rosetta (Rohl et al. 2004). For proteins of less than about 100 residues, these procedures may produce one or a few approximately correct structures (4–6 Å RMSD on Ca atoms). Selecting the most accurate structures from the large set of candidates is currently not a fully solved problem, and most methods rely on clustering procedures, selecting representative structures at the centre of the largest clusters of generated candidates (Skolnick et al. 2001). Reliable identification of accurate models will require the use of refined all-atom models. Thus, in this class of modelling too, the development of atomic level refinement methods is likely crucial to major progress.
In CASP1, all new fold models were close to random. There has been steady improvement over the CASPs, and by CASP6 most non-homology targets less than 100 residues have models that visual inspection shows to resemble experiment. An example is shown in figure 3. Models for larger proteins or domains are still rarely usefully accurate. Thus, while there is very impressive progress for small proteins, there is still a long way to go before all proteins can be modelled at that level. Also, although topologically pleasing, these models often have significant alignment and other errors. Nevertheless, progress over the decade of CASP has been very impressive.
Figure 3.
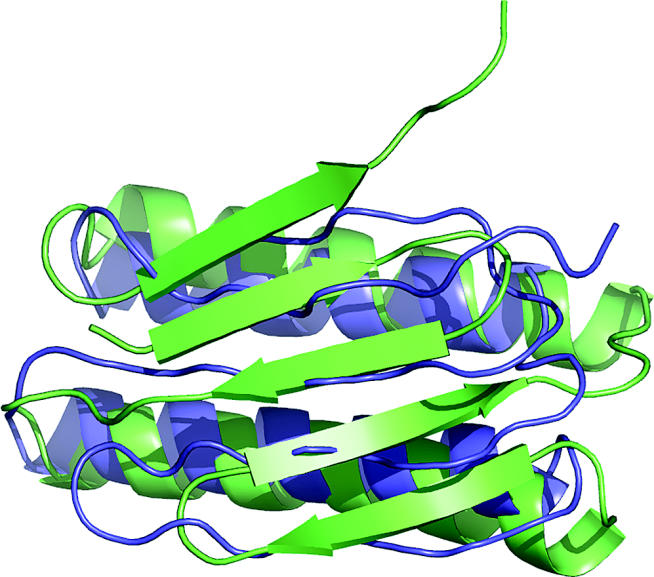
CASP6 Target 201, an example of modelling a previously unknown fold. The best model is blue, and the experimental structure (1s12) is green. The helical regions are accurately modelled, and the general features of the beta sheet are correct, although there is an error in the order of the strands, and the sheet is slightly mis-oriented. This quality of model is now often obtained for small structures.
7. Major current challenges
Overcoming four of the current major bottlenecks—producing close evolutionary relationship models approaching experimental accuracy, improved alignments, refinement of remote evolutionary relationship models, and reliable discrimination between possible template free models—depends on the development of effective all-atom structure refinement procedures. The ‘refinement’ problem has received increasing attention in recent years (http://www.nigms.nih.gov/psi/reports/comparative_modeling.html). At CASP6, for the first time, there was a report of an initial model refined from a backbone RMSD of about 2.2–1.6 Å, with many of the core side chains correctly oriented (Schueler-Furman et al. 2005). The same technology has been effective in protein design (Schueler-Furman et al. 2005), and in protein–protein docking (Schueler-Furman et al. 2005).
8. Lessons for computational biology
The practical and philosophical principles of experimental science evolved over hundreds of years, and have resulted in a system that ensures rigor and reproducibility. Experience in computational studies of protein structure suggests that these principles are not sufficient for computational modelling in biology. The fundamental difference is that modelling does not deal directly with the real world, instead creating some form of artificial reality. Additional steps are necessary to firmly establish the relationship between the artificial and real worlds. These steps are of two types. First, proper and appropriate statistical procedures must be used. In this respect, the computational biology field has become increasingly technically sophisticated in recent years. Second, care must be taken that the model does indeed represent the real world in all relevant respects. This latter issue has received less attention. The procedures outlined below, if widely adopted, will put computer modelling in biology on a par with the experimental work.
Bona fide predictions of experimental observations. Wherever possible, this mechanism should be used, rather than reproduction of known facts. Implementation requires that new experimental data be available on an appropriate time scale. CASP makes use of the high rate of release of new experimental structures, particularly those generated in structural genomics (http://www.nigms.nih.gov/psi/). CAPRI, a community protein–protein docking experiment (Janin 2005) makes use of new structures of complexes.
Bona fide prediction on test sets derived through human analysis. In areas where new experimental data cannot be used, it is some times possible to generate special test sets for bona fide prediction. This mechanism has been applied to genome sequence analysis (Reese et al. 2000). Human annotators examine a large set of data (genome sequence in this example), providing material that computational methods are then tested against.
Large test sets. Where reproduction of known information is the basis for testing, a large body of data produces more robust evaluation. Large test sets were rare in the early history of structure modelling. When they were used, for example in some cases of secondary structure prediction (Rost & Sander 1993), the results were reliable. The LiveBench system (Rychlewski & Fischer 2005) for evaluating protein structure modelling successfully incorporates this principle, encouraging participants to produce models of all newly released experimentally structures, and so accumulating large amounts of data.
Community agreed test sets. These can be developed in almost all areas of biological modelling. In CASP, participants agree to produce models of the same proteins, making methods comparison much easier. A more general example in the structure modelling field are decoy sets for testing protein structure discrimination methods, developed by a number of groups (http://dd.compbio.washington.edu/).
Independence of training and test sets. Parameterizing a method on the same data used for its evaluation will often lead to overestimates of accuracy, particularly where machine learning is employed. The principle of separate training and test sets is well established in statistics. It is appreciated in computational biology, but so far not always adhered to in practice.
Error estimates. In experimental science, provision of uncertainties in any measured quantity is considered mandatory. In computational work, including structure modelling, this is so far rare. There are striking exceptions, such the establishment of reliability estimates for interpreting DNA gels (Ewing & Green 1998). In this case, a reliability estimate played a critical role in developing high throughput sequence methods.
Independent tests of accuracy. All accuracy evaluation procedures have biases, so independent validation should be performed whenever possible. For example, when two unrelated methods have been developed, it is possible to validate by comparison. The specificity of the two methods predicts the fraction of cases where the two methods should agree, and the sensitivity predicts the expected fraction of all cases where at least one method should be correct.
Open results. All data associated with a method should be released, including full evaluation details and results, rather than just summaries. Ease of distributing information electronically has made this a practical procedure.
Open software. In experimental science, the principle of providing sufficient information to reproduce results has long been accepted and broadly adhered to. The equivalent in computational science includes release of software. There is considerable resistance to this, and it has not so far been possible in CASP. The primary reasons for non-release are protection of intellectual property and trade secrets, the resource commitment required to make software robust enough for distribution, or the dangers of abuse (unacknowledged use, or incorrect use leading to substandard results). These may be legitimate concerns, but without software in some form, it is impossible to rigorously check the performance of a method, and there is massive duplication of effort.
Acknowledgements
CASP is made possible by the participation of the prediction community, the generosity of the experimental community in making new structure information available, and the work of the assessment teams and the organizers. Details of the large number of people involved are available on the CASP web site (predictioncenter.org). CASP has been supported by grants from the National Library of Medicine (LM07085 to K. Fidelis), NIH R13GM/DK61967 (to J.M.) and R13GM072354 (to B. Rost).
Footnotes
One contribution of 15 to a Discussion Meeting Issue ‘Bioinformatics: from molecules to systems’.
References
- Aloy P, Stark A, Hadley C, Russell R.B. Predictions without templates: new folds, secondary structure, and contacts in CASP5. Proteins. 2003;53(Suppl. 6):436–456. doi: 10.1002/prot.10546. 10.1002/prot.10546 [DOI] [PubMed] [Google Scholar]
- Altschul S.F, Gish W, Miller W, Myers E.W, Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. 10.1006/jmbi.1990.9999 [DOI] [PubMed] [Google Scholar]
- Altschul S.F, Madden T.L, Schaffer A.A, Zhang J, Zhang Z, Miller W, Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An S, Musier-Forsyth K. Trans-editing of Cys-tRNAPro by Haemophilus influenzae YbaK protein. J. Biol. Chem. 2004;279:42 359–42 362. doi: 10.1074/jbc.C400304200. 10.1074/jbc.C400304200 [DOI] [PubMed] [Google Scholar]
- Bates P.A, Kelley L.A, MacCallum R.M, Sternberg M.J. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins. 2001;(Suppl. 5):39–46. doi: 10.1002/prot.1168. 10.1002/prot.1168 [DOI] [PubMed] [Google Scholar]
- Bystroff C, Simons K.T, Han K.F, Baker D. Local sequence–structure correlations in proteins. Curr. Opin. Biotechnol. 1996;7:417–421. doi: 10.1016/s0958-1669(96)80117-0. 10.1016/S0958-1669(96)80117-0 [DOI] [PubMed] [Google Scholar]
- Bystroff C, Shao Y, Yuan X. Five hierarchical levels of sequence–structure correlation in proteins. Appl. Bioinformatics. 2004;3:97–104. doi: 10.2165/00822942-200403020-00004. [DOI] [PubMed] [Google Scholar]
- Chung S.Y, Subbiah S. The use of side-chain packing methods in modeling bacteriophage repressor and cro proteins. Protein Sci. 1995;4:2300–2309. doi: 10.1002/pro.5560041107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson A.F, Moult J. A unifold, mesofold, and superfold model of protein fold use. Proteins. 2002;46:61–71. doi: 10.1002/prot.10011. 10.1002/prot.10011 [DOI] [PubMed] [Google Scholar]
- DeWeese-Scott C, Moult J. Molecular modeling of protein function regions. Proteins. 2004;55:942–961. doi: 10.1002/prot.10519. 10.1002/prot.10519 [DOI] [PubMed] [Google Scholar]
- Du P, Andrec M, Levy R.M. Have we seen all structures corresponding to short protein fragments in the Protein Data Bank? An update. Protein Eng. 2003;16:407–414. doi: 10.1093/protein/gzg052. 10.1093/protein/gzg052 [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Janin J. Assessing predictions of protein–protein interaction: the CAPRI experiment. Protein Sci. 2005;14:278–283. doi: 10.1110/ps.041081905. 10.1110/ps.041081905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D.T, McGuffin L.J. Assembling novel protein folds from super-secondary structural fragments. Proteins. 2003;53(Suppl. 6):480–485. doi: 10.1002/prot.10542. 10.1002/prot.10542 [DOI] [PubMed] [Google Scholar]
- Kahsay R.Y, Wang G, Gao G, Liao L, Dunbrack R. Quasi-consensus based comparison of profile hidden Markov models for protein sequences. Bioinformatics. 2005 doi: 10.1093/bioinformatics/bti374. [DOI] [PubMed] [Google Scholar]
- Karplus K, Hu B. Evaluation of protein multiple alignments by SAM-T99 using the BAliBASE multiple alignment test set. Bioinformatics. 2001;17:713–720. doi: 10.1093/bioinformatics/17.8.713. 10.1093/bioinformatics/17.8.713 [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. 10.1093/bioinformatics/14.10.846 [DOI] [PubMed] [Google Scholar]
- Karplus K, Karchin R, Draper J, Casper J, Mandel-Gutfreund Y, Diekhans M, Hughey R. Combining local-structure, fold-recognition, and new fold methods for protein structure prediction. Proteins. 2003;53(Suppl. 6):491–496. doi: 10.1002/prot.10540. 10.1002/prot.10540 [DOI] [PubMed] [Google Scholar]
- Kinch L.N, Wrabl J.O, Krishna S.S, Majumdar I, Sadreyev R.I, Qi Y, Pei J, Cheng H, Grishin N.V. CASP5 assessment of fold recognition target predictions. Proteins. 2003;53(Suppl. 6):395–409. doi: 10.1002/prot.10557. 10.1002/prot.10557 [DOI] [PubMed] [Google Scholar]
- Marti-Renom M.A, Madhusudhan M.S, Sali A. Alignment of protein sequences by their profiles. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. 10.1110/ps.03379804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J, Jones D.T. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. 10.1093/bioinformatics/btg097 [DOI] [PubMed] [Google Scholar]
- Moult J, Fidelis K, Zemla A, Hubbard T. Critical assessment of methods of protein structure prediction (CASP)-round V. Proteins. 2003;53(Suppl. 6):334–339. doi: 10.1002/prot.10556. 10.1002/prot.10556 [DOI] [PubMed] [Google Scholar]
- Przybylski D, Rost B. Improving fold recognition without folds. J. Mol. Biol. 2004;341:255–269. doi: 10.1016/j.jmb.2004.05.041. 10.1016/j.jmb.2004.05.041 [DOI] [PubMed] [Google Scholar]
- Reese M.G, Hartzell G, Harris N.L, Ohler U, Abril J.F, Lewis S.E. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. 10.1101/gr.10.4.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C.A, Strauss C.E, Misura K.M, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Rost B, Sander C. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 1993;232:584–599. doi: 10.1006/jmbi.1993.1413. 10.1006/jmbi.1993.1413 [DOI] [PubMed] [Google Scholar]
- Rychlewski L, Fischer D. LiveBench-8: the large-scale, continuous assessment of automated protein structure prediction. Protein Sci. 2005;14:240–245. doi: 10.1110/ps.04888805. 10.1110/ps.04888805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Progress in modeling of protein structures and interactions. Science. 2005;310:638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- Sippl M.J. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. 10.1002/prot.340170404 [DOI] [PubMed] [Google Scholar]
- Skolnick J, Kolinski A, Kihara D, Betancourt M, Rotkiewicz P, Boniecki M. Ab initio protein structure prediction via a combination of threading, lattice folding, clustering, and structure refinement. Proteins. 2001;53(Suppl. 6):149–156. doi: 10.1002/prot.1172. 10.1002/prot.1172 [DOI] [PubMed] [Google Scholar]
- Tramontano A, Morea V. Assessment of homology-based predictions in CASP5. Proteins. 2003;53(Suppl. 6):352–368. doi: 10.1002/prot.10543. 10.1002/prot.10543 [DOI] [PubMed] [Google Scholar]
- Venclovas C. Comparative modeling in CASP5: progress is evident, but alignment errors remain a significant hindrance. Proteins. 2003;53(Suppl. 6):380–388. doi: 10.1002/prot.10591. 10.1002/prot.10591 [DOI] [PubMed] [Google Scholar]
- Venclovas C, Zemla A, Fidelis K, Moult J. Assessment of progress over the CASP experiments. Proteins. 2003;53(Suppl. 6):585–595. doi: 10.1002/prot.10530. 10.1002/prot.10530 [DOI] [PubMed] [Google Scholar]
- von Grotthuss M, Pas J, Wyrwicz L, Ginalski K, Rychlewski L. Application of 3D-Jury, GRDB, and Verify3D in fold recognition. Proteins. 2003;53(Suppl. 6):418–423. doi: 10.1002/prot.10547. 10.1002/prot.10547 [DOI] [PubMed] [Google Scholar]
- Wrabl J.O, Grishin N.V. Gaps in structurally similar proteins: towards improvement of multiple sequence alignment. Proteins. 2004;54:71–87. doi: 10.1002/prot.10508. 10.1002/prot.10508 [DOI] [PubMed] [Google Scholar]
- Zemla A, Venclovas, Moult J, Fidelis K. Processing and evaluation of predictions in CASP4. Proteins. 2001;(Suppl. 5):13–21. doi: 10.1002/prot.10052. 10.1002/prot.10052 [DOI] [PubMed] [Google Scholar]