

Abstract
I would like to thank the Royal Society for inviting me to deliver the Croonian Lecture. In so doing, the Society is adding my name to a list of very distinguished scientists who, since 1738, have preceded me in this task. This is, indeed, a great honour.
For most of my research career my main interest has been the understanding of the normal functioning of the blood vessel wall and the way this is affected in pathology. During this time, our knowledge of these subjects has grown to such an extent that many people now believe that the conquering of vascular disease is a real possibility in the foreseeable future.
My lecture concerns the discovery of two substances, prostacyclin and nitric oxide. I would like to describe the moments of insight and some of the critical experiments that contributed significantly to the uncovering of their roles in vascular biology. The process was often adventurous, hence the title of this lecture. It is the excitement of the adventure that I would like to convey in the text that follows.
Keywords: prostacyclin, aspirin, nitric oxide, oxidative stress, free radicals, cardiovascular pathology
1. The mechanism of action of aspirin-like drugs
When I arrived at the Department of Pharmacology at the Royal College of Surgeons in England in early 1971 to do my PhD under the supervision of John Vane, the laboratory had three notable characteristics. These were the widespread use of the bioassay cascade technique as described by John (Vane 1964), a clearly focused interest in the potential metabolic function of the lungs (Piper et al. 1970), and a highly interactive and intellectually stimulating atmosphere where discussion of science was actively encouraged. It was an exciting time; the idea that aspirin-like drugs might be inhibiting prostaglandin biosynthesis had been taking shape and experiments were being carried out to investigate this hypothesis.
My introduction to the bioassay technique was to learn to prepare the bioassay tissues normally used for the detection of the inflammatory mediators known as prostaglandins (Vane 1964), to superfuse their biological precursor (arachidonic acid) over the tissues and to see whether the responses could be blocked by either aspirin or indomethacin. The results showed that the responses to arachidonic acid were indeed inhibited by both anti-inflammatory compounds. At the same time, other experiments in the laboratory were also showing that aspirin and indomethacin were inhibiting the generation of prostaglandins in lung homogenates.
A few weeks into the project I was asked by John to abandon those bioassay experiments and to join him and Sergio Ferreira in a project using the spleen of the dog in vivo to investigate the release of prostaglandins by catecholamines and to look for inhibition of this release by aspirin-like drugs. The experiments were successful and, together with those carried out on lung homogenates, resulted in two of the landmark papers on the mechanism of action of aspirin-like drugs (Ferreira et al. 1971; Vane 1971). A third paper, published at the same time by Brian Smith & Jim Willis, showed that the generation of prostaglandins by platelets obtained from humans was inhibited following the ingestion of aspirin (Smith & Willis 1971).
These findings have had very significant consequences and ramifications which continue to unfold after more than 30 years. For me, as a PhD student, they represented the starting point of a fascinating three years of hard work and excitement. Besides investigating the mechanisms by which prostaglandins modulate sympathetic neurotransmission (Ferreira & Moncada 1971; Ferreira et al. 1973), Sergio and I started to collaborate on a project dedicated to understanding the way in which prostaglandins participate in pain and inflammation. This led to the finding that prostaglandins, instead of behaving like the classical mediators of the inflammatory process such as bradykinin or histamine, were acting as modulators of the actions of these mediators. This was first shown in experiments on pain and later on oedema formation (see Ferreira 1972; Moncada et al. 1973). The latter experiments were published at the same time as results from Williams & Morley (1973), who essentially described the same phenomenon but in relation to vascular permeability. Thus, prostaglandins were a kind of amplification system which was removed when aspirin or its congeners were administered.
In the early 1970s, our knowledge of the pathway of biotransformation of arachidonic acid was scanty (see figure 1a), and the discovery of the mode of action of aspirin-like drugs simply indicated that they inhibit the generation of the E and F-type prostaglandins. These had been identified in inflammatory exudates (Willis 1969; Di Rosa et al. 1971) and were able to reproduce inflammatory signs and symptoms, including fever, when injected into animals or man (Juhlin & Michaelsson 1969; Crunkhorn & Willis 1971). Since E-type prostaglandins were also known to be released from the stomach (Bennett et al. 1967) and aspirin-like drugs all induce gastrointestinal symptoms, including peptic ulceration, it was proposed (Vane 1971) that prostaglandin synthesis somehow protects the mucosa from damage and that this protective mechanism is removed by aspirin-like drugs.
Figure 1.

Metabolic pathway of arachidonic acid as it was known in (a) 1971, (b) 1975 and (c) 1976.
There was, however, an effect that could not be explained at the time and this related to the fact that, from its very early use, aspirin was known to cause a bleeding disorder (Binz 1891). Over the years this clinical observation was made repeatedly (Singer 1945), leading to the suggestion by Craven (1953) that aspirin might be used for the prevention of thrombosis. The paper by Craven is a meticulous description of clinical observations with remarkable insight into the potential therapeutic value of this compound. It was presented to the Mississippi Valley Medical Society and received the third prize in an essay contest. I have not found out which essays were considered to be better than this one and wonder whether either of them was as clear or prophetic as that of Craven.
The solution to the mystery of the action of aspirin on bleeding came from discoveries made between 1973 and 1975 on the unstable metabolites of arachidonic acid. A cyclic endoperoxide intermediate in the synthesis of prostaglandins had been proposed as early as 1967 (Nugteren et al. 1967; Samuelsson et al. 1967). A few years later such a compound was isolated from incubations of arachidonic acid with sheep vesicular glands (Hamberg & Samuelsson 1973; Nugteren & Hazelhof 1973). The enzyme responsible for the synthesis of prostaglandins, known as cyclo-oxygenase (COX), has in effect two enzymic activities, the COX proper which cyclizes and introduces oxygen into the unesterified precursor fatty acid to form prostaglandin G2 (PGG2) and is the one blocked by aspirin-like drugs, and a peroxidase activity which reduces PGG2 to PGH2. These cyclic endoperoxides are unstable and are further converted into different products by a variety of enzymes (figure 1b). Furthermore, they were shown to be released during platelet aggregation (Hamberg et al. 1974; Smith et al. 1974; Willis et al. 1974; Svensson et al. 1975) and to induce this process at low concentrations (Hamberg et al. 1974; Willis et al. 1974; Svensson et al. 1975). Another property of these compounds was their ability to contract an isolated strip of rabbit aorta, thus mimicking the action of a mysterious unstable substance described a few years earlier by Piper & Vane (1969), the release of which during anaphylactic shock was inhibited by aspirin-like drugs. They named this substance Rabbit Aorta-Contracting Substance or RCS. Since the release of RCS from lungs could also be observed following an infusion of arachidonic acid, it was suggested to be an intermediate in the biotransformation of arachidonic acid (Vargaftig & Dao 1971).
In 1975, a new labile intermediate formed during the conversion of PGG2 into its stable hemiacetal derivative named thromboxane B2 was discovered. The intermediate, named thromboxane A2 (TXA2), was found to be even more unstable than the prostaglandin endoperoxides and a more potent inducer of platelet aggregation and contractor of the rabbit aortic strip (Hamberg et al. 1975). It was postulated that this compound, which was also generated during platelet aggregation, accounted for the activity of RCS. However, it remained unclear whether RCS comprised TXA2 alone, a mixture of TXA2 and prostaglandin endoperoxides, or, in some cases, only the latter.
In the summer of 1975, following Samuelsson's presentation of the above data on TXA2 at an International Prostaglandin Conference in Florence, we decided to investigate further the conversion of PGG2 into TXA2. By this time both John Vane and I had moved to the Wellcome Research Laboratories in Beckenham. The project was carried out with Stuart Bunting, who was a sandwich student doing a BSc at Chelsea College, and we were joined by Phil Needleman who was on sabbatical leave in our laboratory. Those studies, which were possible due to the availability of prostaglandin endoperoxides from Samuelsson's laboratory, led us to the identification in platelet ‘microsomes’ of an enzyme (Needleman et al. 1976), which we later called thromboxane synthase, involved in the conversion of endoperoxides into the more potent TXA2 (figures 2 and 3). We found this enzyme in platelets (Needleman et al. 1976), lungs (Nijkamp et al. 1976; Gryglewski et al. 1976) and polymorphonuclear leukocytes (Higgs et al. 1976). These studies identified the differences in the pharmacological behaviour of TXA2 and prostaglandin endoperoxides and confirmed that the main product of arachidonic acid in platelets was TXA2. This further clarified the mechanism by which aspirin might exert its anti-thrombotic action. Other consequences of this work were the identification of the first series of inhibitors of the enzyme thromboxane synthase (Moncada et al. 1977a) and our gaining of a great deal of experience in developing bioassay techniques for unstable substances such as TXA2, whose half life was approximately 30 s. This experience was going to be crucial for much of the work that I pursued over the next 20 years.
Figure 2.
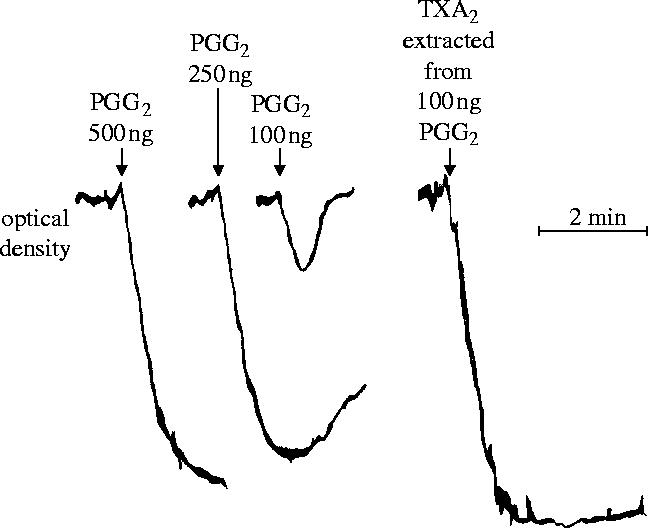
Aggregation of indomethacin-treated human platelet-rich plasma induced by prostaglandin endoperoxide (PGG2) and by thromboxane A2 (TXA2). TXA2 is far more potent than PGG2.
Figure 3.
Differential bioassay of rabbit aorta-contracting substances. PGG2 (200 ng) was added to 500 μl of Tris buffer at 0 °C and a 50 μl sample (equivalent to 20 ng) was tested on the bioassay tissues. Immediately after testing, horse platelet microsomes were added to the PGG2 solution and 50 μl was tested 2 min later. The responses to a high dose of PGG2 and to PGE2 are also shown for comparison. Reprinted, with permission, from Needleman et al. (1976).
2. The discovery of prostacyclin
The discovery of a novel prostaglandin (PGX), which we later called prostacyclin, came about from our interest in ‘mapping’ the distribution of thromboxane synthase in the body, more specifically, from my own interest in some unrelated experiments which were being carried out in our laboratory. Measurements of cutaneous bleeding time in the rat tail were proving difficult not only because the readings were erratic but mostly because there was sometimes an almost immediate stoppage of bleeding. It was as if a vasospastic phenomenon was aiding the platelet aggregation which is responsible for the formation of the haemostatic plug and which should normally take place about 3 min following a cut. This, and my reading of an article by Morrison & Baldini (1969) suggesting that platelets and vascular endothelium share some common proteins, gave me the idea that we should look for TXA2 generation in the vasculature. When Richard Gryglewski joined us soon after for a short stay in Beckenham, I suggested that we should undertake such a project, again with Stuart Bunting, who by then had become an expert in the methodologies required to carry out this work.
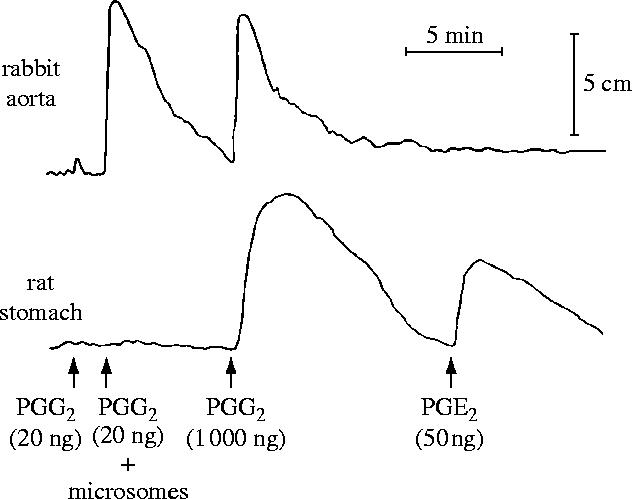
The initial experiments were frustrating for, although other tissues generated TXA2 as well as prostaglandins from the endoperoxides, the vessel wall was not making anything that we could detect by the usual bioassay tissues. In fact, it looked totally inert. It was Stuart Bunting who one day noticed that, although there was no detectable product, something unusual was happening to the activity of the prostaglandin endoperoxides. When he showed me the tracing of the experiment he had just carried out we came to the conclusion that the endoperoxide seemed to be disappearing on incubation with the vascular preparation (figure 4). I suggested two experiments: one was to boil the vascular wall preparation to see whether the disappearance of the vasoconstrictor activity of the prostaglandin endoperoxide on incubation was indeed enzymatic, and the other was to incubate PGE2 with our vessel wall preparation in case this compound was being made but also being metabolized to an inactive end-product. By the following morning we knew that we had most probably discovered a new product of arachidonic acid, which we started to call prostaglandin X (PGX).
Figure 4.

Conversion of PGG2 by aortic microsomes. In a bioassay using (a) rabbit aortic strip and (b) rat colon, PGE2 and PGF2α contracted rat colon, whereas PGG2 contracted rabbit aorta. The spontaneous disappearance of PGG2 following incubation for the times indicated resulted in the appearance of PGE- and PGF-like activity. In the presence of aortic microsomes (AM), however, the contractile activity of PGG2 disappeared within 0.5 min but no PGE- or PGF-like activity was formed, even after 20 min. This did not occur when the AM were boiled. Reprinted, with permission, from Moncada et al. (1976a).
For the next few weeks we carried out many experiments trying to elucidate what this mysterious substance might be. One possibility was that it was 12-hydroxy-5,8,10-heptadecatrienoic acid (HHT), another product of the breakdown of prostaglandin endoperoxides; however, experiments comparing PGX with HHT showed that they were different. The situation was, in addition, bedevilled by the fact that thin-layer radiochromatography of the products of the conversion of arachidonic acid in the vasculature yielded a product with an approximate Rf of 0.48, which was very similar to that of PGE2 (Rf=0.51). There was, however, a glimmer of hope and an indication that PGX was different from PGE2 and might have some vasodilator properties not shared by the latter. This came from studies on strips of bovine coronary arteries, which Stuart and I had been working with and which contracted to PGE2 while they clearly relaxed to PGX (figure 5); furthermore, and very importantly, while PGE2 was a stable compound, PGX was unstable in physiological solutions. So we were in the situation in which we had found a new unstable vasodilator derived from arachidonic acid in the vasculature and no clue as to where exactly to go next.
Figure 5.
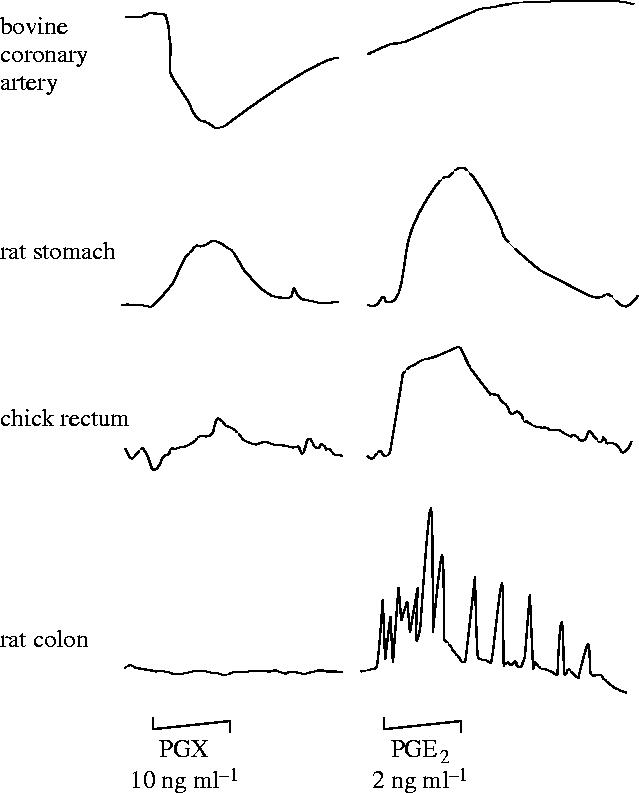
Differential bioassay of prostacyclin (PGX) and PGE2. The tissues were arranged in a cascade, one above the other, and PGX (10 ng ml−1) and PGE2 (2 ng ml−1) were infused over the tissues for 1 min. PGX, but not PGE2, caused a relaxation of the bovine coronary artery.
The breakthrough came during a casual conversation with Richard Gryglewski and Stuart Bunting when it occurred to me that since we had been looking for TXA2, a constrictor and pro-aggregating substance, and we had found a vasodilator, perhaps the vessel wall had a system to oppose TXA2, and PGX might also have platelet anti-aggregating properties. It sounded far-fetched; nevertheless that same afternoon Stuart and I carried out the first experiment which demonstrated the powerful anti-aggregating effect of PGX (figure 6; Moncada et al. 1976a) and opened the door to most of what happened in our laboratory over the following years.
Figure 6.
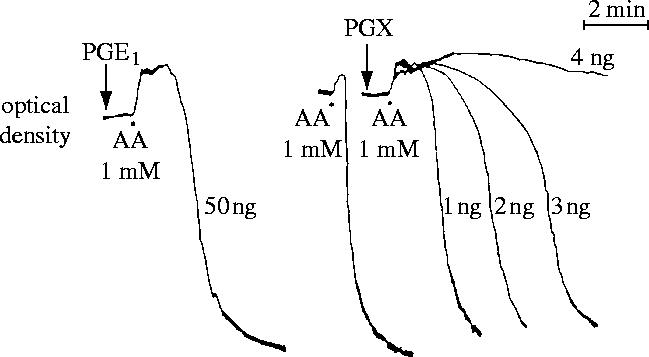
Comparison of the anti-aggregatory potencies of prostacyclin (PGX) and PGE1. PGX was obtained by incubation of 100 ng PGH2 with 500 μg of aortic microsomes in 100 μl of 0.05 M Tris buffer for 2 min at 22 °C and then stored on ice. PGX and PGE1 were added to human platelet-rich plasma 1 min before the platelets were aggregated with arachidonic acid (AA, 1 mM). In this experiment, PGX is at least 25 times more potent as an anti-aggregating agent than PGE1. Reprinted, with permission, from Moncada et al. (1976a).
We were, however, still no closer to the actual identity of this newly discovered pathway until John Vane and I attended the Prostaglandin Conference in Vail, Colorado in 1976 and heard Cecil Pace-Asciak presenting data about a compound he had identified in the rat stomach derived from arachidonic acid, namely 6-oxo-PGF1α (Pace-Asciak & Wolfe 1971). On hearing the talk we agreed that it would be worth investigating whether this had anything to do with our work. We contacted Richard and Stuart, who had stayed behind in the UK, and suggested that they make a homogenate of rat stomach to see whether they could find in it an unstable intermediate which would inhibit platelet aggregation with the characteristics of PGX. Within a couple of days we knew not only that our PGX was most probably an intermediate in the pathway to 6-oxo-PGF1α, but also that it was present in the rat stomach!
The identification of the structure of PGX was the result of a collaboration with the Upjohn Company, at that time the main company dedicated to prostaglandin research. This collaboration proved to be very successful since it led to the elucidation soon afterwards of the structure of PGX, which we then named prostacyclin (PGI2) (Johnson et al. 1976). We announced the structure of prostacyclin at a meeting in Santa Monica, California on the 3rd December 1976. Shortly afterwards we demonstrated that human arterial and venous tissues generate prostacyclin both spontaneously and from arachidonic acid (Moncada et al. 1977b). We also showed that the actions of prostacyclin are mediated via activation of adenylate cyclase and the subsequent increase in cyclic AMP concentrations (Tateson et al. 1977). Prostacyclin has turned out to play a variety of roles in the cardiovascular system beyond its vasodilator and anti-platelet-aggregating effects, the discussion of which is beyond the scope of this review (see Moncada & Vane 1979).
We, at Wellcome, made the decision to develop prostacyclin as a medicine and also to initiate a programme of drug discovery in collaboration with the Upjohn Company. This led to the synthesis and testing of about 1000 compounds, looking for a stable, hopefully orally active, anti-thrombotic and gastroprotective agent. Of all of those, to my knowledge only one, treprostinil sodium, has been developed successfully and is marketed as Remodulin, a stable analogue for subcutaneous or intravenous administration to patients with pulmonary arterial hypertension (Horn & Barst 2002). Prostacyclin has been used in cardiopulmonary operations and in transplant surgery, but its most widespread and life-saving use has been in patients with primary pulmonary hypertension where its efficacy or that of its analogues, as intravenous, oral or inhaled preparations, is now clearly established (Gibbs et al. 2004).
3. The homeostatic balance between prostacyclin and TXA2
The discovery of prostacyclin was of major significance to vascular biology because, among other things, it led to the concept that a balance between prostacyclin and TXA2 was important for vascular homeostasis and that disturbances of this balance might underlie some forms of vascular pathology (Moncada et al. 1976a). In addition, the pivotal role of the prostaglandin endoperoxides in relation to both pathways (see figure 1c) suggested the possibility that this balance could be manipulated.
Some of the experiments that we carried out to investigate this possibility included the demonstration that we could ‘feed’ authentic endoperoxides to aortic microsomes in a platelet suspension and inhibit platelet aggregation (Moncada et al. 1976b) and that indomethacin-treated arterial rings, when added to platelet-rich plasma were able to generate prostacyclin from the prostaglandin endoperoxides released by the aggregating platelets and so prevent their further aggregation (figure 7; Bunting et al. 1976).
Figure 7.
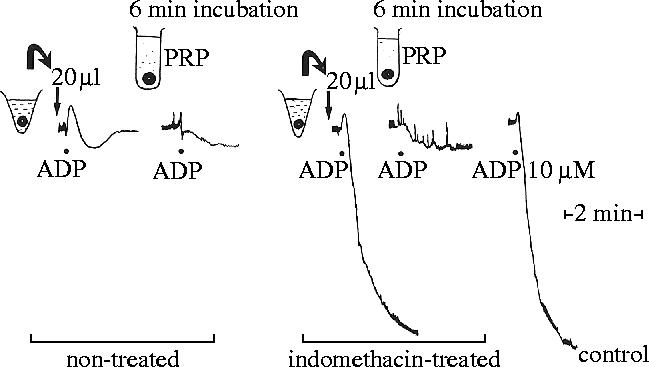
Inhibition of platelet aggregation by prostacyclin (PGX) generated from rings of mesenteric artery from indomethacin-treated and control rabbits. Cut rings of artery (6 mg) from a control rabbit were incubated in 1 ml Tris buffer for 3 min. An aliquot (20 μl) of the supernatant from the incubation mixture added to the platelet-rich plasma (PRP) inhibited the aggregation induced by adenosine diphosphate (ADP; 10 mM). Addition of 6 mg of the rings themselves also prevented platelet aggregation. Supernatant from rings of mesenteric artery (6 mg), removed from an indomethacin-treated rabbit and incubated in Tris buffer as above, did not inhibit platelet aggregation. However, when the rings were added to the PRP, ADP-induced aggregation was prevented. Thus, the rings were able to use endoperoxides generated by the platelet to synthesize PGX. Reprinted from Bunting et al. (1976). Copyright (1976) with permission from Elsevier.
The concept of the ‘balance’ was further strengthened by an entirely serendipitous finding made in the course of those studies. This was the observation that peroxides of arachidonic acid and other polyunsaturated fatty acids inhibit the enzyme, prostacyclin synthase, which converts the prostaglandin endoperoxides into prostacyclin. We discovered this as a result of a break of a few days in our laboratory work when we used a solution of arachidonic acid that had been left in the fridge, rather than a fresh solution, as a substrate for the generation of prostacyclin by vascular tissue. We observed that the tissues could not generate prostacyclin from this solution, nor were they then able to make prostacyclin from freshly prepared arachidonic acid. We surmised that the original solution, which was ‘somewhat yellow’ contained peroxides of arachidonic acid—something likely to happen when a polyunsaturated fatty acid is left in a bottle—and that those were responsible for the observed inhibitory action. This turned out to be correct when we prepared peroxides of arachidonic acid and they prevented the synthesis of prostacyclin (Moncada et al. 1976b; Gryglewski et al. 1976). These findings suggested that inhibition of prostacyclin synthase would not only inhibit the production of the anti-thrombotic prostacyclin but would also boost—by transfer of prostaglandin endoperoxides—the aggregation of platelets leading to a pro-thrombotic situation (Moncada et al. 1976b).
A significant clue to the way in which the balance might be operating in vascular disease came from studies which suggested that both prostacyclin and thromboxane metabolites in urine were increased in patients with unstable angina or atherosclerosis in which platelet activation was increased (FitzGerald et al. 1984; Fitzgerald et al. 1986). This led to the suggestion that the enhanced prostacyclin was a compensatory response initiated by the platelet aggregation, in which case the prostacyclin might be playing an active defence role. As I will discuss later, this is most probably the case in view of the overwhelming evidence indicating that atherosclerosis is an inflammatory disease (Ross 1999), in which case prostacyclin would be upregulated by both platelet aggregation and the underlying inflammatory process.
4. Therapeutic implications of the prostacyclin/TXA2 balance
Our studies led us to propose that thromboxane synthase inhibitors would not only inhibit thromboxane formation but would enhance the generation of prostacyclin, resulting in a superior anti-thrombotic situation (Moncada et al. 1976b; Moncada et al. 1977a). Such compounds were synthesized and investigations on their potential therapeutic effects were carried out. This led to the synthesis of thromboxane receptor antagonists, some of which would also possess thromboxane synthase inhibitory activity. Several of these compounds were developed and launched as drugs and others are still under clinical evaluation (Ishizuka et al. 2003; Dogne et al. 2004; Hanson et al. 2005). However, their potential superiority over aspirin has never been fully investigated due, at least in part, to the great success of the use of small doses of aspirin in anti-thrombotic therapy.
The finding that small doses of aspirin might be an effective anti-thrombotic treatment owes a great deal to research originating from the concept of the balance between prostacyclin and TXA2 (for review see Moncada & Amezcua 1979). The demonstration of the differential sensitivity of platelet versus vessel wall COX (Burch et al. 1978a,b), coupled with the earlier demonstration of the irreversible acetylation of platelet COX by aspirin (Roth & Majerus 1975) and the suggestion that high doses of aspirin might have pro-thrombotic actions (Kelton et al. 1978) led us to carry out a series of experiments both in animals (Korbut & Moncada 1978) and humans (O'Grady & Moncada 1978). In the latter, we measured cutaneous bleeding time and showed that low doses of aspirin are more effective than high doses in prolonging the bleeding time. Furthermore, we found that a high dose of aspirin would increase the bleeding time only after several hours, at which time the vessel wall COX had recovered and could generate prostacyclin while the platelets remained inhibited (Korbut & Moncada 1978; Amezcua et al. 1979). Following these discoveries the actions of aspirin were extensively studied and the effectiveness of low doses of aspirin established. Aspirin at low doses, i.e. 75 mg a day, blocks the platelet COX permanently and spares the vessel wall (Patrono et al. 1985; De Caterina et al. 1985). This differential effect is partly due to the fact that aspirin at low oral doses only reaches the pre-systemic circulation, where it slowly acetylates the platelet enzyme during repeated administration (Pedersen & FitzGerald 1984). The anti-thrombotic effect of low-dose aspirin has now been firmly established in a number of clinical trials (Worrall & Johnston 2000; Antithrombotic Trialists' Collaboration 2002; Landolfi et al. 2004; Ridker et al. 2005) and its prophylactic use is widespread.
Interestingly, further investigations on the way in which continuous administration of therapeutic doses of other non-steroidal anti-inflammatory drugs (NSAIDs) affect the balance between prostacyclin and TXA2 concentrations have never been carried out and as a result only very fragmentary information exists (e.g. Cullen et al. 1998; Capone et al. 2004). The possibility that drugs which lack the differential effect of aspirin on platelet and vessel wall COX might have a prothrombotic action has only been raised very recently (Hippisley-Cox & Coupland 2005) as a result of the problems related to the use of the newest type of NSAIDs, the coxibs, which I will now discuss.
In the early 1990s, a novel isoform of the COX enzyme was identified in cells such as monocytes and macrophages (Fu et al. 1990; Xie et al. 1991). This enzyme, which became known as cyclo-oxygenase 2 (COX-2), was initially thought to be expressed only during inflammation and to generate the prostaglandins that participate in this process. COX-2 therefore differed from the COX enzyme that was already known (COX-1), which was constitutive and generated prostaglandins involved in physiological mechanisms (Morita 2002). Based on this, it was suggested that selective inhibitors of COX-2, if developed, would have anti-inflammatory effects without the side effects of gastrointestinal ulceration, bleeding and platelet dysfunction which characterize the use of the classical NSAIDs and result from inhibition of the house-keeping enzyme COX-1 (see FitzGerald & Patrono 2001).
Over the next 10 years or so, a large effort was mounted by the pharmaceutical industry to develop such compounds, leading to the introduction in 1999 of the first selective COX-2 inhibitors, namely celecoxib and rofecoxib, followed by valdecoxib (FitzGerald & Patrono 2001) and others. The success of coxibs was so great that by the year 2000 they represented three-quarters of the value of total sales of NSAIDs in the USA (FitzGerald 2003). The compounds proved to possess analgesic and anti-inflammatory activity (see FitzGerald & Patrono 2001) and have a slightly reduced capacity to induce serious adverse gastrointestinal effects (Bombardier et al. 2000; Silverstein et al. 2000; Baigent & Patrono 2003); however, the VIGOR study suggested an increase in cardiovascular side effects (Bombardier et al. 2000; Mukherjee et al. 2001). This was later confirmed in a trial designed to investigate the hypothesis that COX-2 inhibitors could prevent recurrent colonic polyps (Bresalier et al. 2005) and in two other studies (Solomon et al. 2005; Nussmeier et al. 2005). These results led to the withdrawal of rofecoxib from the market. As a result there is significant debate in relation to the hypothesis which led to the development of these drugs, to the currently accepted design of clinical trials and to ethical considerations relating to modern marketing procedures of pharmaceuticals. This debate is still ongoing at the time of writing this manuscript (Frantz 2004; Usdin 2005; Drazen 2005).
The fact that diverse compounds from the same class show the same type of toxicity points to a class effect and thus to a common mechanism of action. This became clear from studies showing that oral administration of celecoxib to human volunteers substantially inhibited systemic prostacyclin biosynthesis while it only slightly reduced the synthesis of TXA2 and did not inhibit platelet aggregation (McAdam et al. 1999). Similar results were obtained using rofecoxib (Catella-Lawson et al. 1999). This led the authors to speculate that, in the vasculature, COX-2 was the predominant source of prostacyclin. Thus, inhibition of COX-2 might mediate a risk of thrombosis in predisposed individuals (see FitzGerald 2003).
The pharmacological and clinical evidence in favour of the TXA2/prostacyclin balance hypothesis in the cardiovascular system (Moncada et al. 1976a) had until that time come mainly from the selective inhibition of TXA2 in platelets by aspirin. However, it had not been established whether the anti-thrombotic effect of aspirin is entirely due to inhibition of TXA2 or whether some of it is attributable to the spared prostacyclin. If the cardiovascular side effects of the coxibs are due to inhibition of vascular prostacyclin, as now seems likely, this would provide definitive evidence that the latter plays an important role in vascular protection and that the balance hypothesis is correct. However, this leads to further questions. For example, the cardiovascular side effects of the coxibs have been observed in adults who might have other risk factors, and only following long-term administration. Thus, the ‘protection’ of the vessel wall seems to be provided by the larger amounts of prostacyclin generated by a COX-2 in relatively advanced disease. It is, therefore, still not clear how much of an independent risk factor in cardiovascular disease is the simple decrease or abolition of constitutive prostacyclin synthesis. In addition, it remains to be investigated in detail what is the status of a cardiovascular system in which both prostacyclin and TXA2 are absent, a situation which is likely to occur following continued administration of therapeutic doses of the majority of the classical NSAIDs (see table 1).
Table 1.
Effect of anti-inflammatory drug treatment on the balance between prostacyclin and TXA2.
| low-dose aspirin | more prostacyclin | less TXA2 |
| coxibs | less prostacyclin | more TXA2 |
| classical NSAIDs at therapeutic doses | less prostacyclin | less TXA2 |
5. The discovery of nitric oxide
In 1980, Furchgott & Zawadzki published results describing endothelium-dependent relaxation, a phenomenon whereby acetylcholine relaxes isolated preparations of blood vessels only if the vascular endothelium lining the vessels is undamaged (Furchgott & Zawadzki 1980). Subsequent studies revealed that acetylcholine and other agents release a transferable factor (endothelium-derived relaxing factor, EDRF) which is unstable, acts via stimulation of the soluble guanylate cyclase and is inhibited by haemoglobin and methylene blue (Furchgott et al. 1984). My interest in this area was stimulated in 1984 when Bob Furchgott visited our laboratories in Beckenham to request a lipoxygenase inhibitor, BW755C, which had been synthesized at Wellcome (Higgs et al. 1979). The hypothesis that EDRF might be a product of arachidonic acid metabolism via the lipoxygenase pathway (Singer & Peach 1983) was one of several at that time, thus a selective inhibitor of its synthesis would help to test this possibility. Nevertheless, within a few months Furchgott informed us that the experiments had not been successful and that EDRF must be something else.
By early 1985, we started to work on this ephemeral substance. Due to my previous experience, I was convinced that if more detailed quantitative pharmacology was to be carried out and if the structure of EDRF was to be elucidated we required bioassay tissues mounted in cascades, similar to those that had been used for the identification of thromboxane synthase and the discovery of prostacyclin. We therefore decided not to follow the main trend of comparing vascular strips with and without endothelium but instead proceeded to culture vascular endothelial cells on microcarrier beads, perfuse them inside a modified chromatography column and use the effluent to superfuse vascular tissues denuded of endothelium as a detection system for EDRF (figure 8). Angus and colleagues developed a similar system in which the effluent of a volume of endothelialized beads packed in the barrel of a syringe was used to perfuse a single arterial ring (Cocks et al. 1985). In order to carry out this work, I invited Richard Palmer, who had a great deal of experience working with cell cultures, to join me in developing such a system. I also invited Richard Gryglewski who was again visiting us for another sabbatical leave.
Figure 8.

Bioassay system used to detect the release of EDRF from endothelial cells. Porcine aortic endothelial cells were grown in culture on microcarrier beads (approx. 70 μm), which were packed into a modified chromatographic column. Inset shows an electronmicrograph of a bead covered in endothelial cells. The beads were perfused with Krebs buffer at 37 °C and the perfusate was allowed to flow over the bioassay tissues (four rabbit aortae denuded of endothelium). The time taken for the superfusate to reach the first tissue was 1 s and the gap between each subsequent tissue was 3 s. Reprinted, with permission, from Gryglewski et al. (1986a).
The combination of cell culture with a bioassay cascade was a success and its first use was for the online detection of the vasorelaxant activities of prostacyclin and EDRF (Gryglewski et al. 1986a). This had not been tried before and was the first comparative study of the two mediators, obviously linking our past with our present work. More importantly, perhaps, it led to two observations which, in the long run turned out to be significant; first, that superoxide (O2−) anions are involved in the inactivation of EDRF (Gryglewski et al. 1986b) and second, that several of the described inhibitors of EDRF were redox compounds which generated O2− in solution and this was the mechanism by which they inhibited EDRF (Moncada et al. 1986). The significance of the first observation was twofold; in the first instance, destruction of EDRF or NO by O2− was later used as a test for the identity between these two substances by both Furchgott (see Furchgott 1990) and ourselves (Palmer et al. 1987) and second, generation of O2− in the vasculature is now recognized as a key mechanism in the development of vascular dysfunction and disease. The second observation showed that all the claims about the nature of EDRF based on results with pharmacological inhibitors using an assigned mechanism of action were most probably wrong. All those compounds, independent of their pharmacological classification, shared a property, i.e. redox activity, which explained their activity as EDRF inhibitors/blockers. Furthermore, our experiments led us to suspect that EDRF might be a free radical and therefore focused the quest to elucidate its chemical structure.
In 1987, based on the observations that superoxide dismutase (SOD, which removes O2−) protected EDRF from rapid inactivation (Gryglewski et al. 1986b; Rubanyi & Vanhoutte 1986), that haemoglobin selectively inhibited EDRF (Furchgott et al. 1984) and a study of the characteristics of the transient relaxations of endothelium-denuded rings of rabbit aorta to ‘acidified’ inorganic nitrite solutions, Bob Furchgott proposed that EDRF might be nitric oxide (NO) at a symposium in Rochester, Minnesota on Mechanisms of Vasodilatation (Furchgott 1988). Lou Ignarro and colleagues also made a similar suggestion (Ignarro 1987; Vanhoutte 1987; Ignarro et al. 1988). I thought that the proposal sounded extremely interesting, since the possibility of NO being generated in mammals was unknown, let alone being synthesized for a specific biological purpose. I contacted Richard Palmer back in Beckenham and suggested that he obtained some authentic NO, tried to make an aqueous solution, and compared its actions on the bioassay tissues with those produced by the material that we were releasing from the vascular endothelial cells in culture. He obtained a bottle of NO from British Oxygen Corporation and within a few days it was evident to us that the two materials resembled each other in several pharmacological tests, including their half life down the bioassay cascade (figure 9), their stabilization by SOD and inhibition by haemoglobin (Palmer et al. 1987).
Figure 9.
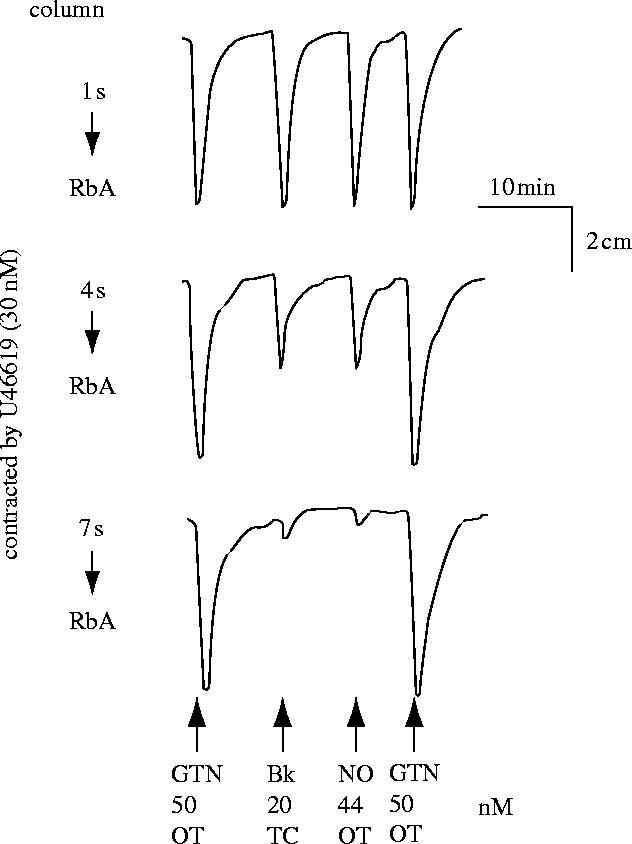
Relaxation of rabbit aortae by EDRF and NO. A column packed with endothelial cells cultured on microcarrier beads (as described in figure 8) was perfused with Krebs buffer. The effluent was used to perfuse three spiral strips of rabbit aorta denuded of endothelium (RbA) in a cascade. The tissues were pre-contracted submaximally. Glyceryl trinitrate (GTN) over the tissues (OT) was used to standardize the sensitivity of the tissues. EDRF was released from the cells by an infusion (1 min) through the column (TC) of bradykinin (Bk). Nitric oxide (NO) was administered OT as a 1 min infusion. Reprinted, with permission, from Palmer et al. (1987).
There were technical difficulties to be solved in the use of NO in biological experiments; these were related to the low solubility of NO in water and its reactivity with O2. Richard solved this problem by deoxygenating water by bubbling it with helium. The deoxygenated water was then used to dilute NO in glass containers into which NO was slowly added until no N2O4—a brown gas—could be seen. Those containers would keep the NO solution stable provided entry of O2 was prevented. The other difficulty was the bioassay of such an unstable substance, after all, EDRF had a much shorter half life than TXA2! Nevertheless, the experience I had accumulated in the prostacyclin/TXA2 project stood us in good stead.
The comparative pharmacology of EDRF/NO on vascular strips convinced us about the identity of EDRF; however, we were interested in measuring NO using methods other than bioassay. Although there are several chemical methods to measure nitrite (NO2−) or nitrate (NO3−), we wanted to measure not its breakdown products but NO itself as it was released from the cells following stimulation. We identified a potential method, used in the car industry and also in the food industry, based on a specific chemiluminescent signal which is generated when NO interacts with ozone. Using this technique, conveniently modified to detect very low quantities of NO, we demonstrated that NO was indeed generated from vascular endothelial cells when stimulated with bradykinin, an autacoid that produces endothelium-dependent relaxation. Furthermore, the quantities released were sufficient to account for the actions of EDRF (figure 10; Palmer et al. 1987). Our results were confirmed a few months later by Ignarro's group using different chemical reactions to identify NO (Ignarro et al. 1987).
Figure 10.

Detection of exogenous and endogenous NO. (a) Bioassay. The rabbit aorta was relaxed in a concentration-dependent manner by EDRF released from the endothelial cells by bradykinin (BK, TC) and by NO (OT), as in figure 9. (b) Chemiluminescence. EDRF was released by bradykinin from a replicate column of the cells used in the bioassay. The amounts of both EDRF (endogenously produced NO) and of exogenously applied authentic NO which relaxed the bioassay tissue were also detectable by chemiluminescence. Reprinted, with permission, from Palmer et al. (1987).
It had been known for some years that NO inhibits platelet aggregation (Mellion et al. 1981) and in 1986 it was shown that EDRF has platelet anti-aggregating properties (Azuma et al. 1986). Platelet studies were very significant in confirming the nature of EDRF. Comparative pharmacological studies between EDRF from vascular tissues and authentic NO demonstrated the resemblance between the two compounds in their actions on platelets (figure 11; Radomski et al. 1987a). Moreover, we examined the interactions between prostacyclin, EDRF and authentic NO on platelets and found that the anti-aggregating and the disaggregating effects of both EDRF and authentic NO were potentiated by sub-threshold concentrations of prostacyclin and vice versa (Radomski et al. 1987b). Furthermore, prostacyclin and NO released from vascular endothelial cells by bradykinin synergized with each other to inhibit platelet aggregation, an effect that could be blocked by treatment with indomethacin and partially reversed by treatment with haemoglobin.
Figure 11.

Anti-aggregatory action of EDRF (NO) and its potentiation by prostacyclin. (a) Sub-threshold concentrations of NO (0.1 μM) and (b) amounts of EDRF released from 0.5 ml of endothelial cells, treated with indomethacin and stimulated with bradykinin, are both potentiated by a sub-threshold concentration of prostacyclin (PGI2, 0.1 nM). The inhibition of aggregation induced by a combination of these sub-threshold concentrations of prostacyclin and NO was reversed by haemoglobin (Hb, 100 nM). Aggregation was induced by collagen (Coll); C represents control aggregation. Reprinted, with permission, from Radomski et al. (1987b).
The origin of NO remained a question of great interest and over the following months we carried out a variety of experiments to investigate this. There were a number of options, including the suggestion that NO2− or NO3− was reduced enzymically to NO or that ammonia was the biological precursor (Vanhoutte 1987). However, the most interesting possibility was that NO originated from the conversion of an amino acid. All these were tested without success so we decided to stop and concentrate on other things. A few weeks later, however, we came back to the project after realizing, first, that the endothelial cells were being grown in a medium which was rich in amino acids and were therefore swamped with them when we gave additional amounts in the bioassay cascade and, second, we came across two papers published a few months earlier, showing that activated macrophages generate NO2− and NO3− from the amino acid l-arginine (Hibbs et al. 1987; Iyengar et al. 1987). Hibbs et al. suggested that NO2− and NO3− were generated by the activity of a deiminase. However, we surmised that NO might be an unstable intermediate in the synthesis of the stable NO2− and NO3−; this was an attractive hypothesis reminiscent of the research into prostacyclin ten years earlier. We therefore prepared a culture medium without l-arginine—hard work in those days when you could not buy such things ready made—cultured the cells in it for the 24 h before the actual experiment, put them in our bioassay system, infused l-arginine, and found that, as a result, not only could we detect NO biologically but also chemically by chemiluminescence! We identified l-citrulline as the co-product of the generation of NO and published those results in 1988 (Palmer et al. 1988a).
There was an important additional finding associated with this work, namely the identification of NG-monomethyl-l-arginine (l-NMMA) as an inhibitor of NO generation and endothelium-dependent relaxation. The idea for this also came from the work of John Hibbs (Hibbs et al. 1987), who had shown that l-NMMA inhibits the generation of NO2− in macrophages. We found that l-NMMA does indeed inhibit endothelium-dependent relaxation and the release of NO (Palmer et al. 1988b). Over the next decade this compound became the most important pharmacological tool to investigate the presence and the roles of NO in biological systems. We also attempted to identify the enzyme responsible for the generation of NO and in 1989 identified NO synthase, able to generate NO and l-citrulline from l-arginine (Palmer & Moncada 1989). Soon afterwards we showed that the enzyme incorporated molecular oxygen into two steps of the process including the formation of l-citrulline (Leone et al. 1991). Those experiments were published after similar results by Kwon et al. (1990). At around this time we started calling this pathway the l-arginine : NO pathway (figure 12; Moncada et al. 1989).
Figure 12.
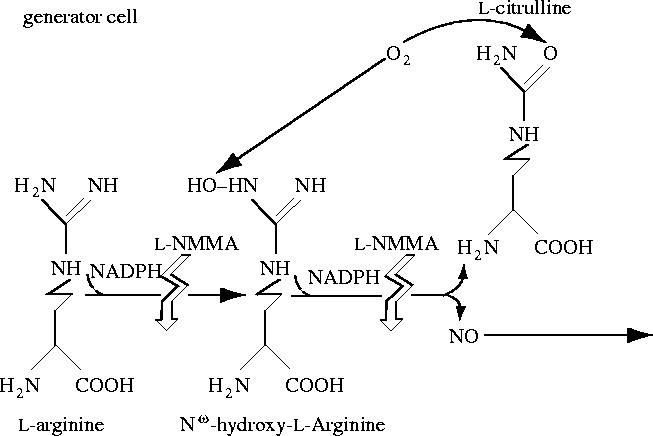
The l-arginine : nitric oxide pathway.
6. Physiological roles of the l-arginine : NO pathway
By 1987, it had become evident to us that the generation of NO was not only taking place in the vascular wall, but was probably a widespread mechanism with far-reaching biological significance. There was, of course, the work of Hibbs (Hibbs et al. 1987) and Marletta (Iyengar et al. 1987) which suggested that NO was formed in macrophages. In addition, an extensive literature search that I asked for on both l-arginine and NO showed an intriguing cross-reference between the two fields; in the late 1970s a group in Japan had found that brain homogenates contain a low molecular weight stimulator of the soluble guanylate cyclase which they later identified as l-arginine (Deguchi 1977; Deguchi & Yoshioka 1982). This work suggested the existence of the l-arginine : NO pathway in the central nervous system, where we then identified the NO synthase and determined its dependency on calcium (Knowles et al. 1989). This paper was preceded by a publication from John Garthwaite and his group demonstrating the release of EDRF from cerebellar cells following activation with N-methyl-d-aspartate (NMDA; Garthwaite et al. 1988). We later teamed up with John to demonstrate that NMDA receptor activation induces the synthesis of NO from l-arginine in rat brain slices (Garthwaite et al. 1989).
An additional area of research was created with the identification of NO as the inhibitory mediator of non-adrenergic, non-cholinergic (NANC) neurotransmission in peripheral nerves (Gibson & Mirzazadeh 1989; Gillespie et al. 1989; Li & Rand 1989; Ramagopal & Leighton 1989). This ‘nitrergic’ neurotransmission has been demonstrated in the gastrointestinal tract (Bult et al. 1990), where it is responsible for NANC-mediated relaxations of the gastric fundus (Li & Rand 1989) and effects adaptive relaxation in the stomach to increases in intragastric pressure (Desai et al. 1991), in the corpus cavernosum where it brings about relaxation of this tissue, resulting in penile erection (Ignarro et al. 1990; Holmquist et al. 1991), and in the trachea (Li & Rand 1991) and the bladder (Persson et al. 1992) where it contributes to NANC-induced relaxation. This area of research not only represents the discovery of a widespread system of peripheral nerves in the body, acting alongside the classical adrenergic and cholinergic systems, but has also led to the explanation of penile erection in animals and humans and its pharmacological manipulation and use in therapy (Cellek 2000). In this context, we have shown that selective degeneration of nitrergic nerves occurs in diabetes (Cellek et al. 1999), thus accounting for the erectile dysfunction and gastropathy of diabetes. Damage of these nerves seems to be the direct result of the endogenous NO interacting with oxygen-derived free radicals or their products, a subject which I will discuss below.
It is difficult to overstate what followed from this early work—an explosion of research into the investigation of the role of the l-arginine : NO pathway in every possible biological system. As I am writing, a review of the literature shows that since 1987 more than 32 000 papers have been written with NO in the title and there are more than 67 000 which in one way or another discuss NO. There are three main fields of research in this area, namely the cardiovascular system, the nervous system and inflammation/immunology. These are loosely based on the activity of three isoforms of the NO synthase, which were originally called endothelial and neuronal after the tissues in which they were first identified, and inducible for the isoform which is expressed in macrophages following immunological stimulation. These enzymes are also known as type III, II and I, respectively, and a great deal of structural and biochemical information has been generated about them. Inhibitors of the different isoforms have been developed and are being investigated for potential clinical use. All of that is beyond the scope of this review. There are very extensive reviews on all these subjects to which the reader is referred (Moncada et al. 1991; Alderton et al. 2001; Fleming & Busse 2004). I will concentrate here on some of the most significant actions of NO in the cardiovascular system and their potential relevance.
To my mind one of the most significant experiments we carried out in relation to the cardiovascular system was the demonstration that l-NMMA, when administered intravenously to an animal, causes an immediate increase in blood pressure (Rees et al. 1989a). This experiment originated from an observation made using isolated rings of aorta while we were blocking the acetylcholine-dependent relaxation with l-NMMA. We noticed that l-NMMA by itself was producing a dose-dependent, endothelium-dependent initial contraction of the vascular tissue, suggesting the abrogation of a continuous NO-dependent relaxing tone (Rees et al. 1989b and figure 13a). Therefore, I decided to investigate whether l-NMMA was also a constrictor of the coronary circulation in the isolated heart of the rabbit (Amezcua et al. 1989) and the demonstration of this effect led us to studies in the whole animal. Still, it was immensely surprising and satisfying to see the immediate elevation of blood pressure (figure 13b; Rees et al. 1989a). It is one of those single, simple experiments which revealed a fundamental mechanism and, because of this, is an experiment whose results, when seen with hindsight, look as if they were entirely predictable. However, my first presentation of these data to the British Society of Hypertension was followed by a question made by a senior researcher in the field saying ‘how do you think you have increased the blood pressure of these animals without affecting any of the important mechanisms that regulate blood pressure?’
Figure 13.
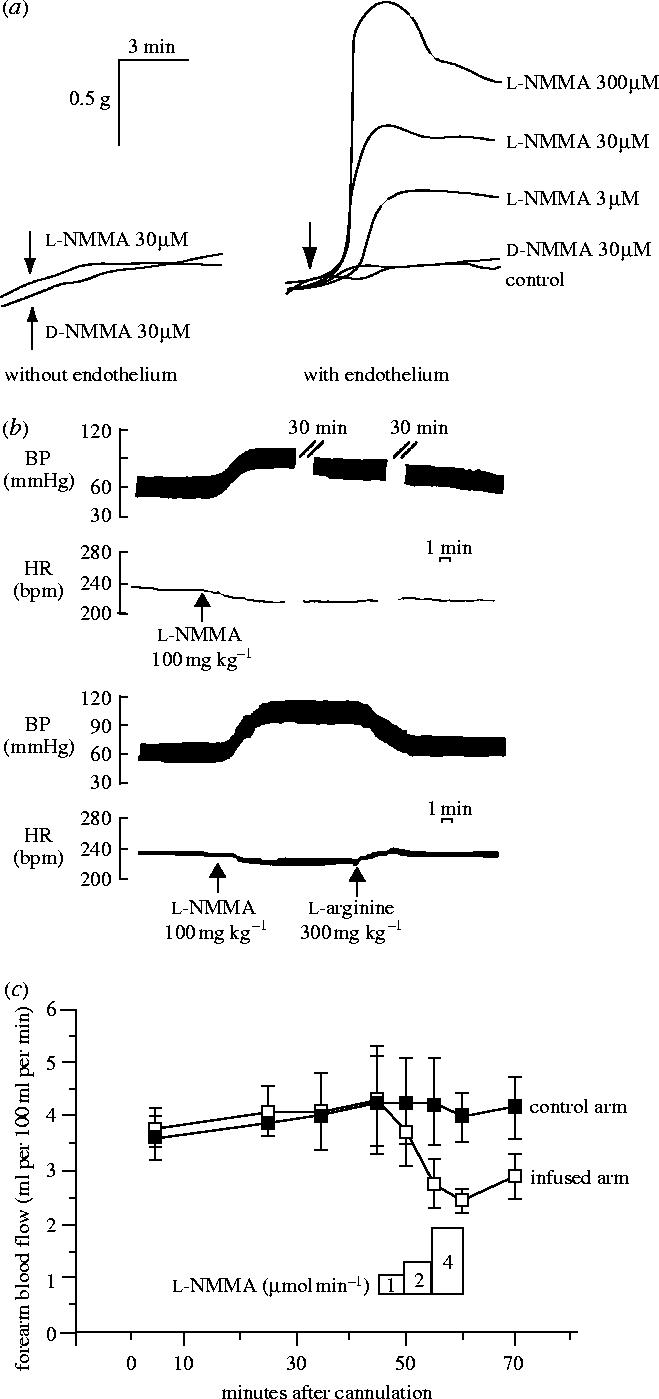
The effect of l-NMMA on vascular tone, blood pressure and blood flow. (a) The effect of NG-monomethyl l-arginine (l-NMMA) and its inactive isomer (d-NMMA) on the basal tone of pre-contracted rabbit aortic rings, with and without endothelium. (B) Long-lasting effect of l-NMMA on blood pressure in the anaesthetized rabbit. The lower trace shows the reversal of this effect by l-arginine. The heart rate is also shown. (c) Effect of l-NMMA on blood flow in the brachial artery in five healthy human subjects. Reprinted, with permission, from (a) Rees et al. (1989b), (b) Rees et al. (1989a) and (c) Vallance et al. (1989). Copyright (1989) with permission from Elsevier.
These experiments, indeed, turned our understanding of the mammalian cardiovascular system the right way round. Prior to our results, it had never been suspected that blood pressure regulation depended so much on a continuously generated local vasodilator tone, the lack of which would lead to such a significant vasoconstrictor response. Not even the discovery of EDRF and then NO could predict that the system operated in that way. This led us to suggest that conductance and not resistance was the main determinant of blood pressure regulation and that at least some forms of hypertension could be considered not as an increased resistance but as a decreased conductance of the system (figure 14). We carried out further experiments demonstrating a vasoconstrictor effect of l-NMMA in many vascular beds including the human forearm circulation (figure 13c; Vallance et al. 1989). Our findings were confirmed in many laboratories and later received ‘modern’ endorsement when it was demonstrated that a model of genetic manipulation in which the endothelial NO synthase (eNOS) is knocked out exhibits a hypertensive phenotype (Huang et al. 1995) and that over-expression of eNOS in endothelial cells reduces the blood pressure (Ohashi et al. 1998). The investigation into the role of NO in the regulation of physiological vascular tone and in hypertension continues to be one of the fastest expanding areas of research in the NO field. Judging from the extensive literature that exists on the subject I wonder whether the idea that NO-dependent vasodilatation might be the main factor regulating blood flow and pressure has indeed been accepted. For it is one thing to investigate how NO fits into a system in which resistance-inducing mechanisms are determinant and a very different one to investigate in which way resistance-inducing mechanisms affect the main NO-dependent vasodilator tone. The difference, although trivial in appearance, might be substantial for understanding how the cardiovascular system in mammals operates.
Figure 14.
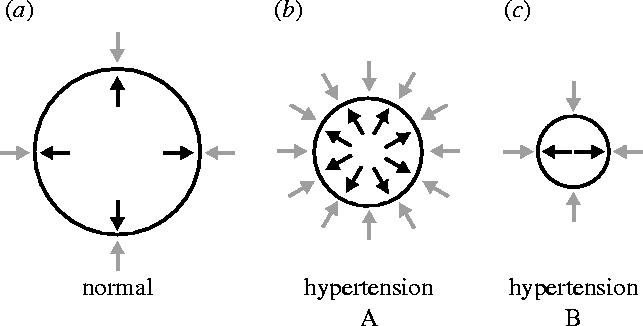
Schematic of two types of hypertension. In the normal situation, vasoconstrictor influences from outside the blood vessel (grey arrows) are counterbalanced by basal production of nitric oxide (black arrows) by endothelial NO synthase. Hypertension can occur in a situation in which vasoconstrictor activity is increased and more NO is generated in an attempt to compensate (Type A). In Type B, reduced synthesis of NO would result in hypertension even in the presence of normal amounts of vasoconstrictor activity. Modified and reprinted, with permission, from Rees et al. (2000).
7. Endothelial Dysfunction
In addition to its vasodilator and platelet inhibitory actions, NO was shown to inhibit vascular smooth muscle proliferation (Garg & Hassid 1989) and to regulate white cell/vessel wall interactions (Kubes et al. 1991). These findings, which suggested an uncanny resemblance to prostacyclin, established NO as a homeostatic regulator in the vasculature, the absence of which plays a role in a number of conditions and pathological states such as hypertension, atherosclerosis and vasospasm (for review see Moncada & Higgs 1993).
Interestingly, the early stages of a number of these conditions have in common a specific pathophysiological feature, namely endothelial dysfunction, an entity which has been investigated over the last 25 years (see Stemerman 1981; Luscher et al. 1993). Because of the variety of functions carried out by the vascular endothelium, endothelial dysfunction is likely to include a number of abnormalities both vascular and haemostatic. However, at present the accepted definition is that of a reduction in endothelial NO, which is measured as a decrease in endothelium-dependent vasodilatation induced either by appropriate agonists (Schachinger et al. 2000) or by flow (Neunteufl et al. 2000). Endothelial dysfunction described in this way occurs prior to any other evidence of cardiovascular disease and can be detected in subjects with a family history of essential hypertension or other risk factors for atherosclerosis (Reddy et al. 1994; Taddei et al. 1996). Furthermore, it has also been associated with smoking (Heitzer et al. 1996) and in general its presence is predictive of cardiovascular disease (for review see Asselbergs et al. 2005). Decreases in NO formation may result either from reduced expression of eNOS or from changes in its substrates or cofactors such as l-arginine or tetrahydrobiopterin (BH4). However, the most likely mechanism for endothelial dysfunction is that of a reduced bioavailability of NO as a result of its interactions with oxygen-derived species, specifically O2−.
Ever since the discovery of the interaction between NO and O2− in the mid-1980s (Gryglewski et al. 1986b; McCall et al. 1989) we have been suggesting that this might be a way in which the actions of NO are limited in disease (Moncada et al. 1991). The subsequent finding that the interaction between the two free radicals leads to the formation of powerful oxidant species including peroxynitrite (Beckman et al. 1990), followed by the identification of nitrated proteins associated with its generation in many tissues including the vasculature (Schopfer et al. 2003) added strength to this concept. Inactivation of NO by O2− is part of what has been called oxidative stress, a loose term used to describe various deleterious processes resulting from an imbalance between excessive formation of reactive oxygen species (ROS) (and/or the oxidants derived from NO) and limited antioxidant defences (Turrens 2003).
There is substantial evidence for increased ROS formation in vessels in disorders such as hypercholesterolaemia, diabetes and coronary artery disease (Guzik et al. 2000; Hink et al. 2001; Spiekermann et al. 2003), and also for the fact that treatment with antioxidants enhances endothelium-dependent vasodilatation in both the forearm and the coronary circulation of individuals with coronary artery disease and diabetes (Levine et al. 1996; Ting et al. 1996; Solzbach et al. 1997). Peroxynitrite has also been implicated in these conditions once they are established (see Brosnan 2004).
Since an early intervention may afford the greatest benefit in terms of preventing vascular disease, it is important to determine how early in the pathophysiological sequence of these conditions oxidative stress takes place and what is the involvement of inactivation of NO by O2−. Recent experiments suggest that O2− generation may indeed be involved in the genesis of hypertension induced by the powerful vasoconstrictor angiotensin II (Rajagopalan et al. 1996; Dzau 2001; Virdis & Schiffrin 2003). This adds support to the concept that increases in blood pressure and hypertension might in some cases be the result of a decrease in NO-dependent vasodilator tone rather than increases in vasoconstrictor activity. Moreover, it strongly suggests the possibility that a free radical-dependent breakdown of NO might in some cases be the pathophysiological basis of such conditions, a hypothesis further supported by a recent report showing that a transgenic mouse which generates more free radicals from mitochondria has a hypertensive phenotype which can be reversed by antioxidants (Bernal-Mizrachi et al. 2005).
There has been a great deal of research investigating the origin of O2− in the vasculature. So far, the activation of enzymes such as NADPH oxidases and xanthine oxidase has been implicated and substantial evidence now exists showing that the activity as well as the expression of these enzymes can be enhanced by pathological stimuli (see Cai & Harrison 2000; Mueller et al. 2005). In addition, vascular cytochrome P450 enzymes that can generate O2− have been described (Fleming 2001), and their inhibition appears to improve endothelium-dependent NO-mediated vasodilatation in patients with coronary artery disease (Fichtlscherer et al. 2004). Another potential source of O2− is what has been recognized as uncoupling the NO synthase, a situation in which eNOS has the capacity to generate O2− under the specific circumstances of low l-arginine or low BH4 (see Vasquez-Vivar et al. 1998; Stuehr et al. 2001). The uncoupling of eNOS has been reported to occur in several pathological conditions such as diabetes, hypercholesterolaemia and hypertension (Stroes et al. 1997; Hink et al. 2001; Landmesser et al. 2003). Uncoupling of eNOS due to depletion of both l-arginine and BH4, however is not likely to be an early mechanism of O2− generation, since the lowering of the substrate or the cofactor to critical levels probably requires drastic changes in the vasculature. However, the recent suggestion that uncoupling of the eNOS may result from subtle changes in the biochemical functioning of the enzyme is intriguing (Pritchard et al. 2001; Lin et al. 2003). Because of our interest in the early events, we have concentrated in the last few years on the possibility that an initiating step in pathophysiology may be the release of free radicals from mitochondria.
8. Nitric oxide, mitochondria and free radicals
In the mid-1990s, we and two other groups independently reported that NO inhibits the activity of the cytochrome c oxidase (complex IV), the terminal enzyme in the mitochondrial oxidative phosphorylation chain which catalyses the reduction of O2 to water (Cleeter et al. 1994; Brown & Cooper 1994; Schweizer & Richter 1994). The inhibitory effect was shown to be reversible, in competition with O2, and to occur at concentrations of NO likely to be present physiologically. Indeed, the affinity of the cytochrome c oxidase for NO is greater than that for O2, such that, for example, at 30 μM O2 (a physiological concentration of O2) the IC50 of NO is 30 nM (Brown & Cooper 1994). Later we demonstrated in vascular endothelial cells that endogenous concentrations of NO modulate cell respiration in an oxygen-dependent manner (figure 15; Clementi et al. 1999). This suggests that NO might, on the one hand, regulate cellular bioenergetics by regulating O2 consumption (Brown 1999; Clementi et al. 1999) and on the other, by inhibiting cytochrome c oxidase, decrease electron flux through the electron transport chain and favour the generation of O2− (Poderoso et al. 1996; Moncada & Erusalimsky 2002). It was further suggested that this mechanism might contribute to cell signalling through the subsequent formation of hydrogen peroxide (H2O2) (Moncada & Erusalimsky 2002). Increases in NO production were also shown to inhibit cellular respiration irreversibly (Cassina & Radi 1996; Lizasoain et al. 1996) by selectively inhibiting complex I through a process dependent on the increased generation of O2− and probably peroxynitrite (Clementi et al. 1998; Beltran et al. 2000).
Figure 15.
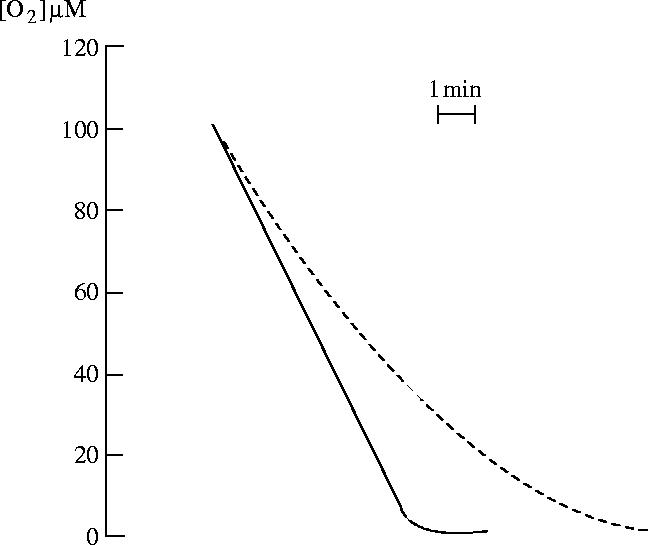
Effect of pretreatment with the NO synthase inhibitor l-NAME (NG-nitro-l-arginine methyl ester) on oxygen consumption by endothelial cells. The dotted line shows oxygen consumption by control cells, which were not treated with l-NAME. The solid trace shows that cells pretreated for 20 min with l-NAME respire at a constant rate at all oxygen concentrations. Modified and reprinted, with permission, from Clementi et al. (1999). Copyright (1999) National Academy of Sciences, USA.
For many years it has been believed that a small percentage of the O2 utilized by mitochondria is not completely reduced to water and escapes as O2− (Chance et al. 1979) and, although it is not clear whether this actually occurs in endothelial cells in vivo, at physiological O2 concentration there is the possibility that the redox status of the mitochondrial respiratory chain is a determinant in the escape of electrons required to generate O2− from O2. We have recently shown that NO, by favouring the reduction of the cytochrome c oxidase, facilitates the release of O2− from mitochondria (figure 16). This is subsequently converted into H2O2 with the resulting signalling consequences (Palacios-Callender et al. 2004). It is likely that such a mechanism, which is just an extension of the physiological action of NO on the cytochrome c oxidase, might provide clues to the understanding of the early origins of oxidative stress in the vasculature, specifically in endothelial cells. Of particular interest is the fact that, if indeed this is the case, there is no requirement for an independent mechanism for generation of O2−; instead, its generation is a consequence of an action of NO (Moncada & Erusalimsky 2002).
Figure 16.

Effect of nitric oxide on the redox state of the electron transport chain (ETC). (a,b) The change in RE (reducing equivalents; a measure of the reduction state of the ETC cytochromes) at different oxygen concentrations, in the presence and absence of l-NMMA. (a) The results in RAW 246.7 (murine monocytic) cells and (b) the results in HUVEC (human vascular endothelial cells). (c,d) The effect of NO on the production of superoxide anion (O2−). Fluorescence, measured by flow cytometry, indicates intracellular O2− production from intact cells incubated with DHE in the presence (shaded) or absence (clear) of l-NMMA at 21% oxygen (c) and 3% oxygen (d). (e) A Western blot of nuclear extracts of cells incubated at 21 and 3% oxygen detecting NFκB in the absence or presence of l-NMMA. Reprinted, with permission, from Palacios-Callender et al. (2004). Copyright (2004) National Academy of Sciences, USA.
A number of questions arise from all this work, such as the nature and function of this signalling mechanism activated by NO in mitochondria. Superoxide and its dismutation product H2O2 have for some time been recognized as signalling molecules acting on transcription factors which activate genes involved in defence mechanisms (Droge 2002). If their release occurs under physiological circumstances one wonders whether their main function is to maintain an ‘antioxidant tone’ in the cells, thus providing a general defence for the endothelial cells which exist in direct contact with the oxygen-carrying blood. Whether, as we and others have suggested, imbalances in this system such as high NO or low O2 are enough to initiate pathology (Poderoso et al. 1996; Beltran et al. 2000, 2002), requires further investigation. Specifically, it needs to be investigated whether increases in O2− or deficiencies in its normal scavenging mechanisms might be an early step in a sequence leading to the activation of other systems that generate O2− such as those described above (Li et al. 2001, 2003; Schafer et al. 2003).
In this context, we have also shown that NO, by regulating mitochondrial behaviour, plays other intriguing roles related to general homeostatic/defence mechanisms. These include the initiation of a Ca2+ related interaction between mitochondria and endoplasmic reticulum leading to the activation of grp78-dependent stress response (Xu et al. 2005) and the activation of a AMPkinase-dependent glycolytic response (Almeida et al. 2004). Both mechanisms might be relevant in the vasculature. Furthermore, we have recently shown that the inhibitory action of NO on O2 consumption in the mitochondria has the further consequence of diverting O2 away from the mitochondria to other areas of the cell and probably to surrounding tissues (Mateo et al. 2003; Hagen et al. 2003). Whether this is a mechanism utilized to divert O2 from the vascular endothelium, which is known to be glycolytic (see Mann et al. 2003), to the vascular smooth muscle which requires O2 is also unclear at present. A further finding in this area of research in which we are unravelling links between cell bioenergetics and signalling mechanisms is the observation that NO plays a role in mitochondriogenesis (Nisoli et al. 2003). This is an additional indication that NO might be involved in the regulation of the balance between glycolysis and oxidative phosphorylation in cells. Interestingly, this latter effect is not the result of NO interacting with the cytochrome c oxidase but, unexpectedly, with the soluble guanylate cyclase!
9. The two stories converge
Although the research fields of prostacyclin/thromboxane and NO are now mature, they have developed mostly as parallel research activities with few points of contact between them. Thus, our understanding of how both might operate in relation to each other in physiology and pathophysiology remains to be developed. Table 2 shows some of the similarities between prostacyclin and NO. Both mediators, from very different biochemical pathways, play a variety of roles in the modulation and protection of the vascular wall. The release of both mediators is dependent on constitutive enzymes, the activity of which seems to be regulated locally, predominantly by the shear stress caused by the blood passing over the endothelial surface (Grabowski et al. 1985; Frangos et al. 1985; for review see Boo & Jo 2003). However, while the constitutive eNOS—localized only in the vascular endothelium—is the enzyme that responds to shear stress, the generation of prostacyclin is dependent on the activity of two enzymes, COX-1 and COX-2, in relation to which several questions remain unanswered. These include whether COX-2 is a constitutive as well as an inducible enzyme, and whether COX-1 or COX-2, or both, respond to shear stress by increases in their mRNA, their activity, or both (Topper et al. 1996; Okahara et al. 1998; McCormick et al. 2000; Garcia-Cardena et al. 2001). Prostacyclin, unlike NO, is constitutively generated throughout the vessel wall (Moncada et al. 1977c) and at this stage we also do not know whether the ratio between COX-1 and COX-2 changes in the different layers. In addition, the similarities and differences between regulation of NO and prostacyclin by shear stress are only now being investigated (Osanai et al. 2000; McAllister et al. 2000; Walshe et al. 2005).
Table 2.
Comparison of the properties of nitric oxide and prostacyclin.
| NO | prostacyclin | |
|---|---|---|
| physiology | ||
| enzyme | eNOS | COX-1, COX-2 |
| main target | ↑cGMP/↓ cyt. c oxidase | ↑cAMP |
| shear stress | eNOS upregulated | COX-2 upregulated, COX-1 upregulated? |
| vasodilator | ✓ | ✓ |
| inhibition of platelet aggregation | ✓ | ✓ |
| inhibition of platelet adhesion | ✓ | only at high concentrations |
| inhibition of vascular smooth muscle proliferation | ✓ | ✓ |
| inhibition of white cell adhesion | ✓ | ✓ |
| main phenotype of knockout | hypertensive, prothrombotic | prothrombotic, hypertensive? |
| pathology | ||
| immunological stimuli | iNOS induced | COX-2 induced |
| oxidative stress | decreases NO bioavailability | inhibits COX-2 activity |
| drug interactions | ||
| glucocorticoids | prevent iNOS induction | prevent COX-2 induction |
| statins | ↑NO | ↑COX? |
| NSAIDs | no effect | inhibit COX |
| oestrogens | ↑NO | ↑PGI2 |
| NOS inhibitors | inhibit NOS | no effect |
A clear synergism between NO and prostacyclin has been demonstrated in regard to inhibition of platelet aggregation; however, only one of them (NO) plays a role in inhibiting platelet adhesion. The significance of this difference remains to be understood. Many years ago a physiological role for platelets in repairing the vessel wall was investigated (for discussion see Higgs et al. 1978). This subject has not been re-evaluated in the light of all this new knowledge about the roles of NO and prostacyclin in platelet/vessel wall interactions. Both mediators also regulate vascular smooth muscle proliferation and white cell vessel wall interactions through similar mechanisms which include, at least in part, the activation of adenylate cyclase and the soluble guanylate cyclase. The interactions between NO and prostacyclin in the control of these functions are not fully understood.
Both mediators are further increased by inflammatory stimuli; however, while in the case of prostacyclin the same COX-2 which responds to shear stress responds to such stimuli by a further increase in its expression, NO is generated during inflammation by a specific ‘inducible’ NO synthase which is not normally present physiologically in the vessel wall. The induction of both is inhibited by anti-inflammatory glucocorticoids (Axelrod 1983; Knowles et al. 1990). It is remarkable that both compounds possess antioxidant properties (Wink et al. 1995; Egan et al. 2004) but are themselves affected by oxidative stress, which inhibits the synthesis of prostacyclin and decreases the bioavailability of NO. This mechanism might be relevant to the ‘malfunctioning’ of the constitutive generation of both mediators and therefore to the genesis of endothelial dysfunction. This, however, is an early phenomenon. In advanced disease the situation is far more complex, akin to chronic inflammation in other parts of the body and, as such, probably varies significantly in the different stages of the disease. A simple hypothesis would suggest that any amount of prostacyclin which is bioavailable, although pro-inflammatory, will provide anti-thrombotic protection, while in the case of NO the balance will vary between bioavailable NO which is protective and cytotoxic peroxynitrite formed from the interaction of NO with O2−. Currently, however, the results are not clear and on the crucial question of the role of both mediators in the progression of atherosclerosis, the information in relation to prostacyclin is contradictory (Burleigh et al. 2002; Olesen et al. 2002; Rott et al. 2003). The evidence in relation to NO, on the other hand, seems to suggest that, while constitutive NO generated by eNOS is protective (e.g. Kawashima & Yokoyama 2004), NO generated by the inducible enzyme favours the development of atherosclerosis (Chyu et al. 1999). Studies of genetically manipulated animals are providing some important clues. For example, knockout of the prostacyclin receptor (IP) leads to mice with normal blood pressure but an increased tendency to thrombosis when the endothelium is damaged (Murata et al. 1997) These animals also exhibit an increased platelet activation and proliferative response to injury that can be prevented by deletion or antagonism of the TXA2 receptor (Cheng et al. 2002). Furthermore, deletion of the IP receptor in animals prone to spontaneous atherosclerosis accelerates the development of the disease (Egan et al. 2004; Kobayashi et al. 2004). On the other hand, knocking out the thromboxane receptor or the thromboxane synthase gives rise to a mild bleeding tendency and a resistance to platelet aggregation and sudden death induced by arachidonic acid infusion (Thomas et al. 1998; Yu et al. 2004). Deletion of the thromboxane receptor also seems to retard atherogenesis in murine models of atherosclerosis (Cayatte et al. 2000; Egan et al. 2005).
Although the lack of either mediator has been shown to increase the risk of thrombosis and atherosclerosis, especially in animals with additional risk factors such as ApoE deficiencies (Kuhlencordt et al. 2001; Belton et al. 2003), there seems to be a certain specialization in their actions, so that NO has a more significant role in the regulation of blood pressure and blood flow, while prostacyclin has a clearer role in regulating platelet/vessel wall interactions. For example, inhibition of NO generation has an immediate and dramatic effect on blood flow and blood pressure and the eNOS−/− animal exhibits a clear hypertensive phenotype. On the other hand, inhibition of prostacyclin synthesis by the coxibs leads to a slow effect on blood pressure and apparently to a more thrombotic situation (Muscara et al. 2000; FitzGerald 2003). Similarly, COX-1−/− and COX-2−/− animals show no change in blood pressure (Norwood et al. 2000; Cheung et al. 2002) and manipulation of COX or IP results in a prothrombotic phenotype.
Protection against decreases in the generation of constitutive NO and prostacyclin in the vasculature may prevent the development of vascular disease. In relation to NO, the most often tried interventions relate to the use of antioxidants (see Carr & Frei 2000) and the manipulation of eNOS expression by genetic means (Von der Leyen & Dzau 2001). Each of these interventions has shown promise in both animal experiments and in humans. An unexpected and highly interesting development relates to the effects of statins which, in the last few years, have been shown to increase the production of endothelial NO in endothelial cell cultures and in animals (for review see Laufs 2003). Many mechanisms have been claimed for this action. However, of interest in the context of our discussion is the fact that statins have been claimed to reduce oxidative stress by increasing the synthesis of BH4 (Hattori et al. 2002), increasing the coupling of the eNOS (Brouet et al. 2001) or reducing the activation of NADPH oxidase (Wagner et al. 2000). Reduction of oxidative stress is likely to preserve the generation of prostacyclin, and to our knowledge there is at least one report suggesting that statins also increase prostacyclin in endothelial cell cultures of human coronary arteries (Mueck et al. 2001). Studies on the transfection of COX-1 or COX-2 into endothelial and other cells, on the other hand, are at an early stage and clear results are not conclusive (Murakami et al. 1999; Shyue et al. 2001). The full consequences of overexpression of both NO and prostacyclin in the vasculature remain to be investigated.
Also relevant to this discussion are studies of the role that NO and prostacyclin play in the protection of the cardiovascular system provided by oestrogens, and therefore in the difference between genders in susceptibility to cardiovascular disease. Oestrogens increase the expression and the activity of eNOS (Weiner et al. 1994; Yang et al. 2000) and the activity of the COX-2 enzyme (Akarasereenont et al. 2000; Egan et al. 2004). They could therefore reduce oxidative stress by simply increasing both mediators. Alternatively, it has been claimed that oestrogens increase the efficiency of the NO synthase, thus reducing free radical formation (Barbacanne et al. 1999).
In summary, the concept of the balance between prostacyclin and TXA2 has to be expanded to include NO. Furthermore, although not discussed in this review, the way in which these compounds interact with many other systems known to be involved in vessel wall physiology and pathophysiology requires further investigation. Both prostacyclin and NO synergize in the protection of the vessel wall. TXA2, however, lies on the negative side of this balance being responsible for, among other things, platelet aggregation and vasoconstriction. The investigation into the interplay between these three molecules is just beginning. This is a sobering thought when one is contemplating probably close to 100 000 papers and over 30 years of research! However, it is clear that the discoveries of prostacyclin and NO have transformed our comprehension of vascular physiology and opened avenues for further understanding of pathophysiological processes. This knowledge has already benefited clinical medicine and no doubt will continue providing clues that will guide future therapy and prevention of vascular disease. I have had the good fortune to be intimately involved with both discoveries. More importantly, many of the colleagues that I have interacted with in the process of doing this work have become life-long personal friends. To those with whom I have managed to combine scientific excitement with friendship I owe a double debt of gratitude.
Acknowledgments
Annie Higgs made a significant contribution to the preparation of this manuscript, I gratefully acknowledge her help. I would also like to thank Arnold Herman, Jorge Erusalimsky and Massimo Di Rosa for helpful discussions.
References
- Akarasereenont P, Techatraisak K, Thaworn A, Chotewuttakorn S. The induction of cyclooxygenase-2 by 17beta-estradiol in endothelial cells is mediated through protein kinase C. Inflamm. Res. 2000;49:460–465. doi: 10.1007/s000110050617. 10.1007/s000110050617 [DOI] [PubMed] [Google Scholar]
- Alderton W.K, Cooper C.E, Knowles R.G. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. 10.1042/0264-6021:3570593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Moncada S, Bolanos J.P. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat. Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. 10.1038/ncb1080 [DOI] [PubMed] [Google Scholar]
- Amezcua J.L, O'Grady J, Salmon J.A, Moncada S. Prolonged paradoxical effect of aspirin on platelet behaviour and bleeding time in man. Thromb. Res. 1979;16:69–79. doi: 10.1016/0049-3848(79)90270-6. 10.1016/0049-3848(79)90270-6 [DOI] [PubMed] [Google Scholar]
- Amezcua J.-L, Palmer R.M.J, de Souza B.M, Moncada S. Nitric oxide synthesized from l-arginine regulates vascular tone in the coronary circulation of the rabbit. Br. J. Pharmacol. 1989;97:1119–1124. doi: 10.1111/j.1476-5381.1989.tb12569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antithrombotic Trialists' Collaboration Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br. Med. J. 2002;321:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselbergs F.W, van der Harst P, Jessurun G.A.J, Tio R.A, van Gilst W.H. Clinical impact of vasomotor function assessment and the role of ACE-inhibitors and statins. Vasc. Pharmacol. 2005;42:125–140. doi: 10.1016/j.vph.2005.01.009. 10.1016/j.vph.2005.01.009 [DOI] [PubMed] [Google Scholar]
- Axelrod L. Inhibition of prostacyclin production mediates permissive effect of glucocorticoids on vascular tone. Perturbations of this mechanism contribute to pathogenesis of Cushing's syndrome and Addison's disease. Lancet. 1983;1:904–906. doi: 10.1016/s0140-6736(83)91330-2. 10.1016/S0140-6736(83)91330-2 [DOI] [PubMed] [Google Scholar]
- Azuma H, Ishikawa M, Sekizaki S. Endothelium-dependent inhibition of platelet aggregation. Br. J. Pharmacol. 1986;88:411–415. doi: 10.1111/j.1476-5381.1986.tb10218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baigent C, Patrono C. Selective cyclooxygenase 2 inhibitors, aspirin, and cardiovascular disease. Arthritis Rheum. 2003;48:12–20. doi: 10.1002/art.10738. 10.1002/art.10738 [DOI] [PubMed] [Google Scholar]
- Barbacanne M.A, Rami J, Michel J.B, Souchard J.P, Philippe M, Besombes J.P, Bayard F, Arnal J.F. Estradiol increases rat aorta endothelium-derived relaxing factor (EDRF) activity without changes in endothelial NO synthase gene expression: possible role of decreased endothelium-derived superoxide anion production. Cardiovasc. Res. 1999;41:672–681. doi: 10.1016/s0008-6363(98)00254-5. 10.1016/S0008-6363(98)00254-5 [DOI] [PubMed] [Google Scholar]
- Beckman J.S, Beckman T.W, Chen J, Marshall P.A, Freeman B.A. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton O.A, Duffy A, Toomey S, Fitzgerald D.J. Cyclooxygenase isoforms and platelet vessel wall interactions in the apolipoprotein E knockout mouse model of atherosclerosis. Circulation. 2003;108:3017–3023. doi: 10.1161/01.CIR.0000104565.78013.AD. 10.1161/01.CIR.0000104565.78013.AD [DOI] [PubMed] [Google Scholar]
- Beltran B, Orsi A, Clementi E, Moncada S. Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharmacol. 2000;129:953–960. doi: 10.1038/sj.bjp.0703147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran B, Quintero M, Garcia-Zaragoza E, O'Connor E, Esplugues J.V, Moncada S. Inhibition of mitochondrial respiration by endogenous nitric oxide: a critical step in Fas signaling. Proc. Natl Acad. Sci. USA. 2002;99:8892–8897. doi: 10.1073/pnas.092259799. 10.1073/pnas.092259799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett A, Friedmann C.A, Vane J.R. Release of prostaglandin E1 from the rat stomach. Nature. 1967;216:873–876. doi: 10.1038/216873a0. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi C, et al. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature. 2005;435:502–506. doi: 10.1038/nature03527. 10.1038/nature03527 [DOI] [PubMed] [Google Scholar]
- Binz C. 2nd edn. 1891. Vorlesungen Ueber Pharmakologie. Berlin, Germany. [Google Scholar]
- Bombardier C, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. 10.1056/NEJM200011233432103 [DOI] [PubMed] [Google Scholar]
- Boo Y.C, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am. J. Cell Physiol. 2003;285:C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- Bresalier R.S, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. 10.1056/NEJMoa050493 [DOI] [PubMed] [Google Scholar]
- Brosnan J. Vascular NAD(P)H oxidase as a novel therapeutic target in vascular disease. Drug News Perspect. 2004;17:429–434. doi: 10.1358/dnp.2004.17.7.863701. 10.1358/dnp.2004.17.7.863701 [DOI] [PubMed] [Google Scholar]
- Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand J.L, Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ. Res. 2001;89:866–873. doi: 10.1161/hh2201.100319. [DOI] [PubMed] [Google Scholar]
- Brown G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- Brown G.C, Cooper C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. 10.1016/0014-5793(94)01290-3 [DOI] [PubMed] [Google Scholar]
- Bult H, Boeckxstaens G.E, Pelckmans P.A, Jordaens F.H, Van-Maercke Y.M, Herman A.G. Nitric oxide as an inhibitory non-adrenergic, non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. 10.1038/345346a0 [DOI] [PubMed] [Google Scholar]
- Bunting S, Gryglewski R, Moncada S, Vane J.R. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and celiac arteries and inhibits platelet aggregation. Prostaglandins. 1976;12:897–913. doi: 10.1016/0090-6980(76)90125-8. 10.1016/0090-6980(76)90125-8 [DOI] [PubMed] [Google Scholar]
- Burch J.W, Baenziger N.L, Stanford N, Majerus P.W. Sensitivity of fatty acid cyclooxygenase from human aorta to acetylation by aspirin. Proc. Natl Acad. Sci. USA. 1978a;75:5181–5184. doi: 10.1073/pnas.75.10.5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch J.W, Stanford N, Majerus P.W. Inhibition of platelet prostaglandin synthetase by oral aspirin. J. Clin. Invest. 1978b;61:314–319. doi: 10.1172/JCI108941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh M.E, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–1823. doi: 10.1161/01.cir.0000014927.74465.7f. 10.1161/01.CIR.0000014927.74465.7F [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Capone M.L, et al. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation. 2004;109:1468–1471. doi: 10.1161/01.CIR.0000124715.27937.78. 10.1161/01.CIR.0000124715.27937.78 [DOI] [PubMed] [Google Scholar]
- Carr A, Frei B. The role of natural antioxidants in preserving the biological activity of endothelium-derived nitric oxide. Free Radic. Biol. Med. 2000;28:1806–1814. doi: 10.1016/s0891-5849(00)00225-2. 10.1016/S0891-5849(00)00225-2 [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch. Biochem. Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. 10.1006/abbi.1996.0178 [DOI] [PubMed] [Google Scholar]
- Catella-Lawson F, et al. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics and vasoactive eicosanoids. J. Pharmcol. Exp. Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- Cayatte A.J, Du Y, Oliver-Krasinski J, Lavielle G, Verbeuren T.J, Cohen R.A. The thromboxane receptor antagonist S18886 but not aspirin inhibits atherogenesis in apoE-deficient mice: evidence that eicosanoids other than thromboxane contribute to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000;20:1724–1728. doi: 10.1161/01.atv.20.7.1724. [DOI] [PubMed] [Google Scholar]
- Cellek S. Nitrergic–noradrenergic interaction in penile erection: a new insight into erectile dysfunction. Drugs Today. 2000;36:135–146. doi: 10.1358/dot.2000.36.2-3.568787. [DOI] [PubMed] [Google Scholar]
- Cellek S, Rodrigo J, Lobos E, Fernandez P, Serrano J, Moncada S. Selective nitrergic neurodegeneration in diabetes mellitus—a nitric oxide-dependent phenomenon. Br. J. Pharmacol. 1999;128:1804–1812. doi: 10.1038/sj.bjp.0702981. 10.1038/sj.bjp.0702981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Austin S.C, Rocca B, Koller B.H, Coffman T.M, Grosser T, Lawson J.A, FitzGerald G.A. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. 10.1126/science.1068711 [DOI] [PubMed] [Google Scholar]
- Cheung R.T, Pei Z, Feng Z.H, Zou L.Y. Cyclooxygenase-1 gene knockout does not alter middle cerebral artery occlusion in a mouse stroke model. Neurosci. Lett. 2002;330:57–60. doi: 10.1016/s0304-3940(02)00738-3. 10.1016/S0304-3940(02)00738-3 [DOI] [PubMed] [Google Scholar]
- Chyu K.Y, Dimayuga P, Zhu J, Nilsson J, Kaul S, Shah P.K, Cercek B. Decreased neointimal thickening after arterial wall injury in inducible nitric oxide synthase knockout mice. Circ. Res. 1999;85:1192–1198. doi: 10.1161/01.res.85.12.1192. [DOI] [PubMed] [Google Scholar]
- Cleeter M.W, Cooper J.M, Darley-Usmar V.M, Moncada S, Schapira A.H. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994;345:50–54. doi: 10.1016/0014-5793(94)00424-2. 10.1016/0014-5793(94)00424-2 [DOI] [PubMed] [Google Scholar]
- Clementi E, Brown G.C, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl Acad. Sci. USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. 10.1073/pnas.95.13.7631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi E, Brown G.C, Foxwell N, Moncada S. On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc. Natl Acad. Sci. USA. 1999;96:1559–1562. doi: 10.1073/pnas.96.4.1559. 10.1073/pnas.96.4.1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks T.M, Angus J.A, Campbell J.H, Campbell G.R. Release and properties of endothelium-derived relaxing factor (EDRF) from endothelial cells in culture. J. Cell. Physiol. 1985;123:310–320. doi: 10.1002/jcp.1041230304. 10.1002/jcp.1041230304 [DOI] [PubMed] [Google Scholar]
- Craven L.L. Experiences with aspirin (acetylsalicylic acid) in the nonspecific prophylaxis of coronary thrombosis. Mississippi Valley Med. J. 1953;75:38–44. [PubMed] [Google Scholar]
- Crunkhorn P, Willis A.L. Cutaneous reactions to intradermal prostaglandins. Br. J. Pharmacol. 1971;41:49–56. doi: 10.1111/j.1476-5381.1971.tb09934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen L, Kelly L, Connor S.O, Fitzgerald D.J. Selective cyclooxygenase-2 inhibition by nimesulide in man. J. Pharmacol. Exp. Ther. 1998;287:578–582. [PubMed] [Google Scholar]
- De Caterina R, et al. Low-dose aspirin in patients recovering from myocardial infarction. Evidence for a selective inhibition of thromboxane-related platelet function. Eur. Heart J. 1985;6:409–417. doi: 10.1093/oxfordjournals.eurheartj.a061879. [DOI] [PubMed] [Google Scholar]
- Deguchi T. Endogenous activating factor for guanylate cyclase in synaptosomal-soluble fraction of rat brain. J. Biol. Chem. 1977;252:7617–7619. [PubMed] [Google Scholar]
- Deguchi T, Yoshioka M. l-Arginine identified as an endogenous activator for soluble guanylate cyclase from neuroblastoma cells. J. Biol. Chem. 1982;257:10 147–10 151. [PubMed] [Google Scholar]
- Desai K.M, Zembowicz A, Sessa W.C, Vane J.R. Nitroxergic nerves mediate vagally induced relaxation in the isolated stomach of the guinea pig. Proc. Natl Acad. Sci. USA. 1991;88:11 490–11 494. doi: 10.1073/pnas.88.24.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa M, Giroud J.P, Willoughby D.A. Studies on the mediators of the acute inflammatory response induced in rats in different sites by carrageenan and turpentine. J. Pathol. 1971;104:15–29. doi: 10.1002/path.1711040103. 10.1002/path.1711040103 [DOI] [PubMed] [Google Scholar]
- Dogne J.M, et al. New developments on thromboxane and prostacyclin modulators part I: thromboxane modulators. Curr. Med. Chem. 2004;11:1223–1241. doi: 10.2174/0929867043365260. [DOI] [PubMed] [Google Scholar]
- Drazen J.M. COX-2 inhibitors—a lesson in unexpected problems. N. Engl. J. Med. 2005;352:1131–1132. doi: 10.1056/NEJMe058038. 10.1056/NEJMe058038 [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Dzau V.J. Theodore Cooper lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- Egan K.M, Lawson J.A, Fries S, Koller B, Rader D.J, Smyth E.M, FitzGerald G.A. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–1957. doi: 10.1126/science.1103333. 10.1126/science.1103333 [DOI] [PubMed] [Google Scholar]
- Egan K.M, Wang M, Lucitt M.B, Zukas A.M, Pure E, Lawson J.A, FitzGerald G.A. Cyclooxygenases, thromboxane and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation. 2005;111:334–342. doi: 10.1161/01.CIR.0000153386.95356.78. 10.1161/01.CIR.0000153386.95356.78 [DOI] [PubMed] [Google Scholar]
- Ferreira S.H. Prostaglandins, aspirin-like drugs and analgesia. Nat. New Biol. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- Ferreira S.H, Moncada S. Inhibition of prostaglandin synthesis augments the effects of sympathetic nerve stimulation on the cat spleen. Br. J. Pharmacol. 1971;43:419P–420P. [PMC free article] [PubMed] [Google Scholar]
- Ferreira S.H, Moncada S, Vane J.R. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nat. New Biol. 1971;231:237–239. doi: 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- Ferreira S.H, Moncada S, Vane J.R. Some effects of inhibiting endogenous prostaglandin formation on the responses of the cat spleen. Br. J. Pharmacol. 1973;47:48–58. doi: 10.1111/j.1476-5381.1973.tb08157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher A.M, Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109:178–183. doi: 10.1161/01.CIR.0000105763.51286.7F. 10.1161/01.CIR.0000105763.51286.7F [DOI] [PubMed] [Google Scholar]
- FitzGerald G.A. Parsing an enigma: the pharmacodynamics of aspirin resistance. Lancet. 2003;361:542–544. doi: 10.1016/S0140-6736(03)12560-3. 10.1016/S0140-6736(03)12560-3 [DOI] [PubMed] [Google Scholar]
- FitzGerald G.A, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. 10.1056/NEJM200108093450607 [DOI] [PubMed] [Google Scholar]
- FitzGerald G.A, Smith B, Pedersen A.K, Brash A.R. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N. Engl. J. Med. 1984;310:1065–1068. doi: 10.1056/NEJM198404263101701. [DOI] [PubMed] [Google Scholar]
- Fitzgerald D.J, Roy L, Catella F, FitzGerald G.A. Platelet activation in unstable coronary disease. N. Engl. J. Med. 1986;315:983–999. doi: 10.1056/NEJM198610163151602. [DOI] [PubMed] [Google Scholar]
- Fleming I. Cytochrome p450 and vascular homeostasis. Circ. Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. The physiology of nitric oxide: control and consequences. Curr. Med. Chem. 2004;3:189–205. [Google Scholar]
- Frangos J.A, Eskin S.G, McIntire L.V, Ives C.L. Flow effects on prostacyclin production by cultured human endothelial cells. Science. 1985;227:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- Frantz S. Vioxx risk could signify trouble in class. Nat. Rev. Drug Discov. 2004;3:899–900. doi: 10.1038/nrd1560. 10.1038/nrd1560 [DOI] [PubMed] [Google Scholar]
- Fu J.Y, Masferrer J.L, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J. Biol. Chem. 1990;265:16 737–16 740. [PubMed] [Google Scholar]
- Furchgott R.F. Studies on relaxation of rabbit aorta by sodium nitrite: the basis for the proposal that the acid-activatable inhibitory factor from retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide. In: Vanhoutte P.M, editor. Vasodilatation: vascular smooth muscle, peptides, autonomic nerves and endothelium. Raven Press; New York, NY: 1988. pp. 401–414. [Google Scholar]
- Furchgott R.J. The 1989 Ulf von Euler lecture. Studies on endothelium-dependent vasodilation and the endothelium-derived relaxing factor. Acta Physiol. Scand. 1990;139:257–270. doi: 10.1111/j.1748-1716.1990.tb08923.x. [DOI] [PubMed] [Google Scholar]
- Furchgott R.F, Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. 10.1038/288373a0 [DOI] [PubMed] [Google Scholar]
- Furchgott R.F, Cherry P.D, Zawadzki J.V, Jothianandan D. Endothelial cells as mediators of vasodilation of arteries. J. Cardiovasc. Pharmacol. 1984;6:S336–S343. doi: 10.1097/00005344-198406002-00008. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Comander J, Anderson K.R, Blackman B.R, Gimbrone M.A., Jr Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl Acad. Sci. USA. 2001;98:4478–4485. doi: 10.1073/pnas.071052598. 10.1073/pnas.071052598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg U.C, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Invest. 1989;83:1774–1777. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Charles S.L, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. 10.1038/336385a0 [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Garthwaite G, Palmer R.M, Moncada S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur. J. Pharmacol. 1989;172:413–416. doi: 10.1016/0922-4106(89)90023-0. 10.1016/0922-4106(89)90023-0 [DOI] [PubMed] [Google Scholar]
- Gibbs J.S, Broberg C.S, Gatzoulis M.A. Idiopathic pulmonary arterial hypertension: current state of play and new treatment modalities. Int. J. Cardiol. 2004;97:7–10. doi: 10.1016/j.ijcard.2004.08.003. 10.1016/j.ijcard.2004.08.003 [DOI] [PubMed] [Google Scholar]
- Gibson A, Mirzazadeh S. Does an endogenous nitrovasodilator mediate NANC relaxations of the mouse anococcygeus muscle? Br. J. Pharmacol. 1989;98:617P. doi: 10.1111/j.1476-5381.1989.tb11863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie J.S, Liu X.R, Martin W. The effects of l-arginine and NG-monomethyl l-arginine on the response of the rat anococcygeus muscle to NANC nerve stimulation. Br. J. Pharmacol. 1989;98:1080–1082. doi: 10.1111/j.1476-5381.1989.tb12650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski E.F, Jaffe E.A, Weksler B.B. Prostacyclin production by cultured endothelial cell monolayers exposed to step increases in shear stress. J. Lab. Clin. Med. 1985;105:36–43. [PubMed] [Google Scholar]
- Gryglewski R.J, Bunting S, Moncada S, Flower R.J, Vane J.R. Arterial walls are protected against deposition of platelet thrombi by a substance (prostaglandin X) which they make from prostaglandin endoperoxides. Prostaglandins. 1976;12:685–713. doi: 10.1016/0090-6980(76)90047-2. 10.1016/0090-6980(76)90047-2 [DOI] [PubMed] [Google Scholar]
- Gryglewski R.J, Moncada S, Palmer R.M.J. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF) from porcine aortic endothelial cells. Br. J. Pharmacol. 1986a;87:685–694. doi: 10.1111/j.1476-5381.1986.tb14586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryglewski R.J, Palmer R.M.J, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986b;320:454–456. doi: 10.1038/320454a0. 10.1038/320454a0 [DOI] [PubMed] [Google Scholar]
- Guzik T.J, West N.E, Black E, McDonald D, Ratnatunga C, Pillai R, Channon K.M. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- Hagen T, Taylor C.T, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1α. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. 10.1126/science.1088805 [DOI] [PubMed] [Google Scholar]
- Hamberg M, Samuelsson B. Detection and isolation of an endoperoxide intermediate in prostaglandin biosynthesis. Proc. Natl Acad. Sci. USA. 1973;70:899–903. doi: 10.1073/pnas.70.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Svensson J, Wakabayashi T, Samuelsson B. Isolation and structure of two prostaglandin endoperoxides that cause platelet aggregation. Proc. Natl Acad. Sci. USA. 1974;71:345–349. doi: 10.1073/pnas.71.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamberg M, Svensson J, Samuelsson B. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl Acad. Sci. USA. 1975;72:2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J, et al. In vitro and in vivo pharmacological characterization of BM-613 ([N-n-pentyl-N′-[2-(2-(4′-methylphenylamino)-5-nitrobenzenesulfonyl]urea], a novel dual thromboxane synthase inhibitor and thromboxane receptor antagonist. J. Pharmacol. Exp. Ther. 2005;313:293–301. doi: 10.1124/jpet.104.079301. 10.1124/jpet.104.079301 [DOI] [PubMed] [Google Scholar]
- Hattori Y, Nakanishi N, Kasai K. Statin enhances cytokine-mediated induction of nitric oxide synthesis in vascular smooth muscle cells. Cardiovasc. Res. 2002;54:649–658. doi: 10.1016/s0008-6363(02)00266-3. 10.1016/S0008-6363(02)00266-3 [DOI] [PubMed] [Google Scholar]
- Heitzer T, Yla-Herttuala S, Luoma J, Kurz S, Munzel T, Just H, Olschewski M, Drexler H. Cigarette smoking potentiates endothelial dysfunction of forearm resistance vessels in patients with hypercholesterolemia. Role of oxidized LDL. Circulation. 1996;93:1346–1353. doi: 10.1161/01.cir.93.7.1346. [DOI] [PubMed] [Google Scholar]
- Hibbs J.B, Jr, Taintor R.R, Vavrin Z. Macrophage cytotoxicity: role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science. 1987;235:473–476. doi: 10.1126/science.2432665. [DOI] [PubMed] [Google Scholar]
- Higgs G.A, Bunting S, Moncada S, Vane J.R. Polymorphonuclear leukocytes produce thromboxane A2-like activity during phagocytosis. Prostaglandins. 1976;12:749–757. doi: 10.1016/0090-6980(76)90050-2. 10.1016/0090-6980(76)90050-2 [DOI] [PubMed] [Google Scholar]
- Higgs E.A, Moncada S, Vane J.R, Caen J.P, Michel H, Tobelem G. Effect of prostacyclin (PGI2) on platelet adhesion to rabbit arterial subendothelium. Prostaglandins. 1978;16:17–22. doi: 10.1016/0090-6980(78)90197-1. 10.1016/0090-6980(78)90197-1 [DOI] [PubMed] [Google Scholar]
- Higgs G.A, Flower R.J, Vane J.R. A new approach to anti-inflammatory drugs. Biochem. Pharmacol. 1979;28:1959–1961. doi: 10.1016/0006-2952(79)90651-8. 10.1016/0006-2952(79)90651-8 [DOI] [PubMed] [Google Scholar]
- Hink U, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ. Res. 2001;88:E14–E22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. Br. Med. J. 2005;330:1366–1372. doi: 10.1136/bmj.330.7504.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist F, Hedlund H, Andersson K.E. l-NG-nitro arginine inhibits non-adrenergic, non-cholinergic relaxation of human isolated corpus cavernosum. Acta Physiol. Scand. 1991;141:441–442. doi: 10.1111/j.1748-1716.1991.tb09103.x. [DOI] [PubMed] [Google Scholar]
- Horn E.M, Barst R.J. Treprostinil therapy for pulmonary artery hypertension. Expert Opin. Investig. Drugs. 2002;11:1615–1622. doi: 10.1517/13543784.11.11.1615. 10.1517/13543784.11.11.1615 [DOI] [PubMed] [Google Scholar]
- Huang P.L, Huang Z, Mashimo H, Bloch K.D, Moskowitz M.A, Bevan J.A, Fishman M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. doi: 10.1038/377239a0. 10.1038/377239a0 [DOI] [PubMed] [Google Scholar]
- Ignarro L.J. Contributions to a quest. Nature. 1987;330:526. doi: 10.1038/330526c0. 10.1038/330526c0 [DOI] [PubMed] [Google Scholar]
- Ignarro L.J, Buga G.M, Wood K.S, Byrns R.E, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl Acad. Sci. USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignarro L.J, Byrns R.E, Wood K.S. Biochemical and pharmacological properties of endothelium-derived relaxing factor and its similarity to nitric oxide radical. In: Vanhoutte P.M, editor. Vasodilatation: vascular smooth muscle, peptide, autonomic nerves and endothelium. Raven Press; New York, NY: 1988. pp. 427–436. [Google Scholar]
- Ignarro L.J, Bush P.A, Buga G.M, Wood K.S, Fukuto J.M, Rajfer J. Nitric oxide and cyclic GMP formation upon electrical field stimulation cause relaxation of corpus cavernosum smooth muscle. Biochem. Biophys. Res. Commun. 1990;170:843–850. doi: 10.1016/0006-291x(90)92168-y. 10.1016/0006-291X(90)92168-Y [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Matsui T, Kurita A. Ramatroban, a TP receptor antagonist, improves vascular responses to acetylcholine in hypercholesterolemic rabbits in vivo. Eur. J. Pharmacol. 2003;468:27–35. doi: 10.1016/s0014-2999(03)01626-1. 10.1016/S0014-2999(03)01626-1 [DOI] [PubMed] [Google Scholar]
- Iyengar R, Stuehr D.J, Marletta M.A. Macrophage synthesis of nitrite, nitrate and N-nitrosamines: precursors and role of the respiratory burst. Proc. Natl Acad. Sci. USA. 1987;84:6369–6373. doi: 10.1073/pnas.84.18.6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.A, et al. The chemical structure of prostaglandin X (prostacyclin) Prostaglandins. 1976;12:915–918. doi: 10.1016/0090-6980(76)90126-x. [DOI] [PubMed] [Google Scholar]
- Juhlin L, Michaelsson G. Cutaneous vascular reactions to prostaglandins in healthy subjects and in patients with urticaria and atopic dermatitis. Acta Derm. Venereol. 1969;49:251–261. [PubMed] [Google Scholar]
- Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004;24:998–1005. doi: 10.1161/01.ATV.0000125114.88079.96. 10.1161/01.ATV.0000125114.88079.96 [DOI] [PubMed] [Google Scholar]
- Kelton J.G, Hirsch J, Carter C.J, Buchanan M.R. Thrombogenic effect of high-dose aspirin in rabbits. Relationship to inhibition of vessel wall synthesis of prostaglandin I2-like activity. J. Clin. Invest. 1978;62:892–895. doi: 10.1172/JCI109203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles R.G, Palacios M, Palmer R.M.J, Moncada S. Formation of nitric oxide from l-arginine in the central nervous system: a transduction mechanism for stimulation of the soluble guanylate cyclase. Proc. Natl Acad. Sci. USA. 1989;86:5159–5162. doi: 10.1073/pnas.86.13.5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles R.G, Salter M, Brooks S.L, Moncada S. Anti-inflammatory glucocorticoids inhibit the induction by endotoxin of nitric oxide synthase in the lung, liver and aorta of the rat. Biochem. Biophys. Res. Commun. 1990;172:1042–1048. doi: 10.1016/0006-291x(90)91551-3. 10.1016/0006-291X(90)91551-3 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, et al. Roles of thromboxane A2 and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Invest. 2004;114:784–794. doi: 10.1172/JCI21446. 10.1172/JCI200421446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbut R, Moncada S. Prostacyclin (PGI2) and thromboxane A2 interaction in vivo. Regulation by aspirin and relationship with anti-thrombotic therapy. Thromb. Res. 1978;13:489–500. doi: 10.1016/0049-3848(78)90134-2. 10.1016/0049-3848(78)90134-2 [DOI] [PubMed] [Google Scholar]
- Kubes P, Suzuki M, Granger D.N. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl Acad. Sci. USA. 1991;88:4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlencordt P.J, Gyurko R, Han F, Scherrer-Crosbie M, Aretz T.H, Hajjar R, Picard M.H, Huang P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- Kwon N.S, Nathan C.F, Gilker C, Griffith O.W, Matthews D.E, Stuehr D.J. l-Citrulline production from l-arginine by macrophage nitric oxide synthase. The ureido oxygen derives from dioxygen. J. Biol. Chem. 1990;265:13 442–13 445. [PubMed] [Google Scholar]
- Landmesser U, Dikalov S, Price S.R, McCann L, Fukai T, Holland S.M, Mitch W.E, Harrison D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. 10.1172/JCI200314172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, Barbui T. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004;350:114–124. doi: 10.1056/NEJMoa035572. 10.1056/NEJMoa035572 [DOI] [PubMed] [Google Scholar]
- Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur. J. Clin. Pharmacol. 2003;58:719–731. doi: 10.1007/s00228-002-0556-0. [DOI] [PubMed] [Google Scholar]
- Leone A.M, Palmer R.M.J, Knowles R.G, Francis P.L, Ashton D.S, Moncada S. Constitutive and inducible nitric oxide synthases incorporate molecular oxygen into both nitric oxide and citrulline. J. Biol. Chem. 1991;266:23 790–23 795. [PubMed] [Google Scholar]
- Levine G.N, Frei B, Koulouris S.N, Gerhard M.D, Keaney J.F, Jr, Vita J.A. Ascorbic acid reverses endothelial vasomotor dysfunction in patients with coronary artery disease. Circulation. 1996;93:1107–1113. doi: 10.1161/01.cir.93.6.1107. [DOI] [PubMed] [Google Scholar]
- Li C.G, Rand M.J. Evidence for a role of nitric oxide in the neurotransmitter system mediating relaxation of the rat anococcygeus muscle. Clin. Exp. Pharmacol. Physiol. 1989;16:933–938. doi: 10.1111/j.1440-1681.1989.tb02404.x. [DOI] [PubMed] [Google Scholar]
- Li C.G, Rand M.J. Evidence that part of the NANC relaxant response of guinea-pig trachea to electrical field stimulation is mediated by nitric oxide. Br. J. Pharmacol. 1991;102:91–94. doi: 10.1111/j.1476-5381.1991.tb12137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.G, Miller F.J, Jr, Zhang H.J, Spitz D.R, Oberley L.W, Weintraub N.L. H2O2-induced O2 production by a non-phagocytic NAD(P)H oxidase causes oxidant injury. J. Biol. Chem. 2001;276:29 251–29 256. doi: 10.1074/jbc.M102124200. 10.1074/jbc.M102124200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.G, et al. Activation of NAD(P)H oxidase by lipid hydroperoxides: mechanism of oxidant-mediated smooth muscle cytotoxicity. Free Radic. Biol. Med. 2003;34:937–946. doi: 10.1016/s0891-5849(03)00032-7. 10.1016/S0891-5849(03)00032-7 [DOI] [PubMed] [Google Scholar]
- Lin M.I, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard K.A, Jr, Sessa W.C. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of l-arginine metabolism to efficient nitric oxide production. J. Biol. Chem. 2003;278:44 719–44 726. doi: 10.1074/jbc.M302836200. 10.1074/jbc.M302836200 [DOI] [PubMed] [Google Scholar]
- Lizasoain I, Moro M.A, Knowles R.G, Darley-Usmar V, Moncada S. Nitric oxide and peroxynitrite exert distinct effects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem. J. 1996;314:877–880. doi: 10.1042/bj3140877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher T.F, Tanner F.C, Tschudi M.R, Noll G. Endothelial dysfunction in coronary artery disease. Annu. Rev. Med. 1993;44:395–418. doi: 10.1146/annurev.me.44.020193.002143. 10.1146/annurev.me.44.020193.002143 [DOI] [PubMed] [Google Scholar]
- Mann G.E, Yudilevich D.L, Sobrevia L. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev. 2003;83:183–252. doi: 10.1152/physrev.00022.2002. [DOI] [PubMed] [Google Scholar]
- Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S. Regulation of hypoxia-inducible factor-1α by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003;376:537–544. doi: 10.1042/BJ20031155. 10.1042/BJ20031155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdam B.F, Catella-Lawson F, Mardini I.A, Kapoor S, Lawson J.A, FitzGerald G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc. Natl Acad. Sci. USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. 10.1073/pnas.96.1.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T.N, Du T, Frangos J.A. Fluid shear stress stimulates prostaglandin and nitric oxide release in bone marrow-derived preosteoclast-like cells. Biochem. Biophys. Res. Commun. 2000;270:643–648. doi: 10.1006/bbrc.2000.2467. 10.1006/bbrc.2000.2467 [DOI] [PubMed] [Google Scholar]
- McCall T.B, Boughton-Smith N.K, Palmer R.M.J, Whittle B.J.R, Moncada S. Synthesis of nitric oxide from l-arginine by neutrophils. Biochem. J. 1989;261:293–296. doi: 10.1042/bj2610293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick S.M, Whitson P.A, Wu K.K, McIntire L.V. Shear stress differentially regulates PGHS-1 and PGHS-2 protein levels in human endothelial cells. Ann. Biomed. Eng. 2000;28:824–833. doi: 10.1114/1.1289472. 10.1114/1.1289472 [DOI] [PubMed] [Google Scholar]
- Mellion B.T, Ignarro L.J, Ohlstein E.H, Pontecorvo E.G, Hyman A.L, Kadowitz P.J. Evidence for the inhibitory role of guanosine 3′, 5′-monophosphate in ADP-induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood. 1981;57:946–955. [PubMed] [Google Scholar]
- Moncada S, Amezcua J.L. Prostacyclin, thromboxane A2 interactions in haemostasis and thrombosis. Haemostasis. 1979;8:252–265. doi: 10.1159/000214316. [DOI] [PubMed] [Google Scholar]
- Moncada S, Erusalimsky J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002;3:214–220. doi: 10.1038/nrm762. 10.1038/nrm762 [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs E.A. The l-arginine–nitric oxide pathway. N. Engl. J. Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. 10.1056/NEJM199312303292706 [DOI] [PubMed] [Google Scholar]
- Moncada S, Vane J.R. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol. Rev. 1979;30:293–331. [PubMed] [Google Scholar]
- Moncada S, Ferreira S.H, Vane J.R. Prostaglandins, aspirin-like drugs and the oedema of inflammation. Nature. 1973;246:217–219. doi: 10.1038/246217a0. 10.1038/246217a0 [DOI] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R, Bunting S, Vane J.R. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976a;263:663–665. doi: 10.1038/263663a0. 10.1038/263663a0 [DOI] [PubMed] [Google Scholar]
- Moncada S, Gryglewski R.J, Bunting S, Vane J.R. A lipid peroxide inhibits the enzyme in blood vessel microsomes that generates from prostaglandin endoperoxides the substance (prostaglandin X) which prevents platelet aggregation. Prostaglandins. 1976b;12:715–733. doi: 10.1016/0090-6980(76)90048-4. 10.1016/0090-6980(76)90048-4 [DOI] [PubMed] [Google Scholar]
- Moncada S, Bunting S, Mullane K, Thorogood P, Vane J.R, Raz A, Needleman P. Imidazole: a selective inhibitor of thromboxane synthetase. Prostaglandins. 1977a;13:611–618. doi: 10.1016/0090-6980(77)90232-5. 10.1016/0090-6980(77)90232-5 [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs E.A, Vane J.R. Human arterial and venous tissues generate prostacyclin (prostaglandin X), a potent inhibitor of platelet aggregation. Lancet. 1977b;1:18–20. doi: 10.1016/s0140-6736(77)91655-5. 10.1016/S0140-6736(77)91655-5 [DOI] [PubMed] [Google Scholar]
- Moncada S, Herman A.G, Higgs E.A, Vane J.R. Differential formation of prostacyclin (PGX or PGI2) by layers of the arterial wall. An explanation for the anti-thombotic properties of vascular endothelium. Thromb. Res. 1977c;11:323–344. doi: 10.1016/0049-3848(77)90185-2. 10.1016/0049-3848(77)90185-2 [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer R.M.J, Gryglewski R.J. Mechanism of action of some inhibitors of endothelium-derived relaxing factor. Proc. Natl Acad. Sci. USA. 1986;83:9164–9168. doi: 10.1073/pnas.83.23.9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer R.M.J, Higgs E.A. Biosynthesis of nitric oxide from l-arginine. A pathway for the regulation of cell function and communication. Biochem. Pharmacol. 1989;38:1709–1715. doi: 10.1016/0006-2952(89)90403-6. 10.1016/0006-2952(89)90403-6 [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer R.M.J, Higgs E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Morita I. Distinct functions of COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002;68–69:165–175. doi: 10.1016/s0090-6980(02)00029-1. 10.1016/S0090-6980(02)00029-1 [DOI] [PubMed] [Google Scholar]
- Morrison F.S, Baldini M.G. Antigenic relationship between blood platelets and vascular endothelium. Blood. 1969;33:46–56. [PubMed] [Google Scholar]
- Mueck A.O, Seeger H, Deuringer F.U, Wallwiener D. Effect of an estrogen/statin combination on biochemical markers of endothelial function in human coronary artery cell cultures. Menopause. 2001;8:216–221. doi: 10.1097/00042192-200105000-00012. 10.1097/00042192-200105000-00012 [DOI] [PubMed] [Google Scholar]
- Mueller C.F, Laude K, McNally J.S, Harrison D.G. Redox mechanisms in blood vessels. Arterioscler. Thromb. Vasc. Biol. 2005;25:274–278. doi: 10.1161/01.ATV.0000149143.04821.eb. 10.1161/01.ATV.0000149143.04821.eb [DOI] [PubMed] [Google Scholar]
- Mukherjee D, Nissen S.E, Topol E.J. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. 10.1001/jama.286.8.954 [DOI] [PubMed] [Google Scholar]
- Murakami M, Kambe T, Shimbara S, Kudo I. Functional coupling between various phospholipase A2s and cyclooxygenases in immediate and delayed prostanoid biosynthetic pathways. J. Biol. Chem. 1999;274:3103–3115. doi: 10.1074/jbc.274.5.3103. 10.1074/jbc.274.5.3103 [DOI] [PubMed] [Google Scholar]
- Murata T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–682. doi: 10.1038/41780. 10.1038/41780 [DOI] [PubMed] [Google Scholar]
- Muscara M.N, Vergnolle N, Lovren F, Triggle C.R, Elliott S.N, Asfaha S, Wallace J.L. Selective cyclo-oxygenase-2 inhibition with celecoxib elevates blood pressure and promotes leukocyte adherence. Br. J. Pharmacol. 2000;129:1423–1430. doi: 10.1038/sj.bjp.0703232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman P, Moncada S, Bunting S, Vane J.R, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature. 1976;261:558–560. doi: 10.1038/261558a0. 10.1038/261558a0 [DOI] [PubMed] [Google Scholar]
- Neunteufl T, Heher S, Katzenschlager R, Wolfl G, Kostner K, Maurer G, Weidinger F. Late prognostic value of flow-mediated dilation in the brachial artery of patients with chest pain. Am. J. Cardiol. 2000;86:207–210. doi: 10.1016/s0002-9149(00)00857-2. 10.1016/S0002-9149(00)00857-2 [DOI] [PubMed] [Google Scholar]
- Nijkamp F.P, Flower R.J, Moncada S, Vane J.R. Partial purification of rabbit aorta contracting substance-releasing factor and inhibition of its activity by anti-inflammatory steroids. Nature. 1976;263:479–482. doi: 10.1038/263479a0. 10.1038/263479a0 [DOI] [PubMed] [Google Scholar]
- Nisoli E, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. 10.1126/science.1079368 [DOI] [PubMed] [Google Scholar]
- Norwood V.F, Morham S.G, Smithies O. Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int. 2000;58:2291–2300. doi: 10.1046/j.1523-1755.2000.00413.x. 10.1046/j.1523-1755.2000.00413.x [DOI] [PubMed] [Google Scholar]
- Nugteren D.H, Hazelhof E. Isolation and properties of intermediates in prostaglandin biosynthesis. Biochim. Biophys. Acta. 1973;326:448–461. doi: 10.1016/0005-2760(73)90145-8. [DOI] [PubMed] [Google Scholar]
- Nugteren D.H, Beerthuis R.K, van Dorp D.A. In: Proc. Second Nobel Symp. Bergstrom S, Samuelsson B, editors. Nobel Foundation; Stockholm, Sweden: 1967. pp. 45–50. [Google Scholar]
- Nussmeier N.A, Whelton A.A, Brown M.T, Langford R.M, Hoeft A, Parlow J.L, Boyce S.W, Verburg K.M. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med. 2005;352:1081–1091. doi: 10.1056/NEJMoa050330. 10.1056/NEJMoa050330 [DOI] [PubMed] [Google Scholar]
- O'Grady J, Moncada S. Aspirin: a paradoxical effect on bleeding time. Lancet. 1978;2:780. doi: 10.1016/s0140-6736(78)92661-2. 10.1016/S0140-6736(78)92661-2 [DOI] [PubMed] [Google Scholar]
- Ohashi Y, et al. Hypotension and reduced nitric oxide-elicited vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. J. Clin. Invest. 1998;102:2061–2071. doi: 10.1172/JCI4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okahara K, Sun B, Kambayashi J. Upregulation of prostacyclin synthesis-related gene expression by shear stress in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998;18:1922–1926. doi: 10.1161/01.atv.18.12.1922. [DOI] [PubMed] [Google Scholar]
- Olesen M, Kwong E, Meztli A, Kontny F, Seljeflot I, Arnesen H, Lyngdorf L, Falk E. No effect of cyclooxygenase inhibition on plaque size in atherosclerosis-prone mice. Scand. Cardiovasc. J. 2002;36:362–367. doi: 10.1080/140174302762659094. 10.1080/140174302762659094 [DOI] [PubMed] [Google Scholar]
- Osanai T, Fujita N, Fujiwara N, Nakano T, Takahashi K, Guan W, Okumura K. Cross talk of shear-induced production of prostacyclin and nitric oxide in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H233–H238. doi: 10.1152/ajpheart.2000.278.1.H233. [DOI] [PubMed] [Google Scholar]
- Pace-Asciak C, Wolfe L.S. Novel prostaglandin derivative formed from arachidonic acid by rat stomach homogenates. Biochemistry. 1971;10:3657–3664. doi: 10.1021/bi00796a004. 10.1021/bi00796a004 [DOI] [PubMed] [Google Scholar]
- Palacios-Callender M, Quintero M, Hollis V.S, Springett R.J, Moncada S. Endogenous NO regulates superoxide production at low oxygen concentrations by modifying the redox state of cytochrome c oxidase. Proc. Natl Acad. Sci. USA. 2004;101:7630–7635. doi: 10.1073/pnas.0401723101. 10.1073/pnas.0401723101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer R.M.J, Moncada S. A novel citrulline-forming enzyme implicated in the formation of nitric oxide by vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989;158:348–352. doi: 10.1016/s0006-291x(89)80219-0. 10.1016/S0006-291X(89)80219-0 [DOI] [PubMed] [Google Scholar]
- Palmer R.M.J, Ferrige A.G, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. 10.1038/327524a0 [DOI] [PubMed] [Google Scholar]
- Palmer R.M.J, Ashton D.S, Moncada S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature. 1988a;333:664–666. doi: 10.1038/333664a0. 10.1038/333664a0 [DOI] [PubMed] [Google Scholar]
- Palmer R.M.J, Rees D.D, Ashton D.S, Moncada S. l-Arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1988b;153:1251–1256. doi: 10.1016/s0006-291x(88)81362-7. 10.1016/S0006-291X(88)81362-7 [DOI] [PubMed] [Google Scholar]
- Patrono C, Ciabattoni G, Patrignani P, Pugliese F, Filabozzi P, Catella F, Davi G, Forni L. Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation. 1985;72:1177–1184. doi: 10.1161/01.cir.72.6.1177. [DOI] [PubMed] [Google Scholar]
- Pedersen A.K, FitzGerald G.A. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N. Engl. J. Med. 1984;311:1206–1211. doi: 10.1056/NEJM198411083111902. [DOI] [PubMed] [Google Scholar]
- Persson K, Igawa Y, Mattiasson A, Andersson K.E. Effects of inhibition of the l-arginine/nitric oxide pathway in the rat lower urinary tract in vivo and in vitro. Br. J. Pharmacol. 1992;107:178–184. doi: 10.1111/j.1476-5381.1992.tb14483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper P.J, Vane J.R. Release of additional factors in anaphylaxis and its antagonism by anti-inflammatory drugs. Nature. 1969;223:29–35. doi: 10.1038/223029a0. [DOI] [PubMed] [Google Scholar]
- Piper P.J, Vane J.R, Wyllie J.H. Inactivation of prostaglandins by the lungs. Nature. 1970;225:600–604. doi: 10.1038/225600a0. 10.1038/225600a0 [DOI] [PubMed] [Google Scholar]
- Poderoso J.J, Carreras M.C, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch. Biochem. Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. 10.1006/abbi.1996.0146 [DOI] [PubMed] [Google Scholar]
- Pritchard K.A, Jr, Ackerman A.W, Gross E.R, Stepp D.W, Shi Y, Fontana J.T, Baker J.E, Sessa W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric oxide synthase. J. Biol. Chem. 2001;276:17 621–17 624. doi: 10.1074/jbc.C100084200. 10.1074/jbc.C100084200 [DOI] [PubMed] [Google Scholar]
- Radomski M.W, Palmer R.M.J, Moncada S. Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br. J. Pharmacol. 1987a;92:181–187. doi: 10.1111/j.1476-5381.1987.tb11310.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski M.W, Palmer R.M.J, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987b;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman B.A, Griendling K.K, Harrison D.G. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramagopal M.V, Leighton H.J. Effects of NG-monomethyl-l-arginine on field stimulation-induced decreases in cytosolic Ca2+ levels and relaxation in the rat anococcygeus muscle. Eur. J. Pharmacol. 1989;174:297–299. doi: 10.1016/0014-2999(89)90325-7. 10.1016/0014-2999(89)90325-7 [DOI] [PubMed] [Google Scholar]
- Reddy K.G, Nair R.N, Sheehan H.M, Hodgson J.M. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J. Am. Coll. Cardiol. 1994;23:833–843. doi: 10.1016/0735-1097(94)90627-0. [DOI] [PubMed] [Google Scholar]
- Rees D.D, Palmer R.M.J, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl Acad. Sci. USA. 1989a;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D.D, Palmer R.M.J, Hodson H.F, Moncada S. A specific inhibitor of nitric oxide formation from l-arginine attenuates endothelium-dependent relaxation. Br. J. Pharmacol. 1989b;96:418–424. doi: 10.1111/j.1476-5381.1989.tb11833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D.D, Higgs E.A, Moncada S. Nitric oxide and the vessel wall. In: Colman R.W, Hirsh J, Marder V.J, Clowes A.W, George J.N, editors. Hemostasis and thrombosis. Lippincott Williams & Wilkins; Philadelphia, PA: 2000. pp. 673–682. [Google Scholar]
- Ridker P.M, Cook N.R, Lee I.M, Gordon D, Gaziano J.M, Manson J.E, Hennekens C.H, Buring J.E. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N. Engl. J. Med. 2005;352:1293–1304. doi: 10.1056/NEJMoa050613. 10.1056/NEJMoa050613 [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. 10.1056/NEJM199901143400207 [DOI] [PubMed] [Google Scholar]
- Roth G.J, Majerus P.W. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J. Clin. Invest. 1975;56:624–632. doi: 10.1172/JCI108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rott D, Zhu J, Burnett M.S, Zhou Y.F, Zalles-Ganley A, Ogunmakinwa J, Epstein S.E. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on atherosclerosis progression and susceptibility to cytomegalovirus replication in apolipoprotein-E knockout mice. J. Am. Coll. Cardiol. 2003;41:1812–1819. doi: 10.1016/s0735-1097(03)00304-8. 10.1016/S0735-1097(03)00304-8 [DOI] [PubMed] [Google Scholar]
- Rubanyi G.M, Vanhoutte P.M. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am. J. Physiol. 1986;250:H822–H827. doi: 10.1152/ajpheart.1986.250.5.H822. [DOI] [PubMed] [Google Scholar]
- Samuelsson B, Granstrom E, Hamberg M. In: Proc. Second Nobel Symp. Bergstrom S, Samuelsson B, editors. Nobel Foundation; Stockholm, Sweden: 1967. pp. 31–44. [Google Scholar]
- Schachinger V, Britten M.B, Zeiher A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101:1899–1906. doi: 10.1161/01.cir.101.16.1899. [DOI] [PubMed] [Google Scholar]
- Schafer M, Schafer C, Ewald N, Piper H.M, Noll T. Role of redox signaling in the autonomous proliferative response of endothelial cells to hypoxia. Circ. Res. 2003;92:1010–1015. doi: 10.1161/01.RES.0000070882.81508.FC. 10.1161/01.RES.0000070882.81508.FC [DOI] [PubMed] [Google Scholar]
- Schopfer F.J, Baker P.R, Freeman B.A. NO-dependent protein nitration: a cell signaling event or an oxidative inflammatory response? Trends Biochem. Sci. 2003;28:646–654. doi: 10.1016/j.tibs.2003.10.006. 10.1016/j.tibs.2003.10.006 [DOI] [PubMed] [Google Scholar]
- Schweizer M, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem. Biophys. Res. Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. 10.1006/bbrc.1994.2441 [DOI] [PubMed] [Google Scholar]
- Shyue S.K, Tsai M.J, Liou J.Y, Willerson J.T, Wu K.K. Selective augmentation of prostacyclin production by combined prostacyclin synthase and cyclooxygenase-1 gene transfer. Circulation. 2001;103:2090–2095. doi: 10.1161/01.cir.103.16.2090. [DOI] [PubMed] [Google Scholar]
- Silverstein F.E, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib long-term arthritis safety study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. 10.1001/jama.284.10.1247 [DOI] [PubMed] [Google Scholar]
- Singer R. Acetylsalicylic acid, a probable cause for secondary post-tonsillectomy hemorrhage. Arch. Otolaryng. 1945;42:19–20. [Google Scholar]
- Singer H.A, Peach M.J. Endothelium-dependent relaxation of rabbit aorta. II. Inhibition of relaxation stimulated by methacholine and A23187 with antagonists of arachidonic acid metabolism. J. Pharmacol. Exp. Ther. 1983;226:796–801. [PubMed] [Google Scholar]
- Smith J.B, Willis A.L. Aspirin selectively inhibits prostaglandin production in human platelets. Nat. New Biol. 1971;231:235–237. doi: 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- Smith J.B, Ingerman C, Kocsis J.J, Silver M.J. Formation of an intermediate in prostaglandin biosynthesis and its association with the platelet release reaction. J. Clin. Invest. 1974;53:1468–1472. doi: 10.1172/JCI107695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S.D, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. 10.1056/NEJMoa050405 [DOI] [PubMed] [Google Scholar]
- Solzbach U, Hornig B, Jeserich M, Just H. Vitamin C improves endothelial dysfunction of epicardial coronary arteries in hypertensive patients. Circulation. 1997;96:1513–1519. doi: 10.1161/01.cir.96.5.1513. [DOI] [PubMed] [Google Scholar]
- Spiekermann S, et al. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: relation to endothelium-dependent vasodilation. Circulation. 2003;107:1383–1389. doi: 10.1161/01.cir.0000056762.69302.46. 10.1161/01.CIR.0000056762.69302.46 [DOI] [PubMed] [Google Scholar]
- Stemerman M.B. Vascular injury: platelets and smooth muscle cell response. Phil. Trans. R. Soc. B. 1981;294:217–224. doi: 10.1098/rstb.1981.0100. [DOI] [PubMed] [Google Scholar]
- Stroes E, Kastelein J, Cosentino F, Erkelens W, Wever R, Koomans H, Luscher T, Rabelink T. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J. Clin. Invest. 1997;99:41–46. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr D, Pou S, Rosen G.M. Oxygen reduction by nitric oxide synthases. J. Biol. Chem. 2001;276:14 533–14 536. doi: 10.1074/jbc.R100011200. 10.1074/jbc.R100011200 [DOI] [PubMed] [Google Scholar]
- Svensson J, Hamberg M, Samuelsson B. Prostaglandin endoperoxides IX. Characterization of rabbit aorta contracting substance (RCS) from guinea pig lung and human platelets. Acta Physiol. Scand. 1975;94:222–228. doi: 10.1111/j.1748-1716.1975.tb05881.x. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Mattei P, Ghiadoni L, Sudano I, Salvetti A. Defective l-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation. 1996;94:1298–1303. doi: 10.1161/01.cir.94.6.1298. [DOI] [PubMed] [Google Scholar]
- Tateson J.E, Moncada S, Vane J.R. Effects of prostacyclin (PGX) on cyclic AMP concentrations in human platelets. Prostaglandins. 1977;13:389–397. doi: 10.1016/0090-6980(77)90019-3. 10.1016/0090-6980(77)90019-3 [DOI] [PubMed] [Google Scholar]
- Thomas D.W, Mannon R.B, Mannon P.J, Latour A, Oliver J.A, Hoffman M, Smithies O, Koller B.H, Coffman T.M. Coagulation defects and altered hemodynamic responses in mice lacking receptors for thromboxane A2. J. Clin. Invest. 1998;102:1994–2001. doi: 10.1172/JCI5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting H.H, Timimi F.K, Boles K.S, Creager S.J, Ganz P, Creager M.A. Vitamin C improves endothelium-dependent vasodilatation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Invest. 1996;97:22–28. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper J.N, Cai J, Falb D, Gimbrone M.A., Jr Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl Acad. Sci. USA. 1996;93:10 417–10 422. doi: 10.1073/pnas.93.19.10417. 10.1073/pnas.93.19.10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. 10.1113/jphysiol.2003.049478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin S. Calming the COX-2 furor. BioCentury. 2005;13:A1–A6. [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. 10.1016/S0140-6736(89)91013-1 [DOI] [PubMed] [Google Scholar]
- Vane J.R. The use of isolated organs for detecting active substances in the circulating blood. Br. J. Pharmacol. 1964;23:360–373. doi: 10.1111/j.1476-5381.1964.tb01592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P.M. The end of the quest? Nature. 1987;327:459–460. doi: 10.1038/327459a0. 10.1038/327459a0 [DOI] [PubMed] [Google Scholar]
- Vargaftig B.B, Dao N. Release of vasoactive substances from guinea-pig lungs by slow-reacting substance c and arachidonic acid. Its blockade by nonsteroid anti-inflammatory agents. Pharmacology. 1971;6:99–108. doi: 10.1159/000136231. [DOI] [PubMed] [Google Scholar]
- Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters B.S, Karoui H, Tordo P, Pritchard K.A., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl Acad. Sci. USA. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. 10.1073/pnas.95.16.9220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdis A, Schiffrin E.L. Vascular inflammation: a role in vascular disease in hypertension? Curr. Opin. Nephrol. Hypertens. 2003;12:181–187. doi: 10.1097/00041552-200303000-00009. 10.1097/00041552-200303000-00009 [DOI] [PubMed] [Google Scholar]
- Von der Leyen H.E, Dzau V.J. Therapeutic potential of nitric oxide synthase gene manipulation. Circulation. 2001;103:2760–2765. doi: 10.1161/01.cir.103.22.2760. [DOI] [PubMed] [Google Scholar]
- Wagner A.H, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler. Thromb. Vasc. Biol. 2000;20:61–69. doi: 10.1161/01.atv.20.1.61. [DOI] [PubMed] [Google Scholar]
- Walshe T.E, Ferguson G, Connell P, O'Brien C, Cahill P.A. Pulsatile flow increases the expression of eNOS, ET-1 and prostacyclin in a novel in vitro coculture model of the retinal vasculature. Invest. Ophthalmol. Vis. Sci. 2005;46:375–382. doi: 10.1167/iovs.04-0806. 10.1167/iovs.04-0806 [DOI] [PubMed] [Google Scholar]
- Weiner C.P, Lizasoain I, Baylis S.A, Knowles R.G, Charles I.G, Moncada S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc. Natl Acad. Sci. USA. 1994;91:5212–5216. doi: 10.1073/pnas.91.11.5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T.J, Morley J. Prostaglandins as potentiators of increased vascular permeability in inflammation. Nature. 1973;246:215–217. doi: 10.1038/246215a0. 10.1038/246215a0 [DOI] [PubMed] [Google Scholar]
- Willis A.L. Release of histamine, kinin and prostaglandin during carrageenin-induced inflammation in the rat. In: Mantegazza P, Horton E.W, editors. Prostaglandins, peptides and amines. Academic Press; New York, NY: 1969. pp. 31–38. [Google Scholar]
- Willis A.L, Vane F.M, Kuhn D.C, Scott C.G, Petrin M. An endoperoxide aggregator (Lass), formed in platelets in response to thrombotic stimuli: purification, identification and unique biological significance. Prostaglandins. 1974;8:453–507. doi: 10.1016/0090-6980(74)90062-8. 10.1016/0090-6980(74)90062-8 [DOI] [PubMed] [Google Scholar]
- Wink D.A, Cook J.A, Krishna M.C, Hanbauer I, De Graff W, Gamson J, Mitchell J.B. Nitric oxide protects against alkyl peroxide-mediated cytotoxicity: further insights into the role nitric oxide plays in oxidative stress. Arch. Biochem. Biophys. 1995;319:402–407. doi: 10.1006/abbi.1995.1310. 10.1006/abbi.1995.1310 [DOI] [PubMed] [Google Scholar]
- Worrall B.B, Johnston K.C. Antiplatelet therapy in secondary stroke prevention. Curr. Atheroscler. Rep. 2000;2:104–109. doi: 10.1007/s11883-000-0103-3. [DOI] [PubMed] [Google Scholar]
- Xie W.L, Chipman J.G, Robertson D.L, Erikson R.L, Simmons D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl Acad. Sci. USA. 1991;88:2692–2696. doi: 10.1073/pnas.88.7.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Charles I.G, Moncada S. Nitric oxide: orchestrating hypoxia regulation through mitochondrial respiration and the endoplasmic reticulum stress response. Cell Res. 2005;15:63–65. doi: 10.1038/sj.cr.7290267. [DOI] [PubMed] [Google Scholar]
- Yang S, Bae L, Zhang L. Estrogen increases eNOS and NOx release in human coronary artery endothelium. J. Cardiovasc. Pharmacol. 2000;36:242–247. doi: 10.1097/00005344-200008000-00015. 10.1097/00005344-200008000-00015 [DOI] [PubMed] [Google Scholar]
- Yu I.S, et al. TXAS-deleted mice exhibit normal thrombopoiesis, defective hemostasis, and resistance to arachidonate-induced death. Blood. 2004;104:135–142. doi: 10.1182/blood-2003-10-3661. 10.1182/blood-2003-10-3661 [DOI] [PubMed] [Google Scholar]