

Abstract
cIAP2 (cellular inhibitor of apoptosis protein 2) is induced by NF-κB (nuclear factor κB) when cells need to respond quickly to different apoptotic stimuli. A recent study using cDNA microarray technology has suggested that cIAP2 transcription is regulated in a cell cycle-dependent manner, although the mechanism for such regulation is unknown. In this study, we confirmed the cell cycle-dependent regulation of cIAP2 expression at both the mRNA and protein levels. Additionally, we found that a bipartite CDE (cell cycle-dependent element)/CHR (cell cycle gene homology region) element in the cIAP2 promoter mediates cIAP2 gene activation in G2/M phase. Cell cycle-dependent G2/M-phase-specific cIAP2 expression is enhanced by NF-κB activation, and selective down-regulation of cIAP2 causes cells blocked in mitosis with nocodazole to become susceptible to apoptosis, indicating that the G2/M-phase-specific expression of cIAP2 contributes to the survival of mitotically arrested cells. Our studies describing the NF-κB-independent G2/M-phase-specific expression of cIAP2 will help in further understanding the molecular basis of cIAP2 over-expression in a variety of human cancers.
Keywords: apoptosis, cell cycle, cellular inhibitor of apoptosis protein (cIAP), inhibitor of apoptosis protein (IAP), mitosis, nuclear factor κB (NF-κB)
Abbreviations: Ad, adenovirus; BIR, baculovirus inhibitor of apoptosis protein repeat; CDE, cell cycle-dependent element; CDF-1, cell cycle-dependent factor 1; CHR, cell cycle gene homology region; cIAP, cellular inhibitor of apoptosis protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; IAP, inhibitor of apoptosis protein; IκBα, inhibitory κBα; NF-κB, nuclear factor κB; RT-PCR, reverse transcription PCR; siRNA, small interfering RNA; TNF, tumour necrosis factor-α; TNFR, TNF receptor
INTRODUCTION
IAPs (inhibitors of apoptosis proteins) comprise a family of proteins that suppress mitochondria-dependent and -independent apoptosis by inhibiting caspases [1]. Eight human IAP family proteins, characterized by the presence of one to three BIRs (baculovirus IAP repeats), have been identified. Among these family members, cIAP1 [cellular IAP1; also known as HIAP2 (human IAP2)] and cIAP2 (HIAP1) are the only two IAPs that have been implicated in the signal transduction pathways activated by TNF (tumour necrosis factor-α). In fact, they were originally discovered as proteins recruited to the activated TNFR2 (TNF receptor 2) complex [2]. cIAP1 and cIAP2 inhibit not only the terminal effector caspases-3 and -7 via their BIR domain, but also caspase-8 with the help of TRAF (TNFR-associated factor)-1 and -2 during TNFR1-mediated apoptosis [3].
We have previously characterized the promoter region of the cIAP2 gene and identified two NF-κB (nuclear factor κB)-binding sites that are essential for TNF-mediated induction [4]. Subsequent studies indicated that expression of cIAP2 can also be regulated by other promoter elements. For example, a CRE (cAMP response element) plays a role in the cAMP-mediated induction of cIAP2 in T84 colon cancer cells [5]. cIAP2 is also induced by dexamethasone and, to a greater extent, by dexamethasone and interferon-γ in A549 lung cancer cells [6]. This induction likely is mediated through a putative glucocorticoid responsive element [7]. Furthermore, comprehensive cDNA microarray analysis of the cell cycle-dependent gene expression profile of HeLa cells revealed that the expression of cIAP2 is regulated in a cell cycle-dependent manner [8]. Using similar approaches, cIAP2 has been identified as a primary target gene down-regulated by the tumour suppressor p53, an important regulator of cell cycle arrest and apoptosis [9]. Together, these results suggest that cIAP2 expression can be regulated by multiple mechanisms.
The regulation of apoptosis and cell cycle progression are thought to be inter-related processes. Survivin, an IAP family member, is a well-studied example of this. It is selectively expressed in G2/M phase in a cell cycle-dependent manner and associates specifically with the microtubules of the mitotic spindle [10], implying that survivin could function in both the mitotic spindle checkpoint and apoptosis [11]. The cell cycle-dependent expression of survivin is likely mediated by CDE (cell cycle-dependent element) and CHR (cell cycle gene homology region) elements in its promoter [12,13]. Bipartite CDE/CHR elements are found in the promoter regions of many genes that are maximally expressed in late S/G2 or G2/M phases, such as those coding for cdc25C, cyclin A, cdc2, plk (polo-like kinase), and many other genes [14–19].
Although the expression of cIAP2 has been suggested to be regulated in a cell cycle-dependent manner, the exact mechanism and functional consequence(s) of this regulation are unknown. The present study demonstrates that the cell cycle dependence of cIAP2 transcription requires a CDE and a CHR in the cIAP2 promoter. These elements repress cIAP2 transcription in asynchronized cultures, and this repression is relieved in nocodazole-arrested G2/M phase cells. Nocodazole has been shown to activate NF-κB in HeLa cells and to promote the survival of mitotically arrested cells [20]. However, the G2/M-phase-specific transcription of cIAP2 in G2/M phase is independent of NF-κB activation and increased cIAP2 expression contributes to the survival of mitotically arrested cells. Our studies demonstrate that cIAP2 is the only G2/M-phase-specific NF-κB-related survival gene.
EXPERIMENTAL
Cell culture and synchronization
Culture medium was purchased from Life Technologies. HeLa cells were maintained at 37 °C with 5% CO2 in DMEM (Dulbecco's modified Eagle's medium) supplemented with 10% fetal bovine serum. For synchronization, HeLa cells were arrested with a double-thymidine block as described previously [8]. Briefly, cells were blocked for 18 h with 2 mM thymidine, released for 9 h by washing out the thymidine, and then blocked again with 2 mM thymidine for 17 h. This treatment arrested the cells at the beginning of S phase. The cells were released from the block by washing out the thymidine, and total RNA was prepared for each hour post-release from the arrest. To obtain G1/S phase- or G2/M phase-arrested cells, an asynchronous population of HeLa cells was treated with 2 mM thymidine for 24 h (to arrest cells in G1/S phase) or 200 ng/ml nocodazole for 16 h (to arrest cells in G2/M phase). The cell cycle distribution of the population was determined by propidium iodide staining and flow cytometry (FACSCalibur system, BD Biosciences).
Reagents
Recombinant human TNF was kindly supplied by LG Biotech Research Institute (Daejeon, Korea). All other reagents, including nocodazole and thymidine, were purchased from Sigma, unless otherwise stated.
RNA isolation and RT (reverse transcription)-PCR
Total RNA was isolated using Easy-BLUE (Intron, Seoul, Korea), according to the manufacturer's instructions. Total RNA (1 μg) was reverse transcribed using MMLV (Moloney-murine-leukaemia virus) reverse transcriptase (Promega) with random primers. Reaction mixture (1 μl) was then used as template in a PCR amplification using Taq polymerase (Genenmed, Seoul, Korea) in a final total volume of 20 μl. The PCR products were analysed on 1.2% agarose gels. The gene encoding GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a control. Specific primers used for the analysis of candidate genes were as follows. cIAP2: forward, 5′-AAG TTC TTA CCA CTG TGC AAT G-3′, and reverse, 5′-CAA GTA GAT GAG GGT AAC TGG C-3′); cIAP1: forward, 5′-CCT GTG GTT AAA TCT GCC TTG-3′, and reverse, 5′-CAA TTC GGC ACC ATA ACT CTG-3′; XIAP: forward, 5′-GCA GGG TTT CTT TAT ACT GG-3′, and reverse, 5′-TGT CCC TTC TGT TCT AAC AG-3′; survivin: forward, 5′-GGC ATG GGT GCC CCG ACG TTG-3′, and reverse, 5′-CAG AGG CCT CAA TCC ATG GCA-3′; ICAM-1: forward, 5′-CGT GCC GCA CTG AAC TGG AC-3′, and reverse, 5′-CCT CAC ACT TCA CTG TCA CCT-3′; GAPDH: forward, 5′-CTC AGA CAC CAT GGG GAA GGT GA-3′, and reverse, 5′-ATG ATC TTG AGG CTG TTG TCA TA-3′.
Plasmid construction
Luciferase reporter constructs containing various fragments of the cIAP2 promoter region have been described previously [4]. Mutations in the CHR and CDE elements of the cIAP2 promoter were generated by the site-directed mutagenesis of the −247LUC plasmid or its NF-κB mutated version [−247mκB(1,3)LUC]. To construct the CDE/CHR double-deleted reporter, a new NruI restriction site (TCGCGA) was introduced into the CHR-mutated reporter (−247mCHR) by replacing the cytosine at the −17 position with adenosine (see Figure 2B). The resulting plasmid was cut with NruI and MluNI [a unique restriction site created by the introduction of the mutated CHR (TGGCCA) fragment] and re-ligated, to generate the −247ΔCDE/CHR LUC plasmid. The introduction of each of these mutations was confirmed by DNA sequencing.
Figure 2. G2/M-specific activity of the cIAP2 promoter is mediated by the region containing CDE-like and CHR elements.

(A) Increased cIAP2 promoter activity during G2/M phase. A series of cIAP2-promoter-fragment- containing luciferase constructs were transfected into HeLa cells, and luciferase activity was analysed in cultures synchronized in G1/S phase or in G2/M phase as described in Figure 1(B). The G1/S phase- or G2/M phase-specific increase in luciferase activity is expressed as the fold increase relative to the luciferase activity detected in asynchronized cells. The results are expressed as the means±S.D. for three independent experiments. (B) Sequence analysis of the region (−93 downstream) mediating the cell cycle-regulated transcription of the cIAP2 gene (left-hand panel). The CDE-like element and a CHR element are indicated. The alignment of the CDE-like and CHR elements from the cIAP2 promoter with those of other cell cycle-regulated genes is also shown (right-hand panel).
Transfection and luciferase assays
Transfections were carried out using the poly(ethylenimine) technique [21]. Each reporter luciferase (2 μg) construct was co-transfected with 0.5 μg of pCDM8-β-galactosidase when cells seeded into six-well plates reached 50–70% confluence. At 24 h post-transfection, the drugs, such as nocodazole (200 ng/ml) or TNF (10 ng/ml), were added as indicated. Transfectants were lysed in 0.15 ml of lysis buffer (Promega) and centrifuged at 10000 g for 5 min to remove cellular debris. The resulting cleared lysates were assayed for luciferase and β-galactosidase activities. The results of the luciferase assays were normalized to the respective results of the β-galactosidase assays.
Adenoviral infection
Adenoviruses expressing β-galactosidase (Ad-βGal) and the super-repressor form of IκBα (inhibitory κBα) (Ad-IκBαM) in which serine residues 32 and 36 were mutated to alanine were provided by Dr C.-G. Yoo (Department of Internal Medicine, School of Medicine, Seoul National University, Seoul, Korea). Cells were infected with adenovirus at a multiplicity of infection of 100 for 4 h. After the virus was removed, cells were allowed to recover for 20 h in complete medium.
Western blot analysis
After stimulation, cells were washed with cold PBS, scraped off the plate and resuspended in lysis buffer containing 1% Nonidet P40, 50 mM Hepes (pH 7.5), 100 mM NaCl, 2 mM EDTA, 1 mM pyrophosphate, 10 mM sodium orthovanadate, 1 mM PMSF and 100 mM sodium fluoride. Cell lysates were centrifuged at 12000 g for 10 min at 4 °C. The supernatants were resolved by SDS/PAGE. Proteins were transferred on to nitrocellulose membrane (Bio-Rad). The resulting membranes were blocked in Tris-buffered saline/Tween 20 supplemented with 5% BSA for 1 h, and then incubated with the indicated primary antobodies for 1 h and with a 1:5000 dilution of the horseradish-peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG secondary antibodies (Santa Cruz) for 1 h at room temperature. The blots were then treated with enhanced chemiluminescence reagents (Amersham Pharmacia Biotech) and exposed to film. For Western blotting, we purchased commercial antibodies specific for cIAP1 and cIAP2 (R&D systems; rabbit or goat antiserum), cyclin A (Cell Signaling) and IκBα (Santa Cruz).
RNA interference
The target sequences for the cIAP1- and cIAP2-specific siRNA (small interfering RNA) were determined using the Ambion web-based criteria. Using the T7 RNA polymerase-mediated transcription method (Silencer siRNA construction kit; Ambion), cIAP2 (5′-AAT GAT GGT TGA AGG TTA CAT-3′), cIAP1 (5′-AAG GAG TCT TGC TCG TGC TGG-3′) and GFP (green fluorescent protein) (5′-AAG TCC GCC ATG CCC GAA GGC-3′) siRNAs were synthesized. siRNA (20 pmol) was transfected using Lipofectamine 2000™ (Life Technologies) into HeLa cells grown in six-well plates. Post-transfection (60 h), RT-PCR analysis was performed to examine the effectiveness of the siRNAs.
Flow cytometric assessment of apoptosis
Apoptotic cells were identified using M30, an antibody specific for a caspase-cleaved cytokeratin 18. HeLa cells that had been transfected with the indicated siRNAs were treated with nocodazole (2 μg/ml) for 14 h. Mitotic cells were recovered using the mechanical shake-off method. These cells were fixed and labelled with a fluorescein-tagged anti-M30 monclonal antibody [22], according to the manufacturer's instructions (Roche Molecular Biochemicals) to identify the apoptotic cells. In addition, cells were stained with propidium iodide to measure apoptotic cells with sub-G1 DNA content. Flow cytometry was performed using the FACSCalibur and CellQuest software (Becton Dickinson).
RESULTS
Cell cycle-dependent expression of cIAP1 and cIAP2
A previous cDNA microarray study identified cIAP2 as one of the candidates of cell cycle-regulated genes [8]. We initially confirmed this regulation by examining the levels of cIAP2 mRNA in synchronized cell populations using RT-PCR. For these experiments, HeLa cells were synchronized at the G1/S boundary by a double-thymidine block and harvested at various times after release from thymidine block for cIAP2 expression analysis. Figure 1(A) (upper left-hand panel) shows that, from its basal level in G1/S synchronized cells, cIAP2 mRNA expression begins to increase in S phase (2 h post-release, as determined by flow cytometric analysis of cellular DNA content) (Figure 1A, lower panel). cIAP2 mRNA expression level peaked in G2 phase (6 h post-release) and decreased as cells enter G1 phase (8–12 h post-release). The maximal level of cIAP2 mRNA was detected earlier than the maximal level of survivin mRNA, which was detected at 8–10 h post-release. cIAP1 was also regulated in a cell cyclespecific manner, with kinetics similar to that of cIAP2. These results demonstrate that the steady-state level of cIAP2 mRNA is tightly regulated throughout the cell cycle, and its expression peaks in late S/G2 phase. To determine if the cell cycle-dependent accumulation of cIAP1 and cIAP2 mRNA results in corresponding accumulation of proteins, we performed Western blot analysis of HeLa cell lysates prepared at various time points post-release from a thymidine block. Relative to the kinetics of mRNA accumulation, there was a delay in cIAP1 and cIAP2 protein accumulation, which began in G2/M phase (6 h post-release). The protein levels were maintained until cells reached early G1 phase (12 h post-release), then abruptly decreased (Figure 1A, upper right-hand panel). In addition, when compared with the protein accumulation of cyclin A, a cell cycle-regulated gene that is expressed beginning in S-phase and reaches a maximal level of expression in G2-phase, cIAP1 and cIAP2 exhibited delayed accumulation kinetics. To further examine the cell cycle-specific expression of cIAP2, we arrested HeLa cells at G1/S phase by treatment with thymidine for 24 h or at G2/M phase by treatment with nocodazole, a microtubule-depolymerizing drug, for 16 h. The level of cIAP2 in these cells was analysed by RT-PCR. The results show elevated levels of cIAP2 transcripts detected in G2/M-phase-arrested cells, but not in cells arrested in G1/S phase (Figure 1B).
Figure 1. Cell cycle-regulated expression of cIAP1 and cIAP2.

(A) RT-PCR and Western blot analysis of cIAP2 expression in HeLa cells after release from a double-thymidine block-induced cell cycle arrest. Total mRNA was prepared at the indicated times, and RT-PCR was performed to measure steady-state mRNA levels of cIAP2, cIAP1, survivin and XIAP genes, as described in the Experimental section. Survivin mRNA was measured as a positive control for cell cycle-dependent expression and GAPDH was used as a loading control. Cell lysates were also prepared at the indicated times and processed for Western blot analysis using antibodies specific for cIAP2, cIAP1, cyclin A and GAPDH (as a loading control). Flow cytometric analysis of cellular DNA content at the indicated times post-release from the double-thymidine block is shown in the lower panel. (B) HeLa cells were arrested at G1/S or G2/M phases by treatment with thymidine for 24 h or nocodazole for 16 h respectively. Cell cycle arrest at the respective phases was confirmed by flow cytometric analysis of cellular DNA content. RT-PCR analysis of cIAP2 mRNA expression was conducted as described in the text.
Mapping of the promoter region responsible for the cell cycle-regulated expression of cIAP2
We examined whether the accumulation of cIAP2 mRNA during G2/M phase was regulated by the cIAP2 promoter. A series of plasmids in which different cIAP2 promoter fragments drive the expression of luciferase [4] were transfected into HeLa cells, and luciferase activity was assayed in cultures synchronized in G1/S and G2/M phases (Figure 2A). Cells arrested in G2/M phase exhibited luciferase activities which were 4–8-fold greater than that of cells arrested in G1/S phase. The promoter fragment containing a deletion up to nucleotide position −93 (−93LUC) retained the activity for the G2/M-phase-specific expression, indicating the promoter region regulating the cell cycle-dependent transcription of cIAP2 is further downstream of −93. Sequence analysis of this region revealed a putative CDE-like element (at position +6) and a CHR element (at position +20) downstream of a TATA-like box (TTTAAA). Alignment of these promoter elements with the CDE and CHR of other cell cycle-regulated gene promoters [23,24] showed that the CHR site of the cIAP2 promoter perfectly matches with the CHR consensus sequence (TTGAAA). The CDE element of the cIAP2 promoter has one biased base change (at the fifth position) from the CDE consensus sequence (Figure 2B).
Bipartite CDE/CHR promoter element regulates the cell cycle-dependent expression of cIAP2
The presence of a bipartite CDE/CHR element in the cIAP2 promoter suggests that this element may function as a binding site for a G1-phase-specific repressor, a function which it performs in other G2/M-phase-specific genes. To investigate such a repressive function of this element, we constructed luciferase expression plasmids containing cIAP2 promoters with mutations in either the CHR element or in both the CDE and CHR elements or with a deletion of both the CDE and CHR elements (Figure 3A). These three mutant constructs were transfected into HeLa cells, and basal luciferase activity was measured (Figure 3B). Comparison with a wild-type construct (−247LUC), mutation of the CHR resulted in an increase in basal promoter activity, and further gradual increases in promoter activity were observed for the CDE/CHR-mutated construct and the CDE/CHR-deleted construct. Following cell cycle arrest of transfected HeLa cells in G2/M phase with nocodazole treatment for 16 h, the wild-type promoter construct showed 5.4-fold increase in promoter activity relative to the activity of asynchronized cells, and mutating the CHR element modestly reduced this increase to 3-fold. Further decreases in activity were detected in cells transfected with the CDE/CHR-mutated (2.2-fold) and CDE/CHR-deleted (1.4-fold) promoter constructs (Figure 3C, white bars). These results suggest that the CDE and CHR sites in cIAP2 promoter transcriptionally repress expression in cycling cells. This repression is likely to be mediated by the binding of a G1-phase-specific repressor to these sites and is relieved when cells enter G2/M phase. In experiments performed using CDE/CHR mutant promoter constructs containing non-functional NF-κB mutations, similar results were obtained, although the increase in G2/M-phase cells was slightly decreased (Figure 3C, black bars).
Figure 3. Mutational analysis of the CDE-like and CHR elements of the cIAP2 promoter.

(A) Representation of the luciferase reporter constructs containing mutations in either the CHR or both the CDE and CHR elements of the cIAP2 promoter. (B) The reporter plasmids were transfected into HeLa cells, and basal promoter activities of the wild-type reporter (−247 LUC) and each mutant reporter were measured in asynchronously growing transfectants. The results are expressed as the means±S.D. for three independent experiments. (C) Promoter activities of the mutant reporters containing either wild-type (white bars) or mutant (black bars) NF-κB sites were measured in transfectants arrested in G2/M phase by nocodazole treatment. Results are shown as the fold increase relative to the activity detected in untreated transfectants. The results are expressed as the means±S.D. for three independent experiments.
Cell cycle-dependent expression of cIAP2 is independent of NF-κB activation in nocodazole-arrested cells
It has been demonstrated previously that microtubule-depolymerizing compounds, such as nocodazole, can activate NF-κB, especially in HeLa cells [20,25]. Reporter constructs containing a cIAP2 promoter with either mutations of the two NF-κB elements (Figure 3C) or deletions of the two NF-κB elements (Figure 2A) were activated in nocodazole-mediated G2/M phase-arrested cells. This result indicates that cIAP2 gene activation in nocodazole-arrested G2/M phase cells is independent of nocodazole-mediated activation of NF-κB. To confirm this result, we examined whether cIAP2 expression could be activated in nocodazole-arrested HeLa cells in which the NF-κB signalling pathway was inhibited. Initially, we examined cIAP2 mRNA levels following treatment of cells with nocodazole and/or TNF (Figure 4A, upper panel). TNF induced cIAP2 transcription by 2 h post-treatment, and this level decreased after 16 h. Although the nocodazole treatment did not significantly increase cIAP2 transcription after 2 h, cIAP2 expression was increased to similar levels by 16 h. Treatment with both TNF and nocodazole for 16 h increased cIAP2 expression to a level that was the sum of that detected with either treatment alone. These findings were supported by experiments using cells infected with adenoviruses (Figure 4A, lower panel); the control virus (Ad-βGal) did not affect cIAP2 mRNA expression following 16 h treatment with TNF or nocodazole, or both (Figure 4A, upper panel). Immunoblot analysis using a cIAP2-specific antibody demonstrated the additive effects of simultaneous treatment of TNF and nocodazole on cIAP2 expression. NF-κB inhibition, mediated by the infection with adenovirus expressing the IκBα super-repressor (Ad-IκBαM), completely blocked TNF-induced cIAP2 expression at both the mRNA and protein level; however, this effects was not observed in cells treated with nocodazole for 16 h, although the cIAP2 expression level was reduced. In addition, cells treated with both TNF and nocodazole and those treated with only nocodazole showed similar levels of cIAP2 expression upon inhibition of NF-κB activation. This finding indicates that cIAP2 gene activation in G2/M-phase-arrested cells is independent of NF-κB activation.
Figure 4. G2/M-phase-specific transcription of cIAP2 is additively increased by TNF-activated NF-κB.
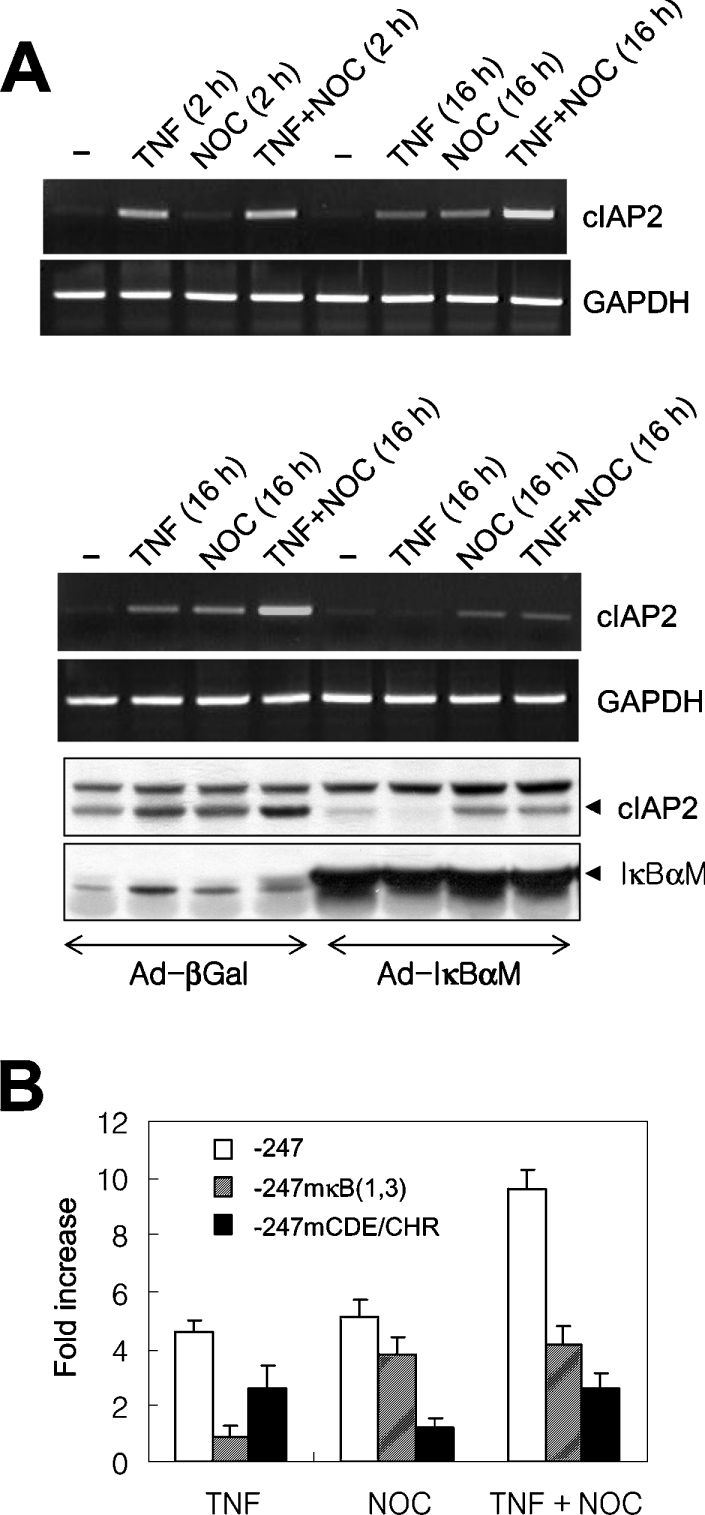
(A) HeLa cells were treated with TNF or nocodazole (NOC), or both TNF and nocodazole, for 2 or 16 h, and the level of cIAP2 mRNA was assessed by RT-PCR. GAPDH mRNA was used as a loading control (upper panel). HeLa cells infected with either a control virus (Ad-βGal) or a virus expressing the IκBα super-repressor (Ad-IκBαM) were treated with TNF or nocodazole, or both TNF and nocodazole, for 16 h. Total RNA was processed, and the levels of cIAP2 and GAPDH mRNA were analysed by RT-PCR. Cell lysates were also prepared to measure levels of cIAP2 and IκBα super-repressor protein by immunoblotting with polyclonal antibodies specific for cIAP2 and IκBα respectively (lower panel). (B) HeLa cells were transfected with the wild-type (−247LUC), the NF-κB mutant [−247mκB(1,3)] or the CDE/CHR mutant (−247mCDE/CHR) reporters. After 24 h, the transfectants were divided into four samples: one was left untreated, and the other three were treated with TNF or nocodazole, or both TNF and nocodazole, for 16 h. Luciferase activities of each transfectant culture were measured, and the results are expressed as the fold increase in luciferase activity relative to the activity of the untreated control. The results are expressed as the means±S.D. for three independent experiments.
To further confirm the NF-κB-independent G2/M phase-specific expression of cIAP2, we analysed the activity of reporter constructs containing cIAP2 of wild-type, mutant CDE/CHR or mutant NF-κB promoters (Figure 4B). In cells transfected with the wild-type reporter (−247LUC), TNF and nocodazole treatment resulted in 4.6- and 5.1-fold increases in luciferase activity respectively relative to untreated controls. The luciferase activity of cells treated with both TNF and nocodazole was increased 9.6-fold, further indicating that the two stimuli have additive effects. Although the cIAP2 promoter with mutations in the two functional NF-κB-binding sites [−247mκB(1,3)] did not respond to TNF treatment, it was responsive to nocodazole treatment. In contrast, the reporter containing the promoter lacking a functional CDE/CHR element (−247mCDE/CHR) was TNF-inducible (2.5-fold induction), but was unresponsive to nocodazole treatment.
Increased levels of cIAP2 in G2/M phase contribute to cell survival during mitotic cell cycle arrest
As mentioned above, nocodazole induces NF-κB activation independent of its mitotic arrest function. This activated NF-κB serves to promote cell survival, possibly through the induction of NF-κB-regulated anti-apoptotic genes [20]. Therefore, we examined whether increased cIAP2 expression in nocodazole-arrested cells provide protection from apoptosis following mitotic arrest. HeLa cells were transfected with cIAP1-specific siRNA, cIAP2-specific siRNA or control GFP-specific siRNA. Post-transfection (60 h), the cells were treated with nocodazole (2 μg/ml) for 14 h. This treatment resulted in the mitotic arrest of more than 80% of cells with a sign of early apoptosis, which can be easily collected using a mechanical shake-off method as described previously [20]. RT-PCR and Western blot analysis showed that mRNA and protein levels of cIAP1 and cIAP2 were significantly reduced from basal levels in their respective siRNA-transfected cells, but not in cells transfected with the GFP siRNA (Figure 5A). To determine whether this selective down-regulation of cIAP1 or cIAP2 expression increased the incidence of nocodazole-induced apoptosis, we stained the cells with the fluorescein-tagged M30 antibody, which is specific for caspase-cleaved cytokeratin 18 products, and examined them by flow cytometry. As shown in Figure 5(B), 16–18% of asynchronized cells undergo spontaneous apoptosis, regardless of transfected siRNA targets. Treatment of siRNA transfectants with nocodazole increased the percentage of cells undergoing apoptosis (GFP siRNA transfectants, 40.2%; cIAP1 siRNA, 59.7%; cIAP2 siRNA, 58.2%; cIAP1 and cIAP2 siRNAs, 73.1%) (Figure 5B). The siRNA-mediated knockdown of both cIAP1 and cIAP2 resulted in a greater increase in the induction of apoptosis than the knockdown of either molecule alone. Treatment of cells with proteasome inhibitors, which block the nocodazole-induced activation of NF-κB, reduced the survival of nocodazole-arrested mitotic cells [20]. When GFP siRNA transfectants were treated with both nocodazole and ALLN (N-acetyl-L-leucyl-L-leucylnorleucinal), a proteasome inhibitor, the percentage of cells undergoing apoptosis increased to 85.2%. This result indicates that, in addition to cIAP1 and cIAP2, other NF-κB-induced anti-apoptotic proteins are involved in the protection from mitotic cell cycle arrest-mediated apoptotic cell death. Therefore, the cell cycle-dependent, G2/M phase-specific expression of cIAP2 and cIAP1 contributes to cell survival during mitotic cell cycle arrest. Representative overlapped M30 antibody-stained profiles of untreated and nocodazole-treated siRNA transfectants are shown in Figure 5(C). We also examined the effect of cIAP1 and cIAP2 knockdown on cell survival by investigating the hypodiploid DNA content of HeLa siRNA transfectants after exposure to nocodazole (400 ng/ml) for 24 h. In the absence of nocodazole, we observed no significant difference in the percentage of cells with sub-G1-phase DNA content, nor were there any differences in the cell cycle profiles compared with the control GFP siRNA-transfected or naïve untransfected cells. This indicates that transfection of each siRNA itself had no effect on normal progression through cell cycle and mitosis. As expected, in the presence of nocodazole, a significant increase in apoptosis was observed in the cells in which cIAP1 or cIAP2 had been silenced, but not in the control GFP siRNA-transfected or naïve cells, confirming that knockdown of cIAP1 or cIAP2 sensitized cells to nocodazole by significantly increasing apoptosis (Figure 5D).
Figure 5. Down-regulation of cIAP1 and/or cIAP2 proteins increases cell death in nocodazole-arrested mitotic cells.

(A) HeLa cells were transfected with cIAP1-specific siRNA, cIAP2-specific siRNA or control GFP-specific siRNA. Post-transfection (60 h), the cells were processed for RT-PCR analysis using primers specific for cIAP1 and cIAP2 mRNA and also for Western analysis. (B, C) HeLa cells were transfected with cIAP1-specific siRNA, cIAP2-specific siRNA or control GFP-specific siRNA. Post-transfection (60 h), cells were collected and separated into two samples: one was left untreated and the other was treated with nocodazole (2 μg/ml) for 14 h. The nocodazole-treated mitotically-arrested cells were collected by mechanical shake-off. These cells, along with the untreated control cells, were processed for flow cytometric analysis after staining with a fluorescein-tagged anti-M30 antibody. GFP-specific siRNA transfectants treated with nocodazole and ALLN (N-acetyl-L-leucyl-L-leucylnorleucinal) for 14 h were used as a positive control for apoptotic cells. Cells in the area marked by M1 are considered to be undergoing apoptosis. The change in the percentage of apoptotic cells following nocodazole treatment is indicated. Three independent experiments were performed, and the mean percentage of apoptotic cells is presented (B). Representative profiles of anti-M30 antibody-stained untreated control (continuous line) and nocodazole-arrested (grey shading) cells transfected with each of the specific siRNAs are shown (C). (D) HeLa cells were transfected with the indicated siRNA and after 48 h of transfection the cells were left untreated or treated with nocodazole (400 ng/ml) for 24 h. Cells were harvested and collected for flow cytometric analysis of cellular DNA content. Cell death was scored by the percentage of cells with a DNA content of less than 2N (marked by M1).
DISCUSSION
In the present study, we examined the cell cycle-dependent regulation of cIAP2 promoter activity and identified the promoter sequence elements responsible for this regulation. Our results showed that the CDE and CHR elements in the cIAP2 promoter control the transcriptional activation of cIAP2 expression in G2/M-phase-synchronized cells. Additionally, we demonstrated that this enhanced cIAP2 expression in mitotically arrest cells contributes to cell survival.
In HeLa cells released from a G1/S-phase arrest, steady-state levels of cIAP2 mRNA rose and peaked in late S-phase/G2. Correspondingly, after a 4 h lag period, cIAP2 and cIAP1 proteins levels increased during G2/M phase. These elevated levels were maintained until early G1 phase and then decreased when cells entered early G1 phase (Figure 1). A previous study reported that the cell cycle-dependent change in cIAP1 protein levels was not detectable in HeLa cells by Western blot analysis [26]; however, in the present study we demonstrated that the cell cycle periodicity of cIAP1 protein expression in the same HeLa cells is detectable. cIAP proteins function as E3 ubiquitin ligases for various target proteins and for themselves through a RING finger domain at their C-terminal ends [27,28]. Therefore, the cellular abundance of cIAPs can be regulated by transcription (regulating protein synthesis) and by ubiquitin-mediated proteasome degradation (regulating protein stability). An intriguing question is whether the sharp decrease in early G1 phase is a result of its intrinsic auto-ubiquitinating activity or the activity of other ubiquitin ligases that are activated during G1 phase. In addition, relative to the kinetics of mRNA accumulation, there is a delay in protein accumulation of cIAP1 and cIAP2, strongly suggesting that post-transcriptional controlling mechanisms involving the action of E3 ubiquitin ligase activity of cIAPs or factors that control such activity towards themselves are involved in cell cycle-dependent expression of these proteins.
The variation in cIAP2 mRNA levels throughout the progression through the cell cycle is based, at least in part, on promoter function. As shown in the reporter gene activation experiments (Figure 2), the region 93 nt upstream of the putative transcription start site directs basal cIAP2 promoter activity in asynchronously growing HeLa cells. A 4–8-fold increase in promoter activity was observed in G2/M phase-synchronized cells relative to G1/S-phase-arrested cells. An alignment of the cIAP2 promoter sequence with the CDE/CHR elements of the known G2/M-phase-specific genes revealed that, although the CHR sequence perfectly matches the consensus sequence, the CDE sequence has one biased base change from the CDE consensus sequence. Also, the spacing of the CDE and CHR elements in the cIAP2 promoter is different from the spacing in the promoters of other G2/M-phase-specific genes. However, mutagenesis experiments revealed that the cIAP2 CDE and CHR sequences functioned as G1 repressor elements, since both the basal expression level and the elevated expression level of the promoter with a mutation in the CHR element were further increased by mutation of the CDR element. Furthermore, reporter gene activation in G2/M-phase-synchronized cells gradually decreased with the sequential introduction of CHR and CDE element mutations, indicating that these elements mediate the cell cycle periodicity of cIAP2 gene expression.
To examine the protein complexes that bind to the CDE/CHR elements of the cIAP2 promoter, we performed gel-shift assays using nuclear extracts from both asynchronously growing and nocodazole-treated, G2/M-phase-arrested HeLa cells. The results demonstrated a specific protein complex binds to the CDE/CHR elements of the cIAP2 promoter in growing HeLa cells. However, these DNA-binding complexes were still detectable in G2/M phase-arrested HeLa nuclear extract (results not shown). In fact, the identity of the repressor complex that binds to various CDE/CHR elements in vivo is controversial. Although the specific DNA-binding activity of protein complexes can be demonstrated using gel-shift assays, the cell cycle-dependent binding of these complexes is not often described, with the exception of the, as yet uncharacterized, CDF-1 (cell cycle-dependent factor 1). CDF-1 binds to the CDE and CHR elements in the cdc2 promoter in quiescent NIH 3T3 cells, and this gradually diminishes following serum stimulation [29]. In addition to CDF-1, other studies have shown that E2F ternary complexes can bind with low affinity to the CDE element of the cdc2 cyclin A promoters [16]. Another uncharacterized protein, called CHF (cyclin A CHR-binding factor), binds to the CHR of the cyclin A promoter [30]. These results suggest the existence of different complexes of CDE/CHR and their binding proteins.
Among the CDE/CHR-regulated S/G2 phase- or G2/M phase-specific genes, cIAP2 is the only gene that can be activated by NF-κB. The connection between NF-κB activation and cell cycle progression has been described for the expression of the G1-phase specific cyclin D1 gene. Cyclin D1 is a key regulator of the G1 phase checkpoint, and the expression of the cyclin D1 gene during G1 phase is controlled by NF-κB in conjunction with other transcription factors, such as AP1, Sp1 or E2F-1 [31,32]. This pathway represents one mechanism by which NF-κB regulates cell growth. Concurrently, NF-κB regulates cell survival, a function which primarily depends upon its ability to induce a variety of anti-apoptotic genes, including cIAP2 [33]. Recently, NF-κB has been shown to promote cell survival in nocodazole-arrested mitotic HeLa cells [20]. Since nocodazole can induce NF-κB, NF-κB-regulated anti-apoptotic gene products have been suggested to be involved in the survival of mitotically arrested cells. Our studies demonstrate that increased cIAP2 expression in nocodazole-synchronized mitotic cells contributes to survival from apoptosis induced by this anti-microtubule drug. cIAP2 gene activation by nocodazole-induced NF-κB is independent of the CDE/CHR-mediated transcriptional de-repression of cIAP2 gene in G2/M- phase cells. The magnitude of cIAP2 expression in nocodazole-arrested G2/M-phase cells is further enhanced by the treatment with TNF, suggesting that certain cancer cells under inflammatory stimuli or with constitutive NF-κB activation may have a better chance of survival during a mitotic cell cycle arrest.
In addition to the suppression of apoptosis in mitotically arrested cells, what function might increased cIAP2 expression during G2/M phase serve? A recent study [26] has suggested a potential role for cIAP molecules in the cell cycle. They showed that cIAP1 is predominantly a nuclear protein in HeLa cells, whereas cIAP2 is localized to both the cytoplasm and nuclear compartments. Over-expression of cIAP1 induces aberrant cell division and accumulation of polyploid cells, possibly due to a defective regulation of mitotic arrest. Furthermore, they showed that cIAP1 can interact with survivin which controls the mitotic checkpoint. Given the structural and functional similarity between cIAP1 and cIAP2, it is likely that cIAP2 could also perform such a cell cycle regulatory function, possibly as part of G2/M-phase checkpoint.
Deregulation of cIAP2 transcription provides a mechanism by which malignant cells could escape cell cycle checkpoints. cIAP1 and cIAP2 have been identified as candidate oncogenes that are overexpressed in lung cancer [34]. Over-expression of cIAP2 is also observed in neutrophilic leukeamia [35] and bladder cancer [36]. It has recently been reported that a genetic defect in the CDE and CHR elements of the human survivin promoter is correlated with the over-expression of survivin in cancer cells [37]. Therefore, our finding that the CDE and CHR elements of the cIAP2 gene promoter functionally regulate the gene's cell cycle-dependent transcription may further our understanding of the molecular consequences of cIAP2 over-expression in some human cancers.
Acknowledgments
This work was supported by Korea Research Foundation grants (KRF-2004-005-C00129, KRF-2000-015-DP0407) and by a grant to the Protein Network Research Center (Yonsei University) from the Ministry of Science and Technology/Korea Science and Engineering Foundation. This work was also partly supported by the Yonsei Biomedical Science and Technology Initiative (YBSTI) program.
References
- 1.Liston P., Fong W. G., Korneluk R. G. The inhibitors of apoptosis: there is more to life than Bcl2. Oncogene. 2003;22:8568–8580. doi: 10.1038/sj.onc.1207101. [DOI] [PubMed] [Google Scholar]
- 2.Rothe M., Pan M. G., Henzel W. J., Ayres T. M., Goeddel D. V. The TNFR2–TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 1995;83:1243–1252. doi: 10.1016/0092-8674(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang C. Y., Mayo M. W., Korneluk R. G., Goeddel D. V., Baldwin A. S., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 4.Hong S. Y., Yoon W. H., Park J. H., Kang S. G., Ahn J. H., Lee T. H. Involvement of two NF-κB binding elements in tumor necrosis factor α -, CD40-, and Epstein-Barr virus latent membrane protein 1-mediated induction of the cellular inhibitor of apoptosis protein 2 gene. J. Biol. Chem. 2000;275:18022–18028. doi: 10.1074/jbc.M001202200. [DOI] [PubMed] [Google Scholar]
- 5.Nishihara H., Kizaka-Kondoh S., Insel P. A., Eckmann L. Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)-2. Proc. Natl. Acad. Sci. U.S.A. 2003;100:8921–8926. doi: 10.1073/pnas.1533221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wen L. P., Madani K., Fahrni J. A., Duncan S. R., Rosen G. D. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-γ and Fas. Am. J. Physiol. 1997;273:L921–L929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- 7.Webster J. C., Huber R. M., Hanson R. L., Collier P. M., Haws T. F., Mills J. K., Burn T. C., Allegretto E. A. Dexamethasone and tumor necrosis factor-α act together to induce the cellular inhibitor of apoptosis-2 gene and prevent apoptosis in a variety of cell types. Endocrinology. 2002;143:3866–3874. doi: 10.1210/en.2002-220188. [DOI] [PubMed] [Google Scholar]
- 8.Whitfield M. L., Sherlock G., Saldanha A. J., Murray J. I., Ball C. A., Alexander K. E., Matese J. C., Perou C. M., Hurt M. M., Brown P. O., Botstein D. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kannan K., Amariglio N., Rechavi G., Jakob-Hirsch J., Kela I., Kaminski N., Getz G., Domany E., Givol D. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene. 2001;20:2225–2234. doi: 10.1038/sj.onc.1204319. [DOI] [PubMed] [Google Scholar]
- 10.Li F., Ambrosini G., Chu E. Y., Plescia J., Tognin S., Marchisio P. C., Altieri D. C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature (London) 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 11.Altieri D. C. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 12.Li F., Altieri D. C. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59:3143–3151. [PubMed] [Google Scholar]
- 13.Otaki M., Hatano M., Kobayashi K., Ogasawara T., Kuriyama T., Tokuhisa T. Cell cycle-dependent regulation of TIAP/m-survivin expression. Biochim. Biophys. Acta. 2000;1493:188–194. doi: 10.1016/s0167-4781(00)00142-1. [DOI] [PubMed] [Google Scholar]
- 14.Zwicker J., Lucibello F. C., Wolfraim L. A., Gross C., Truss M., Engeland K., Muller R. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J. 1995;14:4514–4522. doi: 10.1002/j.1460-2075.1995.tb00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwicker J., Gross C., Lucibello F. C., Truss M., Ehlert F., Engeland K., Muller R. Cell cycle regulation of cdc25C transcription is mediated by the periodic repression of the glutamine-rich activators NF-Y and Sp1. Nucleic Acids Res. 1995;23:3822–3830. doi: 10.1093/nar/23.19.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu N., Lucibello F. C., Engeland K., Muller R. A new model of cell cycle-regulated transcription: repression of the cyclin A promoter by CDF-1 and anti-repression by E2F. Oncogene. 1998;16:2957–2963. doi: 10.1038/sj.onc.1201838. [DOI] [PubMed] [Google Scholar]
- 17.Uchiumi T., Longo D. L., Ferris D. K. Cell cycle regulation of the human polo-like kinase (PLK) promoter. J. Biol. Chem. 1997;272:9166–9174. doi: 10.1074/jbc.272.14.9166. [DOI] [PubMed] [Google Scholar]
- 18.Liu N., Lucibello F. C., Zwicker J., Engeland K., Muller R. Cell cycle-regulated repression of B-myb transcription: cooperation of an E2F site with a contiguous corepressor element. Nucleic Acids Res. 1996;24:2905–2910. doi: 10.1093/nar/24.15.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haugwitz U., Wasner M., Wiedmann M., Spiesbach K., Rother K., Mossner J., Engeland K. A single cell cycle genes homology region (CHR) controls cell cycle-dependent transcription of the cdc25C phosphatase gene and is able to cooperate with E2F or Sp1/3 sites. Nucleic Acids Res. 2002;30:1967–1976. doi: 10.1093/nar/30.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mistry P., Deacon K., Mistry S., Blank J., Patel R. NF-κB promotes survival during mitotic cell cycle arrest. J. Biol. Chem. 2004;279:1482–1490. doi: 10.1074/jbc.M310413200. [DOI] [PubMed] [Google Scholar]
- 21.Boussif O., Lezoualc'h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leers M. P., Kolgen W., Bjorklund V., Bergman T., Tribbick G., Persson B., Bjorklund P., Ramaekers F. C., Bjorklund B., Nap M., Jornvall H., Schutte B. Immunocytochemical detection and mapping of a cytokeratin 18 neo-epitope exposed during early apoptosis. J. Pathol. 1999;187:567–572. doi: 10.1002/(SICI)1096-9896(199904)187:5<567::AID-PATH288>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 23.Badie C., Itzhaki J. E., Sullivan M. J., Carpenter A. J., Porter A. C. Repression of CDK1 and other genes with CDE and CHR promoter elements during DNA damage-induced G(2)/M arrest in human cells. Mol. Cell. Biol. 2000;20:2358–2366. doi: 10.1128/mcb.20.7.2358-2366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y., Saavedra H. I., Holloway M. P., Leone G., Altura R. A. Aberrant regulation of survivin by the RB/E2F family of proteins. J. Biol. Chem. 2004;279:40511–40520. doi: 10.1074/jbc.M404496200. [DOI] [PubMed] [Google Scholar]
- 25.Rosette C., Karin M. Cytoskeletal control of gene expression: depolymerization of microtubules activates NF-κB. J. Cell Biol. 1995;128:1111–1119. doi: 10.1083/jcb.128.6.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuel T., Okada K., Hyer M., Welsh K., Zapata J. M., Reed J. C. cIAP1 localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 2005;65:210–218. [PubMed] [Google Scholar]
- 27.Vaux D. L., Silke J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005;6:287–297. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 28.Park S. M., Yoon J. B., Lee T. H. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004;566:151–156. doi: 10.1016/j.febslet.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 29.Liu N., Lucibello F. C., Korner K., Wolfraim L. A., Zwicker J., Muller R. CDF-1, a novel E2F-unrelated factor, interacts with cell cycle-regulated repressor elements in multiple promoters. Nucleic Acids Res. 1997;25:4915–4920. doi: 10.1093/nar/25.24.4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philips A., Chambeyron S., Lamb N., Vie A., Blanchard J. M. CHF: a novel factor binding to cyclin A CHR corepressor element. Oncogene. 1999;18:6222–6232. doi: 10.1038/sj.onc.1203017. [DOI] [PubMed] [Google Scholar]
- 31.Guttridge D. C., Albanese C., Reuther J. Y., Pestell R. G., Baldwin A. S., Jr NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinz M., Krappmann D., Eichten A., Heder A., Scheidereit C., Strauss M. NF-κB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 1999;19:2690–2698. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perkins N. D. NF-κB: tumor promoter or suppressor? Trends Cell Biol. 2004;14:64–69. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Dai Z., Zhu W. G., Morrison C. D., Brena R. M., Smiraglia D. J., Raval A., Wu Y. Z., Rush L. J., Ross P., Molina J. R., Otterson G. A., Plass C. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 2003;12:791–801. doi: 10.1093/hmg/ddg083. [DOI] [PubMed] [Google Scholar]
- 35.Hasegawa T., Suzuki K., Sakamoto C., Ohta K., Nishiki S., Hino M., Tatsumi N., Kitagawa S. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood. 2003;101:1164–1171. doi: 10.1182/blood-2002-05-1505. [DOI] [PubMed] [Google Scholar]
- 36.Jonsson G., Paulie S., Grandien A. cIAP-2 block apoptotic events in bladder cancer cells. Anticancer Res. 2003;23:3311–3316. [PubMed] [Google Scholar]
- 37.Xu Y., Fang F., Ludewig G., Jones G., Jones D. A mutation found in the promoter region of the human survivin gene is correlated to overexpression of survivin in cancer cells. DNA Cell Biol. 2004;23:527–537. doi: 10.1089/dna.2004.23.527. [DOI] [PubMed] [Google Scholar]