

Abstract
Trypanosoma cruzi, the causative agent of Chagas disease, encodes a number of different cAMP-specific PDE (phosphodiesterase) families. Here we report the identification and characterization of TcrPDEB1 and its comparison with the previously identified TcrPDEB2 (formerly known as TcPDE1). These are two different PDE enzymes of the TcrPDEB family, named in accordance with the recent recommendations of the Nomenclature Committee for Kinetoplast PDEs [Kunz, Beavo, D'Angelo, Flawia, Francis, Johner, Laxman, Oberholzer, Rascon, Shakur et al. (2006) Mol. Biochem. Parasitol. 145, 133–135]. Both enzymes show resistance to inhibition by many mammalian PDE inhibitors, and those that do inhibit do so with appreciable differences in their inhibitor profiles for the two enzymes. Both enzymes contain two GAF (cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coli FhlA) domains and a catalytic domain highly homologous with that of the T. brucei TbPDE2/TbrPDEB2 family. The N-terminus+GAF-A domains of both enzymes showed significant differences in their affinities for cyclic nucleotide binding. Using a calorimetric technique that allows accurate measurements of low-affinity binding sites, the TcrPDEB2 N-terminus+GAF-A domain was found to bind cAMP with an affinity of ∼500 nM. The TcrPDEB1 N-terminus+GAF-A domain bound cAMP with a slightly lower affinity of ∼1 μM. The N-terminus+GAF-A domain of TcrPDEB1 did not bind cGMP, whereas the N-terminus+GAF-A domain of TcrPDEB2 bound cGMP with a low affinity of ∼3 μM. GAF domains homologous with those found in these proteins were also identified in related trypanosomatid parasites. Finally, a fluorescent cAMP analogue, MANT-cAMP [2′-O-(N-methylanthraniloyl)adenosine-3′,5′-cyclic monophosphate], was found to be a substrate for the TcPDEB1 catalytic domain, opening the possibility of using this molecule as a substrate in non-radioactive, fluorescence-based PDE assays, including screening for trypanosome PDE inhibitors.
Keywords: cAMP, cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coliFhlA domain (GAF domain), phosphodiesterase (PDE), Trypanosoma cruzi
Abbreviations: EHNA, erythro-9-(2-hydroxy-3-nonyl)adenine; GAF, cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coliFhlA; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HEK, human embryonic kidney; ITC, isothermal calorimetry; MANT-cAMP, 2′-O-(N-methylanthraniloyl)adenosine-3′,5′-cyclic monophosphate; IBMX, 3-isobutylmethylxanthine; ORF, open reading frame; PDE, cyclic nucleotide phosphodiesterase; RT-PCR, reverse-transcription PCR; Tbr, Trypanosoma brucei; Tcr, Trypanosoma cruzi
INTRODUCTION
The second messenger cAMP is a key regulator of mammalian cell proliferation and differentiation. In trypanosomatid parasites, cAMP also plays a role in these processes and, in addition, is important during cell invasion. Intracellular levels of cAMP vary greatly during the different life-cycle stages of the kinetoplastid protozoa that cause many illnesses, including Chagas disease, leishmaniasis, sleeping sickness in humans, as well as nagana in cattle [1–4]. In kinetoplastids, cAMP levels are regulated by PDE (cyclic nucleotide phosphodiesterase) isoenzymes as well as by adenylate cyclases [1], just as they are in other eukaryotes. PDEs are hydrolytic enzymes that break down cAMP to 5′-AMP, and, by controlling cAMP levels, regulate a large number of cellular processes [5,6]. For example, in mammals and other eukaryotes, PDEs are known to regulate insulin secretion and signalling, olfaction and visual transduction, steroid synthesis and secretion, and cell differentiation and proliferation. Regulation of PDE activities allows fine control of the shape, duration and amplitude of cAMP signals. The different mammalian PDE families can hydrolyse either cAMP or cGMP, or both, despite similarities in their catalytic domains. The mammalian PDEs also have diverse N-terminal domains, allowing for differential localization, kinetics and regulation of activity [5,7,8].
The presence of cAMP PDE activity in lysates of kinetoplasts has been known for many years, but a molecular understanding of the extent of the PDE family members, their structure, regulation and functions in kinetoplasts has just begun. The recently completed genomes of several kinetoplasts [9–11] predict four different Class I PDE families for Trypanosoma cruzi, Trypanosoma brucei and Leishmania mexicana [5], as well as a number of adenylate cyclases.
Unfortunately, the rapid proliferation in the descriptions of kinetoplastid PDEs resulted in diverse nomenclatures among laboratories, making it difficult for investigators to easily compare properties of the enzymes across species. Therefore a comprehensive nomenclature system was recently proposed, based largely on homology comparisons that provides a name for all putative Class I PDEs identified in the recently completed genomes of T. cruzi, T. brucei and Leishmania major [20]. This new system is used in the present paper. In a few places, both are used for clarity of comparison, in which case the original name is put in quotes.
The first kinetoplastid PDEs to be cloned and characterized were the members of the T. brucei ‘TbPDE2’ (T. brucei PDE 2)/TbrPDEB family. RNA interference knock-down studies showed these genes to be essential for proliferation of bloodstream-form T. brucei [12–14]. This observation suggested a role for these enzymes in trypanosome survival and presented these enzymes as prospective drug targets. Another PDE family (‘TbPDE1’/TbrPDEA) also has been characterized in T. brucei [15]. However, this PDE does not appear to be essential for bloodstream forms of the parasite. cAMP-specific PDEs were also recently characterized in the related trypanosomatid L. major [16]. In addition, two different PDE families have recently been characterized in T. cruzi [17–19]. The first was named ‘TcPDE1’, as it was the first identified in T. cruzi. However, owing to its homology with the more thoroughly studied TbrPDEB2, it was renamed TcrPDEB2 in the new nomenclature system [20]. The two PDEs that constitute the PDEB family in trypanosomes appear to be relatively high-affinity cAMP-specific enzymes, structurally most similar to mammalian PDEs 2, 5, 6, 10 and 11. These enzymes all have two N-terminal GAF (cGMP-specific and -stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coli FhlA) domains followed by a catalytic domain. Proteins containing GAF domains can be found in almost all organisms, from cyanobacteria to plants and mammals. In many cases studied, GAF domains appear to function as dimerization or regulatory domains that bind cyclic nucleotides or other small molecules [21]. Whereas most of the mammalian PDE GAF domains (PDEs 2, 5 and 6) are known to bind and/or signal using cGMP [22–24], recently the GAF-A domain of TbrPDEB2 was shown to preferentially bind cAMP over cGMP [25]. Subsequently the GAF domain of human PDE10 also was shown to bind cAMP [26], as were the GAF domains of an adenylate cyclase from the cyanobacterium Anabaena [27].
In the present study we identify and characterize TcrPDEB1, the second member of the PDEB family from T. cruzi, and compare its properties to the recently identified and renamed TcrPDEB2 [18,20]. TcrPDEB1, like TcrPDEB2, is a low-Km, cAMP-specific enzyme, not modulated by cGMP and resistant to most mammalian PDE inhibitors. Additionally, evidence is provided for the expression of the mRNA of both TcrPDEB enzymes in all T. cruzi life-cycle stages and for the existence of homologous-GAF-domain PDEs in numerous other kinetoplastid parasites. The GAF-A domains of both TcrPDEB1 and TcrPDEB2 were found to bind to cAMP. The N-terminus+GAF-A domain of TcrPDEB1 binds cAMP with a relatively low affinity of ∼1 μM. Only the N-terminus+GAF-A of TcrPDEB2 bound cGMP with a measurable affinity. Finally, the catalytic domain of TcrPDEB1 was found to hydrolyse MANT-cAMP [2′-O-(N-methylanthraniloyl)adenosine-3′,5′-cyclic monophosphate], suggesting the possible use of this molecule in non-radioactive fluorescence-based PDE assays.
EXPERIMENTAL
Databases and programs
The expressed-sequence-tag database was searched using BLAST (http://www.ncbi.nlm.nih.gov/BLAST). Conserved domains were identified using the NCBI (National Center for Biotechnology Information) Conserved Domain Search Program (http://www.ncbi.nlm.nih.gov/structure/cdd/wrpsb.cgi), and amino-acid-sequence alignments were performed with ClustalW [28]. For Km or Ki calculations, data from assays were analysed with the PRISM 4.0 program (GraphPad, San Diego, CA, U.S.A.) using a one-site non-linear regression fit or a one-site binding fit. Primers were designed with the help of the Amplify program [29].
Amplification of genomic DNA from T. cruzi
The CL Brenner strain of T. cruzi was obtained from the laboratory of Dr Bianca Zingales (Laboratório de Biologia Molecular de Tripanossomas, Departamento de Bioquimica, Instituto de Quimica, Universidade de São Paulo, São Paulo, Brazil) through Dr José Luis Ramírez (Instituto de Biología Experimental, Universidad Central de Venezuela, Caracas, Venezuela). Parasite genomic DNA was isolated from 1010 parasites, washed twice with 0.85% NaCl, lysed, and the DNA extracted by the method of Medina-Acosta and Cross [30].
Screening of a T. cruzi genomic library, hybridization of a T. cruzi electrokaryotype and generation of the complete ORFs (open reading frames)
A T. cruzi genomic library was originally used to identify and isolate these two enzymes by PCR methods. However, during the process of identification of these two enzymes, the cloning and characterization of TcPDE1/TcrPDEB2 was published [18], and the genome of T. cruzi was published soon after [9], making a detailed description of the methods of limited utility. Nevertheless, for completeness, these methods are provided in the Supplementary data at http://www.BiochemJ.org/bj/399/bj3990305add.htm, along with the primers used.
Expression of T. cruzi PDEs
Full-length TcrPDEB1 and TcrPDEB2 ORFs were amplified by PCR and cloned into the expression vector pCDNA3.1-V5/His (Invitrogen). HEK-293T (human embryonic kidney 293) cells were transiently transfected with 24 μg of DNA using Lipofectamine® 2000 transfection reagent (Invitrogen) according to the manufacturer's protocol, in 100 mm-diameter dishes kept at 37 °C under 5% CO2 for 48 h. Transfected and control (non-transfected) cells were grown in Dulbecco's modified Eagle's medium+10% fetal bovine serum. Cells were harvested, resuspended in 25 mM Tris/HCl, pH 7.5, plus a protease inhibitor cocktail (Sigma; catalogue no. P8340), sonicated (five 3 s bursts) using a Virsonic 100 sonicator (VirTis, Gardiner, NY, U.S.A.) and placed on ice. Homogenates were clarified by a brief centrifugation (∼30 s at 14000 g), and glycerol was added to a final concentration of 25% (v/v). PDE activity in homogenates treated this way was stable for up to 1 week at 4 °C and for over 3 weeks at −20 °C. Protein expression was confirmed by measuring PDE activity compared with non-transfected cells, or cells transfected with the same vector carrying the gene for the green fluorescent protein, and by Western blot analysis using an anti-V5 antibody. The anti-V5 antibody specifically detects the V5 epitope tag (GKPIPNPLLGLDST) present on the N-terminus of the TcrPDEB constructs used in the present study.
Expression and purification of T. cruzi PDE N-terminals+GAF-A domains or the TcrPDEB1 catalytic domain in E. coli
The N-terminus+GAF-A domains of both enzymes, or the catalytic domain of TcrPDEB1 were cloned by PCR into the pET15b vector (Novagen) using the following primers:
 |
They were then transformed into Rosetta™ (DE3) cells (Novagen), grown in Luria–Bertani broth with 75 μg/ml carbenicillin at 37 °C to an attenuance (D600) of 0.6–1.0, induced with 0.2 mM isopropyl β-D-thiogalactoside, and harvested after 22 h of growth at 16 °C. Cells were resuspended and lysed in buffer [25 mM Tris/HCl, pH 7.5, 100 mM NaCl, 10% (v/v) glycerol, 2 mM PMSF and 5 mM β-mercaptoethanol] by microfluidization (10000 lbf/in2; 1 lbf/in2≡6.9 kPa) using a Microfluidizer® high-shear processor (Microfluidics, Newton, MA, U.S.A.), and centrifuged at 16000 g for 30 min. The supernatant was purified on a TALON® metal-affinity resin (Clontech) and eluted with lysis buffer and 150 mM imidazole. Imidazole was removed using a PD10 buffer exchange column, replacing it with the original lysis buffer.
Identification of homologous GAF domains in other trypanosomatid parasites
Genomic DNA from Leishmania amazonensis and Leishmania braziliensis, obtained from the Molecular Genetics Laboratory [IBE-UCV (Instituto de Biología Experimental–Universidad Central de Venezuela), Caracas, Venezuela], Trypanosoma evansi (obtained from Dr Trina Perrone (Grupo de Bioquimica e Inmunologia de Hemoparasitos, Departamento de Biologia Celular, Universidad Simon Bolivar, Caracas, Venezuela), L. mexicana and Crithidia fasciculata, isolated as described in the Supplementary Data at http://www.BiochemJ.org/bj/399/bj3990305add.htm, was digested with XhoI, transferred on to a nylon membrane and hybridized against probe Tc13 (see the Supplementary Data at http://www.BiochemJ.org/bj/399/bj3990305add.htm). Conditions of hybridization were of medium-to-low stringency (25% formamide, 42 °C). As an alternative method to detect PDEs in other parasites, different combinations of oligonucleotides, employed initially for sequencing of T. cruzi PDEs, were used to amplify by PCR genomic DNA from the parasites mentioned above. The following primers were used with the PCR Super Mix High Fidelity Kit (Invitrogen) to amplify the regulatory region, comprising both GAF domains (TcrGAF1):
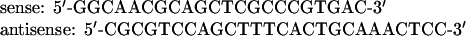 |
The cycling protocol was the same as that used for amplification of T. cruzi DNA, except for lowering the annealing temperature to 60 °C. The sequences amplified were cloned and sequenced.
Generation of antibodies
Antisera against a purified protein fragment encompassing the N-terminal end plus the GAF-A from the T. brucei TbrPDEB2 (Met1–Asp395), denominated ‘anti-NT+GAF-A’, was raised in rabbit. In immunoblots, this antiserum specifically recognizes GAF-A+B and NT+GAF-A protein fragments and holo-enzymes of TbrPDEB1 and TbrPDEB2, as well as the T. cruzi enzymes TcrPDEB1 and TcrPDEB2, but does not detect other (mammalian) GAF domain PDEs.
Protein detection by Western-blot analysis
Homogenates of HEK-293 cells expressing recombinant TcrPDEB1 or TcrPDEB2, were electrophoresed in SDS/10%-(w/v)-polyacrylamide gels and transferred on to PVDF membranes. Following transfer, the membrane was blocked in 5% (w/v) dried skimmed milk/TBST (0.5% Tween 20, 200 mM NaCl and 25 mM Tris/HCl, pH 8.0) and incubated with a 1:1000 dilution of the rabbit NT+GAF-A antiserum (described above) or the anti-V5 antibody. Membranes were washed four times with TBST and further incubated with a 1:2000 dilution of goat anti-rabbit IgG coupled with horseradish peroxidase (Bio-Rad), or rabbit antimouse IgG coupled with horseradish peroxidase (for the anti-V5 antibody). Detection was carried out with the Super Signal West Pico chemiluminescent substrate system (Pierce).
Expression levels of TcrPDEB1 and TcrPDEB2 in T. cruzi morphotypes
Expression levels of TcrPDEB1 and TcrPDEB2 during the parasite life cycle were determined using RT-PCR (reverse-transcription PCR). Total RNA was extracted from different T. cruzi morphotypes (amastigotes, epimastigotes and trypomastigotes) with TRIzol reagent (Gibco) and treated with RQ1-RNAse free DNAse (Promega), and checked for purity and integrity using 0.8% agarose gels. cDNA was synthesized with a Reverse Transcription System (Promega) using 1 μg of RNA. cDNAs were amplified using the same cycling parameters described above, with primers specific for each enzyme, producing fragments of 592 and 569 bp respectively:
 |
for the amplification of sequences belonging to TcrPDEB2 and primers, and
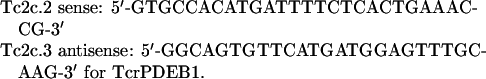 |
Oligonucleotides for amplification of α-tubulin and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) fragments were used as standards. Controls to verify RNA purity, and controls using DNA from Vero cells (guest cells for growing amastigotes), were performed (to rule out amplification of mammalian PDEs with parasite primers). Quantification of the intensities of PCR products was carried out using a Gel Doc 1000 apparatus with the Multi analyst/PC, version 1.1, Build 34 program (Bio-Rad). PCR amplification products were evaluated every five cycles, and amplification for 30 cycles was chosen for further experiments, since band intensities were in the linear range of amplification.
PDE assays
PDE activities were assayed with different concentrations of [3H]cAMP or [3H]cGMP, from 0.1 to 100 μM, using the method of Hansen et al. [31]. Briefly, the assay was performed at 30 °C, in 40 mM Mops, pH 7.5, 0.8 mM EGTA, 15 mM magnesium acetate, 0.2 mg/ml BSA and 100000 c.p.m. of the radiolabelled cyclic nucleotide, in a final volume of 250 μl. The reaction was started by the addition of protein to the substrate/buffer mix in the presence of 15 μM IBMX (3-isobutylmethylxanthine; to inhibit endogenous PDEs in HEK-293 homogenates), after ensuring that IBMX did not affect TcrPDE catalytic activity. Hydrolysis of substrate did not exceed 25%, and PDE activity was proportional to time and enzyme concentration.
For inhibition studies, assays were performed using 1 μM cAMP as substrate, with different concentrations of rolipram (BIOMOL International, L.P., Plymouth Meeting, PA, U.S.A.), enoximone (Marion Merrell Dow Research Institute, Cincinnati, OH, U.S.A.), zaprinast (May & Baker, now part of Rhone-poulenc Rorer, Inc., Collegeville, PA, U.S.A.), dypiridamole, EHNA, cGMP, IBMX, papaverine and pentoxifylline (the latter six obtained from Sigma). ITC (isothermal calorimetry) was also used in some enzyme assays (for both cAMP, as well as MANT-cAMP, hydrolysis measurements), as described below.
cNMP competition binding assays and IC50 curves
To determine IC50 values for cAMP displacement of [3H]cAMP from the purified NT+GAF-A domain of TcrPDEB2, binding assays were conducted using a modification of the assay described previously [25] in a total volume of 1 ml of binding buffer (25 mM NaCl, 5 mM Tris and 5 mM EDTA, pH 7.5). [3H]cAMP at 10 nM and purified bacterial NT+GAF-A protein at ∼8 nM were used for each assay point, with increasing concentrations of unlabelled cNMP (as indicated). Following an incubation of 20–30 min on ice (after determining equilibrium time), sufficient (NH4)2SO4 was added to give a final concentration of 3 M (as described in the Results section). These conditions for the assay were selected by keeping in mind the assumptions of competitive binding curves as described by Motulsky and Christopoulos [32] and as previously described [25]. Some further information is also currently available on the GraphPad Prism website (www.graphpad.com). The solution was filtered on a 0.45-μm-pore-size HA nitrocellulose filter (Millipore), washed twice with the ammonium sulfate solution, dissolved in scintillation fluid (Filter-Count; PerkinElmer), and the counts bound measured in a scintillation counter. Non-linear regression analysis of the data was done using Prism 4.0 (GraphPad) to obtain IC50 values, and presented as means±S.E.M. for four independent experiments. The equilibrium dissociation constant (Ki) was calculated from the IC50 values using the Cheng and Prusoff equation [33]. The binding affinities presented are means±S.E.M for four separate experiments.
ITC
Binding of cAMP to the NT+GAF-A domain of TcrPDEB1 was measured using titration ITC, using a VP-ITC calorimeter from MicroCal (Northampton, MA, U.S.A.). In brief, 1.42 ml of purified TcrPDEB1 NT+GAF-A domain protein at 15–40 μM in 40 mM Mops buffer (pH 7.0) was used for each binding assay, and increasing amounts of cAMP or cGMP (in the same Mops buffer) were titrated (in 5 μl portions) until no more binding occurred (typically 20 injections, with 4 min between each injection). The heat released from each injection was then directly analysed to obtain the binding constant, and the stoichiometry of binding, using the MicroCal/Origin® software (MicroCal) in a method similar to that described previously [34]. The binding affinities presented are the means±S.E.M for four separate experiments done with independently expressed and purified protein. The VP-ITC instrument was also used to determine the heat of hydrolysis of cAMP or MANT-cAMP by the TcrPDEB2 catalytic domain in a continuous assay using a method similar to that described previously for measuring enzyme kinetics using ITC [35]. These data were analysed directly with the Microcal/Origin® software to obtain Km and kcat. The Km and kcat values presented are the means±S.E.M for four separate experiments. Some further information and references are currently available on the MicroCal website (http://www.microcal.com/index.php).
RESULTS
Amplification of ORFs encoding two different PDEs
A novel PDE named TcrPDEB1, conforming to the new proposed nomenclature for cyclic nucleotide PDEs of kinetoplastidae [20], was identified, expressed and characterized. The complete TcrPDEB1 gene codes for a 918-amino-acid protein with a high amino acid homology with (i) the catalytic domain (90%) and (ii) the regulatory domain of T. brucei TbrPDEB1 (93%). TcrPDEB1 has a predicted molecular mass of 102484 Da and a pI of 5.49. The TcrPDEB2 gene codes for a 929-amino-acid protein with high homology at the amino acid level to (i) the catalytic domain (88%) and (ii) the regulatory domain (93%) of T. brucei TbrPDEB2. While this work was in progress, this second gene was reported and designated TcPDE1 by D'Angelo et al. [18]. The name TcrPDEB2 for TcPDE1 is in accordance with the recent proposed kinetoplast nomenclature [20] and is used throughout the present paper.
Expression of TcrPDEB1 and TcrPDEB2
Western-blot analysis of transfected HEK-293 cell lysates with an anti-V5 antibody showed a molecular mass for TcrPDEB1 and TcrPDEB2 of approx. 103 kDa for both recombinant enzymes (Figure 1A), a value in agreement with their predicted molecular masses. The homogenates of HEK-293T cells transfected either with TcrPDEB1 or TcrPDEB2 were also analysed by Western blot, and the recombinant proteins were detected using the polyclonal anti-(NT+GAF-A) antibody, showing that, despite being from different species, they cross-react with their T. cruzi PDE orthologues (Figure 1B). The NT+GAF-A fragment (41 kDa) expressed with an N-terminal GST epitope tag was used as a positive control. The anti-(NT+GAF-A) antibody was also used to detect endogenous TcrPDEBs in Western blots of whole-cell lysates of trypomastigote T. cruzi (with bloodstream form T. brucei as a control) (Figure 1C). The antibody recognizes a single band [both TcrPDEB1 and TcrPDEB2 are about the same molecular mass (∼103 kDa) and cannot easily be distinguished from the recombinant enzyme on an SDS/polyacrylamide gel] and did not have any significant background staining or cross-reactivity with non-specific proteins (in both trypanosome species). The same result was obtained with epimastigote forms of T. cruzi (not shown) and, additionally, this antibody did not recognize other GAF domains from mammalian PDE2 and PDE5. Also, no GAF domains other than the GAF domains present on TcrPDEB1 and TcrPDEB2 can be found in the recently completed T. cruzi, T. brucei and L. major genomes. In immunolocalization studies, a strong signal was seen in the flagellar region of stained typomastigotes, with diffuse cytoplasmic signal (Supplementary data), corroborating previous results obtained for for TcrPDEB2 [18].
Figure 1. Detection of recombinant T. cruzi PDE proteins using specific antibodies, and Western-blot analysis of TcrPDEBs in T. cruzi trypomastigote lysates.

The Figure shows Western blots of homogenates from: H or U, untransfected HEK-293T cells; B1, HEK-293T cells transfected with TcrPDEB1; B2, HEK-293T cells transfected with TcrPDEB2; N, purified NT+GAF-A fragment with a GST epitope tag expressed in E. coli. The antisera used were: (A) anti-V5; (B) anti-(NT+GAF-A). Molecular-mass markers (shown on the left) are given in kDa; 1, 103 kDa; 2, 72 kDa. (C) Western blot of whole-cell lysates from trypomastigote forms of T. cruzi (Tcr), or bloodstream forms of T. brucei (Tbr) detecting native protein using anti-(NT+GAF-A) antibodies. Molecular-mass markers in kDa are shown on the right.
Characterization of TcrPDEB1, and a comparison with TcrPDEB2
When assayed for cyclic nucleotide PDE activity, TcrPDEB1 had a Km of 11.2±0.2 μM, and TcrPDEB2 had a Km of 5.8±0.6 μM with cAMP as substrate (Table 1), corroborating published results for TcrPDEB2 [18]. There was no PDE activity with up to 100 μM cGMP, and cGMP did not stimulate or inhibit the cAMP-hydrolysing activity at concentrations from 1 μM to 1 mM. Both enzymes showed low sensitivity to the non-selective mammalian PDE inhibitors pentoxifylline and IBMX, and to the selective mammalian PDE inhibitors rolipram (which inhibits the PDE4 family), zaprinast (inhibitor for families PDE5 and PDE6) and enoximone (PDE3 inhibitor) (Table 2). However, the enzymes showed substantial differences in their inhibition profiles with papaverine and EHNA. Papaverine (a non-selective mammalian PDE inhibitor) did not inhibit recombinant TcrPDEB1 at concentrations as high as 500 μM. However, an IC50 for papaverine of 105±3 μM was obtained for recombinant TcrPDEB2. EHNA, a selective inhibitor of the mammalian PDE2 family, inhibited TcrPDEB2 with an IC50 of 128±2 μM, but did not inhibit TcrPDEB1 at concentrations up to 200 μM. Dipyridamole was the most effective inhibitor for both recombinant PDEs, with similar IC50 values of 11.3±2 μM (TcrPDEB1) and 15±2 μM (TcrPDEB2) (Table 2). The enzymes, when expressed without a histidine or V5 epitope tag, showed the same activity and inhibitor profiles.
Table 1. Comparison of Km values for cAMP hydrolysis of T. brucei and T. cruzi PDEs.
The Km values for cAMP hydrolysis of the T. cruzi PDEBs were measured, and compared with those of their T. brucei PDEB orthologues, published previously, as well as with the Km of TbrPDEA. Km values for cAMP hydrolysis were calculated using non-linear analysis with the Prism software. The Km values for TbrPDEA, TbrPDEB1 and TbrPDEB2 are from previously published data, as is one of the TcrPDEB2 values.
Table 2. Effect of different compounds on the enzymatic activity of recombinant T. cruzi PDEs.
Inhibition of TcrPDEB1 or B2 cAMP hydrolysis by various known cNMP PDE inhibitors were measured using conventional radioactive PDE assays. The results were also compared with published data for the T. brucei TbrPDEBs as well as for L. mexicana homogenates. A dash (–) indicates values that were not determined, and underlined numbers are from D'angelo et al. [18]. The results are the means±S.E.M. for three independent experiments. The cAMP concentration was 2 μM.
| IC50 (μM) | |||||
|---|---|---|---|---|---|
| Inhibitor | TbrPDEB2/TbPDE2B [12] | TbrPDEB1/TbPDE2C [13] | L. mexicana [39] | TcrPDEB2/TcPDE1 [18] | TcrPDEB1 |
| Dipyridamole | 27 | 14.6 | − | 15.0±2 17±4 | 11.3±2 |
| EHNA | >180 | − | − | 128±2 217±4 | >200 |
| Enoximone | >100 | − | − | >100 | >100 |
| cGMP | >200 | >100 | >1000 | >500 No inhibition | >500 |
| IBMX | >1000 | 1700 | 2000 | >300 >1000 | >300 |
| Papaverine | 304 | − | − | 104.8±3 111±17 | >500 |
| Pentoxifylline | >800 | − | − | >500 | >500 |
| Rolipram | >300 | − | 700 | >500 >500 | >500 |
| Zaprinast | >50 | − | 2800 | >300 | − |
Identification of homologous PDEs in other trypanosomatid parasites
Probe Tc13 was hybridized at medium-low stringency with genomic DNA from L. amazonensis, L. braziliensis, L. mexicana, T. rangeli, T. evansi and C. fasciculata, digested with XhoI. A 5 kb band was observed in the lanes corresponding to members of the Leishmania genus (Figure 2A), whereas at least six bands were obtained with T. cruzi genomic DNA, which strongly suggests the existence of more than one homologous PDE isoform in this parasite. No signal was observed in the case of C. fasciculata or T. rangeli. Amplification of genomic DNA from these parasites with primers designed to amplify the PDE GAF domains (TcGAF1.S–TcGAF1.AS) from T. cruzi resulted in 1 kb PCR products for all the kinetoplastids assayed (Figure 2B). These PCR products were subsequently sequenced and the GAF domain region sequences were compared with each other, and the GAF domains of TbrPDEBs.
Figure 2. Identification of homologous PDEs in different kinetoplastid protozoa.

(A) Autoradiography of probe Tc13 hybridization against genomic DNA from different parasites digested with XhoI. Values alongside arrows indicate sizes in kb. (B) PCR amplification of GAF domains from different parasites: La, L. amazonensis; Lb, L. braziliensis; Tc, T. cruzi; Cf, C. fasciculata; Lm, L. mexicana; Te, T. evans; Tr, T. rangeli. M, molecular-size markers. Arrow indicates 1 kb.
TcrPDEB1 and TcrPDEB2 expression in different T. cruzi morphotypes
T. cruzi PDE mRNA expression was studied using semi-quantitative RT-PCR, by amplifying cDNA obtained from total RNA isolated from three developmental stages from T. cruzi (amastigotes, trypomastigotes and epimastigotes). Primers specific for the N-terminal region of TcrPDEB2 and TcrPDEB1 were used. Figure 3(A) shows nucleotide fragments obtained from RT-PCR amplifications (against TcrPDEB2 and TcrPDEB1 RNA), indicating that transcription occurs in vivo at the different stages of the cell cycle. Figure 3(B) shows the expression levels for each PDE in each morphotype normalized with respect to the intensities of α-tubulin and GAPDH controls (obtained by this method of RT-PCR). Expression of the mRNA of both enzymes was clearly observed in all three developmental stages, suggesting the presence of both PDEs in all T. cruzi life-cycle stages. The data also indicate possible higher levels of TcrPDEB2 message in amastigotes, and TcrPDEB1 mRNA in epimastigotes and trypomastigotes. However, this method is only a semi-quantitative RT-PCR, indicating a possible change in stable message levels, but not providing any absolute estimate of the fold change in RNA transcripts or the post-transcriptional stability of specific mRNAs. It must be noted that higher message levels in trypanosomes need not necessarily translate into higher protein levels, since trypanosomes primarily regulate gene expression post-transcriptionally, unlike many other eukaryotes.
Figure 3. TcrPDEB1 and TcrPDEB2 expression in different T. cruzi morphotypes.

(A) Agarose-gel electrophoresis of the PCR amplification performed after reverse transcription of total RNA from the three different life-cycle stages of T. cruzi. Primers used specifically amplified: 1, TcrPDEB2, 2, TcrPDEB1, 3, a fragment from α-tubulin gene; and 4, fragment of the GAPDH gene. M, size markers. (B) Relative expression of TcrPDEB2 and TcrPDEB1 in the different stages of T. cruzi cell cycle. A, amastigotes; E, epimastigotes; T, trypomastigotes.
Binding of the GAF domains of both enzymes to cNMPs
The GAF-A domains of the two T. cruzi PDEs show considerable homology with the cAMP-binding GAF-A domain of TbrPDE2B and also with the other trypanosomatid GAF-A domains identified that were described above (Figure 4A). This strongly suggested that the GAF-A domains of the T. cruzi PDEs could bind cAMP. To investigate this possibility, the NT+GAF-A domains of TcrPDEB1 and TcrPDEB2 were expressed in E. coli and purified on a TALON column (Figure 4B). Radioactivity binding assays using 3[H]cAMP and cAMP were carried out to determine the3 binding affinity of this domain of TcrPDEB2. As expected, the NT+GAF-A domain was found to bind cAMP with a Kd of 190±5 nM (Figure 5A). Interestingly, when the NT+GAF-A domain of TcrPDEB1 (the orthologue of TbrPDE2C) was tested, no binding could be seen under the same conditions using the radioactivity binding assay, suggesting a lack of binding ability, or at least a substantially lower affinity. Therefore we decided to test for cAMP binding using ITC. This is a highly accurate equilibrium method that has been extensively used to study binding of small molecules to proteins. Binding studies using ITC reconfirmed that cAMP bound to the NT+GAF-A domain of TcrPDEB2 with an affinity of 520±40 nM (Figure 5B), The difference in apparent affinity as measured by the two different methods may be due to the presence of high (NH4)2SO4 in the filter-binding assay. More importantly, this technique revealed that cAMP did indeed bind to the NT+GAF-A domain of TcrPDEB1, albeit with a lower affinity of 0.97±0.1 μM (Figure 5C). The measured stoichiometry of binding was 0.97 mol of cAMP bound/mol of protein. In our experience, this is exceptional, as the best preparations of other PDE GAF-A domains expressed in E. coli (mammalian PDEs 2, 5, and TbrPDE2B) typically show a stoichiometry of ∼0.5 mol of cNMP/mol of protein or less. The protein was extremely stable, retaining full binding ability even after incubation at 30 °C for 15 h, whereas the TcrPDEB2 GAF-A domain showed greatly decreased binding (∼50% of original) when left at 30 °C for the same time period. When tested for binding to cGMP, the NT+GAF-A domain of TcrPDEB2 did bind cGMP, but with a low affinity of 2.5±0.3 μM (Figure 5D). Interestingly, the NT+GAF-A domain of TcrPDEB1 did not show any appreciable binding to cGMP. A representative Figure showing relative heats released for cGMP or cAMP binding to TcrPDEB1 is shown in Figure 5(E).
Figure 4. Alignment of GAF-A domains from multiple trypanosomatid species with the cAMP-binding GAF-A domain of TbPDE2B, and expression of the NT+GAF-A domains of TcrPDEB1 and 2.

(A) Amino-acid-sequence alignment of GAF-A domains from T. brucei TbrPDEB2 (TbrB2), T. cruzi TcrPDEB1 (TcrB1) and TcrPDEB2 (TcrB2) and the GAF domains from the PDEs identified in other kinetoplastids: L. amazonensis (L. amaz), L. braziliensis (L. braz), L. mexicana (L. mex), C. fasciculata (C. fasc), T. evansi (T. evan) and T. rangeli (T. rang). The 11 putative cNMP-binding residues [40] are marked with arrows, residues that differ from the TbPDE2B/TbrPDEB2 cAMP-binding GAF-A domain are in boxes, and the NKFDE motif is shown with asterisks. (B) SDS/PAGE gel stained with Coomassie Blue and showing purified NT+GAF-A domains from TcrPDEB1 (lane 1), TcrPDEB2 (lane 2) and the catalytic domain of TcrPDEB1 (lane 3).
Figure 5. Binding of cAMP to the GAF-A domains of TcrPDEB1 and TcrPDEB2.

(A) Competitive binding curve of cAMP to the NT+GAF-A domain of TcrPDEB2, with cAMP used to displace [3H]cAMP. (B) ITC titration curve showing binding of cAMP to the NT+GAF-A domain of TcrPDEB2. (C) ITC titration curve showing binding of cAMP to the NT+GAF-A domain of TcrPDEB1. (D) Binding of cGMP to the NT+GAF-A domain of TcrPDEB2. (E) Relative heats of binding for cGMP (▲) and cAMP (■) for the NT+GAF-A TcrPDEB1. 1 μcal=4.184 μJ.
TcrPDEB1 catalytic domain hydrolyses MANT-cAMP
ITC was also used to test the kinetics of hydrolysis of both cAMP and MANT-cAMP by the catalytic domain of TcrPDEB1, since ITC does not require labelled substrates. cAMP was hydrolysed by the catalytic domain of TcrPDEB1 with a Km of 2.8±0.6 μM as measured by ITC (Table 3), which compared well with the Km of ∼4 μM for cAMP measured using a conventional radioactivity assay. Importantly, MANT-cAMP was found to be at least as good a substrate for the TcrPDEB1 catalytic domain as cAMP, and was hydrolysed with an apparent Km of 1.8±0.3 μM (Table 3). The kcat/Km values for both substrates were almost identical, with a kcat/Km of 9.8 M−1·s−1 for MANT-cAMP and a kcat/Km of 7.9 M−1·s−1 for cAMP. Similar results were seen with the catalytic domain from the T. brucei TbrPDE2C catalytic domain as well (results not shown). ITC was also used to obtain a Ki of 10.8±0.2 μM for dipyridamole inhibition of cAMP hydrolysis by this enzyme, comparing extremely well with our previously obtained inhibitor data with dipyridamole measured using a conventional radioactive assay (Table 2).
Table 3. cAMP and MANT-cAMP hydrolysis and the Ki of dipyridamole (for cAMP hydrolysis) by the catalytic domain of TcrPDEB1, measured by ITC.
Hydrolysis of cAMP or MANT-cAMP by TcrPDEB1, or inhibition of TcrPDEB1 cAMP hydrolysis, using the PDE inhibitor dipyridamole were measured using ITC. The heats released/absorbed were plotted and analysed using the MicroCal® software to obtain Km or Ki values. N/A, not applicable.
| Substrate | Inhibitor | Km (μM) | Ki (μM) | kcat/Km (M−1·s−1) |
|---|---|---|---|---|
| cAMP | − | 2.8±0.6 | − | 7.9 |
| MANT-cAMP | − | 1.8±0.3 | − | 9.8 |
| cAMP | Dipyridamole | − | 10.8±0.2 | N/A |
MANT derivatives of cAMP and cGMP have been successfully used to measure PDE activity accurately in fluorescence assays for some mammalian PDEs (1, 2 and 5) [36–38]. However, MANT derivatives are not good substrates of all mammalian PDEs, and a number of PDEs hydrolyse these derivatives very inefficiently. Table 3 shows that MANT-cAMP can be used instead of cAMP as a substrate for kinetic studies. It is therefore likely that it can be used in PDE assays for screening new potential inhibitors of the enzyme with results representative of the in vivo substrate, cAMP.
DISCUSSION
The T. cruzi genome encodes two members of the PDEB family. In the present work a novel low-Km GAF domain-containing cAMP-specific PDE was identified and designated TcrPDEB1 according to the new consensus for kinetoplastid PDE nomenclature [20]. The characteristics and properties of TcrPDEB1 were compared with those of the previously identified TcrPDEB2 [18]. Both genes contain two N-terminal GAF domains and a C-terminal catalytic core. The catalytic domains have the conserved motif YHN, and the metal-binding motif HDX2HX4N common to all Class I PDEs. TcrPDEB1 has 32% identity in the catalytic domain with mammalian PDEs and 77% identity with the T. brucei enzyme TbrPDEB1. TcrPDEB2 [18] appears to be the orthologue of TbrPDEB2 [12]. The genomic organization of these two genes (seen in the T. cruzi genome [9]) has TcrPDEB1 (accession number Tc00.1047053508277.100) and TcrPDEB2 (accession number Tc00.1047053508277.110) in tandem, followed by a putative ribosomal protein S6 and a phosphomevalonate kinase-like protein, and this genomic organization seems conserved in the T. brucei orthologues as well. The reasons for this evolutionary conservation of tandem genes are unknown. Our results also indicate the presence of homologous PDEs in other parasites of the Trypanosomatidae family, with the American leishmaniasis-causing parasites, L. amazonensis, L. braziliensis and L. mexicana, containing regions with considerable homology with both the regulatory and catalytic domains of TcrPDEB1 from T. cruzi.
Like TcrPDEB2, TcrPDEB1 was found to be a low-Km cAMP-specific enzyme incapable of hydrolysing cGMP or being modulated by it. The nucleotide substrate specificity is consistent with that reported for the homologous PDEs in T. brucei [12,13] and the PDE activity reported in L. mexicana [39]. TcrPDEB1 (along with TcrPDEB2) was found to be resistant to a number of inhibitors of several mammalian PDEs. However, TcrPDEB2 and TcrPDEB1 showed differences in their inhibition by papaverine and EHNA. The only PDE inhibitor that substantially inhibited both trypanosomatid PDEs was dipyridamole, a relatively broad-acting mammalian PDE inhibitor (inhibits PDEs 5, 6, 7, 8, 10 and 11) which inhibited TcrPDEB1 and TcrPDEB2 with IC50 values of 11.8±2 and 15±2 μM respectively. The inhibition profiles obtained here agree with previously obtained data for TcrPDEB2 alone [18] and other PDEs in T. brucei [12–14] and L. mexicana [39]. Their resistance to common PDE inhibitors is likely due to structural differences in the catalytic site, increasing the possibility of designing novel drugs selective against kinetoplastid PDEs that do not inhibit mammalian PDEs.
RT-PCR data showed expression of the mRNA of both enzymes in all three life-cycle stages of T. cruzi, suggesting possible unique roles of both enzymes in all life-cycle stages. Our localization data corroborated previously published findings for TcrPDEB2. Whether or not each PDE has a unique role is currently unknown, and enzyme-specific antibodies would be required to probe specific localizations of the individual PDEs. TbrPDEB2 (from T. brucei) has previously been shown to bind cAMP through its GAF-A domain with high affinity and selectivity [25]. The GAF domains of the adenylate cyclase CyaB1 from Anabaena also have been shown to bind cAMP [27]. The GAF-A domains of TcrPDEB1 and TcrPDEB2 are highly homologous with the TbrPDEB2 GAF-A domain, especially around the predicted ‘critical’ residues for cNMP binding [40]. Hence we studied the binding of the GAF-A domains from TcrPDEB1 and TcrPDEB2 to cAMP and cGMP using ITC. The NT+GAF-A domain of TcrPDEB2 was found to bind cAMP with an affinity of ∼500 nM. The NT+GAF-A domain of TcrPDEB1 also bound cAMP, but with a lower affinity of approx. 1 μM. Despite the lower affinity, this NT+GAF-A domain from TcrPDEB1 bound cAMP with a stoichiometry of nearly 1 mol of cAMP bound to 1 mol of protein. This is to our knowledge the first PDE GAF domain from any species reported to bind a cyclic nucleotide with such low affinity (the others bind with nanomolar affinities). It probably should be noted, however, that such low-affinity binding sites would not likely be seen by the usual filter-binding assays that are commonly used to measure binding in the mammalian PDEs. Therefore it would probably be worthwhile to re-examine, for example, the mammalian PDE11 by the ITC method. The NT+GAF-A domain of TcrPDEB1 did not appear to bind cGMP (at a measurable affinity), but, interestingly, the TcrPDEB2 GAF-A domain did bind cGMP albeit with a relatively low affinity. Since cGMP activity is still unknown in trypanosomes, this selectivity for cAMP binding is not surprising. These binding properties are different from those observed with the GAF-A domain from the T. brucei TbrPDEB2 [25]. It is likely that the differences in GAF domain–cNMP binding affinities arise from subtle differences in sequence or interactions with other domains. Interestingly, although the GAF-A domains from both T. cruzi PDEB enzymes are very similar, the presence of the N-terminus appears to alter the affinity or selectivity for cNMPs. It is not yet known how the presence of other domains (GAF-B or catalytic domain) influences cAMP binding to these enzymes. It has been observed in other PDEs (such as mammalian PDE5 or TbrPDEB2) that the presence of additional domains alters the affinity of the GAF domains for cNMPs [25,41]. It was also observed for TbrPDEB2 (the T. brucei orthologue of TcrPDEB2) that the presence of additional domains resulted in the GAF domain being highly selective for cAMP over cGMP [25]. The reasons for these differences (both physiological as well as structural) need to be explored, and they may reveal yet another aspect of the versatility and subtlety of PDE regulation by GAF domains. ITC seems particularly suitable for measuring binding affinities between 50 nM and 10 μM, as well as for measuring stoichiometry of binding.
Finally, we found that the catalytic domain of TcrPDEB1 efficiently hydrolysed MANT-cAMP (as did the catalytic domain from the T. brucei orthologue) with the kinetic parameters for this substrate being almost identical with those for cAMP itself. Some mammalian PDEs do not hydrolyse MANT-cNMPs as efficiently as their natural cNMP substrates [38], which restricts their use in PDE assays. However, our data suggest that it should be possible to use these analogues in non-radioactive fluorescence PDE assays [36–38] to measure TcrPDEB1 (and TbrPDE2) catalytic activity. This type of assay is believed to be accurate, sensitive, less time-consuming and less expensive than those based on radioactive substrates. Also, this could be convenient for research groups that do not have facilities to use, or cannot afford, radioactive materials. Importantly, it should be possible to adapt this assay technique for high-throughput screens for trypanosomal PDE inhibitors. As the trypanosomal PDEs have been shown to be essential regulatory enzymes [13], new specific inhibitors have high potential to be therapeutically useful.
Online data
Acknowledgments
We thank Dr Jörg Hoheisel [Functional Genome Analysis (H08000), Deutsches Krebsforschungszentrum, Heidelberg, Germany] for kindly providing the cosmids from the genomic library of T. cruzi, Dr Julio Urbina (Laboratorio de Química Biológica, Instituto Venezolano de Investigaciones Científicas, Caracas, Venezuela) for kindly providing the different life-cycle forms of T. cruzi, Esteban Cordero (Escuela Paulista de Medicina, São Paulo, Brazil) for supplying primers for the amplification of α-tubulin and GAPDH genes, Professor Wesley Van Voorhis (Department of Pathobiology, University of Washington, Seattle, WA, U.S.A.) for T. cruzi CL-Brenner strain trypomastigotes and bloodstream forms of T. brucei, and Dr Jennifer Glick for helpful discussions. We also are grateful to UNESCO-L'Oréal organization for granting a fellowship to R.D.-B. This work was supported by grants to A.R. (FONACIT no. S1-2001000654 and CDCH no. 03-33-4833-01) and J.A.B. (DK 21723 and HL44948).
References
- 1.Seebeck T., Schaub R., Johner A. cAMP signalling in the kinetoplastid protozoa. Curr. Mol. Med. 2004;4:585–599. doi: 10.2174/1566524043360113. [DOI] [PubMed] [Google Scholar]
- 2.Vassella E., Reuner B., Yutzy B., Boshart M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 1997;110:2661–2671. doi: 10.1242/jcs.110.21.2661. [DOI] [PubMed] [Google Scholar]
- 3.Walter R. D. Oxford: Pergamon Press; 1981. Regulation of cAMP metabolism in Leishmania promastigotes and amastigotes. [Google Scholar]
- 4.Gonzales-Perdomo M., Romero P., Goldenberg S. Cyclic AMP and adenylate cyclase activators stimulate Trypanosoma cruzi differentiation. Exp. Parasitol. 1988;66:205–212. doi: 10.1016/0014-4894(88)90092-6. [DOI] [PubMed] [Google Scholar]
- 5.Beavo J. A. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 6.Beavo J. A., Brunton L. L. Cyclic nucleotide research – still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 7.Soderling S. H., Beavo J. A. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions. Curr. Opin. Cell Biol. 2000;12:174–179. doi: 10.1016/s0955-0674(99)00073-3. [DOI] [PubMed] [Google Scholar]
- 8.Francis S. H., Turko I. V., Corbin J. D. Cyclic nucleotide phosphodiesterases: relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 2001;65:1–52. doi: 10.1016/s0079-6603(00)65001-8. [DOI] [PubMed] [Google Scholar]
- 9.El-Sayed N. M., Myler P. J., Bartholomeu D. C., Nilsson D., Aggarwal G., Tran A. N., Ghedin E., Worthey E. A., Delcher A. L., Blandin G., et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 10.Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D. C., Lennard N. J., Caler E., Hamlin N. E., Haas B., et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 11.Ivens A. C., Peacock C. S., Worthey E. A., Murphy L., Aggarwal G., Berriman M., Sisk E., Rajandream M. A., Adlem E., Aert R., et al. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rascon A., Soderling S. H., Schaefer J. B., Beavo J. A. Cloning and characterization of a cAMP-specific phosphodiesterase (TbPDE2B) from Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4714–4719. doi: 10.1073/pnas.002031599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoraghi R., Seebeck T. The cAMP-specific phosphodiesterase TbPDE2C is an essential enzyme in bloodstream form Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4343–4348. doi: 10.1073/pnas.062716599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zoraghi R., Kunz S., Gong K., Seebeck T. Characterization of TbPDE2A, a novel cyclic nucleotide-specific phosphodiesterase from the protozoan parasite Trypanosoma brucei. J. Biol. Chem. 2001;276:11559–11566. doi: 10.1074/jbc.M005419200. [DOI] [PubMed] [Google Scholar]
- 15.Kunz S., Kloeckner T., Essen L. O., Seebeck T., Boshart M. TbPDE1, a novel class I phosphodiesterase of Trypanosoma brucei. Eur. J. Biochem. 2004;271:637–647. doi: 10.1111/j.1432-1033.2003.03967.x. [DOI] [PubMed] [Google Scholar]
- 16.Johner A., Kunz S., Linder M., Shakur Y., Seebeck T. Cyclic nucleotide specific phosphodiesterases of Leishmania major. BMC Microbiol. 2006;6:25. doi: 10.1186/1471-2180-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alonso G. D., Schoijet A. C., Torres H. N., Flawia M. M. TcPDE4, a novel membrane-associated cAMP-specific phosphodiesterase from Trypanosoma cruzi. Mol. Biochem. Parasitol. 2006;145:40–49. doi: 10.1016/j.molbiopara.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 18.D'Angelo M. A., Sanguineti S., Reece J. M., Birnbaumer L., Torres H. N., Flawia M. M. Identification, characterization and subcellular localization of TcPDE1, a novel cAMP-specific phosphodiesterase from Trypanosoma cruzi. Biochem. J. 2004;378:63–72. doi: 10.1042/BJ20031147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kunz S., Oberholzer M., Seebeck T. A FYVE-containing unusual cyclic nucleotide phosphodiesterase from Trypanosoma cruzi. FEBS. J. 2005;272:6412–6422. doi: 10.1111/j.1742-4658.2005.05039.x. [DOI] [PubMed] [Google Scholar]
- 20.Kunz S., Beavo J. A., D'Angelo M. A., Flawia M. M., Francis S. H., Johner A., Laxman S., Oberholzer M., Rascon A., Shakur Y., et al. Cyclic nucleotide specific phosphodiesterases of the kinetoplastida: a unified nomenclature. Mol. Biochem. Parasitol. 2006;145:133–135. doi: 10.1016/j.molbiopara.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Aravind L., Ponting C. P. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem. Sci. 1997;22:458–459. doi: 10.1016/s0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 22.Gillespie P. G., Beavo J. A. cGMP is tightly bound to bovine retinal rod phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 1989;86:4311–4315. doi: 10.1073/pnas.86.11.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martins T. J., Mumby M. C., Beavo J. A. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J. Biol. Chem. 1982;257:1973–1979. [PubMed] [Google Scholar]
- 24.Francis S. H., Lincoln T. M., Corbin J. D. Characterization of a novel cGMP binding protein from rat lung. J. Biol. Chem. 1980;255:620–626. [PubMed] [Google Scholar]
- 25.Laxman S., Rascon A., Beavo J. A. Trypanosome cyclic nucleotide phosphodiesterase 2B binds cAMP through its GAF-A domain. J. Biol. Chem. 2005;280:3771–3779. doi: 10.1074/jbc.M408111200. [DOI] [PubMed] [Google Scholar]
- 26.Gross-Langenhoff M., Hofbauer K., Weber J., Schultz A., Schultz J. E. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J. Biol. Chem. 2006;281:2841–2846. doi: 10.1074/jbc.M511468200. [DOI] [PubMed] [Google Scholar]
- 27.Kanacher T., Schultz A., Linder J. U., Schultz J. E. A GAF-domain-regulated adenylyl cyclase from Anabaena is a self-activating cAMP switch. EMBO J. 2002;21:3672–3680. doi: 10.1093/emboj/cdf375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson J. D., Higgins D. G., Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engels W. R. Contributing software to the internet: the Amplify program. Trends Biochem. Sci. 1993;18:448–450. doi: 10.1016/0968-0004(93)90148-g. [DOI] [PubMed] [Google Scholar]
- 30.Medina-Acosta E., Cross G. A. Rapid isolation of DNA from trypanosomatid protozoa using a simple ‘mini-prep’ procedure. Mol. Biochem. Parasitol. 1993;59:327–329. doi: 10.1016/0166-6851(93)90231-l. [DOI] [PubMed] [Google Scholar]
- 31.Hansen R. S., Charbonneau H., Beavo J. A. Purification of two calcium/calmodulin-dependent forms of cyclic nucleotide phosphodiesterase by using conformation-specific monoclonal antibody chromatography. Proc. Natl. Acad. Sci. U.S.A. 1982;79:2788–2792. doi: 10.1073/pnas.79.9.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motulsky H., Christopoulos A. Oxford: Oxford University Press; 2004. Fitting Models to Biological Data using Linear and Non-linear Regression: A Practical Guide to Curve Fitting. [Google Scholar]
- 33.Cheng Y., Prusoff W. H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 34.Wiseman T., Williston S., Brandts J. F., Lin L. N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 35.Todd M. J., Gomez J. Enzyme kinetics determined using calorimetry: a general assay for enzyme activity? Anal. Biochem. 2001;296:179–187. doi: 10.1006/abio.2001.5218. [DOI] [PubMed] [Google Scholar]
- 36.Hiratsuka T. New fluorescent analogs of cAMP and cGMP available as substrates for cyclic nucleotide phosphodiesterase. J. Biol. Chem. 1982;257:13354–13358. [PubMed] [Google Scholar]
- 37.Johnson J. D., Walters J. D., Mills J. S. A continuous fluorescence assay for cyclic nucleotide phosphodiesterase hydrolysis of cyclic GMP. Anal. Biochem. 1987;162:291–295. doi: 10.1016/0003-2697(87)90039-x. [DOI] [PubMed] [Google Scholar]
- 38.Karuppiah N., Mutus B. Selective adsorption of 2′-O-anthraniloyl-AMP on DEAE-Sephadex: the basis of a direct, fluorescent assay for cyclic nucleotide phosphodiesterase. Anal. Biochem. 1985;149:202–208. doi: 10.1016/0003-2697(85)90496-8. [DOI] [PubMed] [Google Scholar]
- 39.Rascon A., Viloria M. E., De-Chiara L., Dubra M. E. Characterization of cyclic AMP phosphodiesterases in Leishmania mexicana and purification of a soluble form. Mol. Biochem. Parasitol. 2000;106:283–292. doi: 10.1016/s0166-6851(99)00224-8. [DOI] [PubMed] [Google Scholar]
- 40.Martinez S. E., Wu A. Y., Glavas N. A., Tang X. B., Turley S., Hol W. G., Beavo J. A. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13260–13265. doi: 10.1073/pnas.192374899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zoraghi R., Bessay E. P., Corbin J. D., Francis S. H. Structural and functional features in human PDE5A1 regulatory domain that provide for allosteric cGMP binding, dimerization, and regulation. J. Biol. Chem. 2005;280:12051–12063. doi: 10.1074/jbc.M413611200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.