

Abstract
Amines, including those present on proteins, spontaneously react with glucose to form fructosamines in a reaction known as glycation. In the present paper, we have explored, through a targeted gene inactivation approach, the role of FN3K (fructosamine 3-kinase), an intracellular enzyme that phosphorylates free and protein-bound fructose-ϵ-lysines and which is potentially involved in protein repair. Fn3k−/− mice looked healthy and had normal blood glucose and serum fructosamine levels. However, their level of haemoglobin-bound fructosamines was approx. 2.5-fold higher than that of control (Fn3k+/+) or Fn3k+/− mice. Other intracellular proteins were also significantly more glycated in Fn3k−/− mice in erythrocytes (1.8–2.2-fold) and in brain, kidney, liver and skeletal muscle (1.2–1.8-fold), indicating that FN3K removes fructosamines from intracellular proteins in vivo. The urinary excretion of free fructose-ϵ-lysine was 10–20-fold higher in fed mice compared with mice starved for 36 h, and did not differ between fed Fn3k+/+ and Fn3k−/− mice, indicating that food is the main source of urinary fructose-ϵ-lysine in these mice and that FN3K does not participate in the metabolism of food-derived fructose-ϵ-lysine. However, in starved animals, the urinary excretion of fructose-ϵ-lysine was 2.5-fold higher in Fn3k−/− mice compared with Fn3k+/+ or Fn3k+/− mice. Furthermore, a marked increase (5–13-fold) was observed in the concentration of free fructose-ϵ-lysine in tissues of fed Fn3k−/− mice compared with control mice, indicating that FN3K participates in the metabolism of endogenously produced fructose-ϵ-lysine. Taken together, these data indicate that FN3K serves as a protein repair enzyme and also in the metabolism of endogenously produced free fructose-ϵ-lysine.
Keywords: diabetic complication, fructosamine 3-kinase (FN3K) inactivation, fructoselysine, glycated haemoglobin (GlcHb), intracellular glycation, protein repair
Abbreviations: 3-PGK, 3-phosphoglycerate kinase; ES cell, embryonic stem cell; FN3K, fructosamine 3-kinase; GlcHb, glycated haemoglobin; HbA1c, stable minor haemoglobin variant containing glycated N-terminal β chains; PAC, P1-derived artificial chromosome; PGI, phosphoglucose isomerase; RT, reverse transcriptase; siRNA, small interfering RNA; TPI, triose-phosphate isomerase
INTRODUCTION
Glucose spontaneously reacts with amines to form Schiff bases, which slowly undergo an Amadori rearrangement to become more stable, fructose-like compounds known as fructosamines [1,2]. Originally identified in vitro, the formation of fructosamines (known as early glycation), also takes place in living organisms. Many intracellular (e.g., haemoglobin) and extracellular proteins (e.g. collagen and serum albumin) are known to bear fructosamines on lysine residues or on their N-terminal amine. Unlike enzyme-catalysed post-translational modifications, protein glycation is largely substoichiometric. Fructosamine formation is proportional to glucose concentration and glycation is therefore enhanced in diabetic patients [2,3].
Until recently, it was thought that the formation of fructosamines was an irreversible process, which is unavoidably followed by their slow conversion into a series of compounds known as advanced glycation end-products. The latter are thought to participate in the development of diabetic complications [2,4]. However, the recent identification of FN3K (fructosamine 3-kinase), an enzyme that is able to convert free and protein-bound fructose-ϵ-lysine residues [5,6] into unstable fructosamine 3-phosphates, opened the perspective that fructosamines can be physiologically removed from proteins. The finding that in the presence of a competitive inhibitor of FN3K, the accumulation of GlcHb (glycated haemoglobin) in erythrocytes incubated with 200 mM glucose increases, which indicates that at least under these non-physiological conditions FN3K may act to repair haemoglobin [7]. However, it still needs to be proven that this mechanism takes place in vivo at physiological concentrations of glucose and that it also applies to cell types other than erythrocytes. Furthermore, FN3K phosphorylates free fructose-ϵ-lysine in vitro with high affinity [5] and it could also be involved in the metabolism of this compound. To investigate these points, we have carried out the inactivation of the Fn3k gene in mice.
EXPERIMENTAL
Materials
Reagents were of analytical grade. Radiochemicals were from GE Healthcare (Roosendaal, The Netherlands). AG 1X-8 (200–400 mesh) and AG 50W-X4 (100–200 mesh) resins were from BioRad Laboratories (Hercules, CA, U.S.A.). Auxiliary enzymes and enzyme substrates were from Roche Molecular Biochemicals (Mannheim, Germany) and Sigma-Aldrich (St. Louis, MO, U.S.A.). Recombinant Escherichia coli fructoselysine 6-kinase [8] was His6-tagged at the N-terminus and over-expressed in E. coli for 24 h at 19 °C. It was purified to near homogeneity by Ni2+ affinity chromatography after elution by a step-wise imidazole gradient from a HisTrap™ column (GE Healthcare). The active fractions were concentrated, desalted by gel filtration, enriched with 10% (v/v) glycerol and stored at −80 °C. The purified preparation (2.4 mg/ml) did not contain any contaminating phosphatase activities.
Animals
All mice were raised under standard husbandry conditions and were housed in colony cages with a 12 h light/dark cycle. They had free access to water and food, unless otherwise specified. Mice used in this study were the offspring of fourth and fifth generation heterozygous (Fn3k+/−) mice. The experiments were performed with the approval of the ethical committee of the host institution.
Construction of the targeting vector and generation of knockout mice
The targeting vector was obtained by directly subcloning fragments of the Fn3k gene from the RPCI-21 female (129S6/SvEvTac) mouse PAC (P1-derived artificial chromosome) Library (BACPAC Resources, Children's Hospital Oakland Research Institute, Bruce Lyon Memorial Research Laboratory, Oakland, CA, U.S.A.) and the neomycin resistance gene, with a PGK promoter and poly(A) signal from the pPNT vector [9] into the pSKT-NLSLacZ vector (Figure 1A). The heterologous genes were flanked with 5.5 kb of sequence upstream of the Fn3k initiator ATG codon and 2.8 kb of sequence downstream of the second exon. This construct was electroporated in mouse R1 ES cells (embryonic stem cells) (a gift from A. Nagy, Mount Sinai Hospital, Toronto, Ontario, Canada). PCR analysis of 300 neomycin-resistant clones indicated that 12 had undergone a site-specific recombination event. Southern blot analysis (Figure 1B) and karyotyping of five of the clones allowed the selection of an appropriate ES cell clone, which was aggregated to CD1 morulae and the resulting blastocysts were transferred into pseudo-pregnant mice. Of the 26 chimeric mice that were born, 23 were males. From the 23 males, six were mated with CD1 females, of which five transmitted the deletion of the Fn3k gene. Heterozygous offspring were crossed to generate knockout progeny, resulting in a mixed CD1/129 background. The mice were genotyped by PCR with primers corresponding to DNA sequences in exon 2 and in the neo cassette using the primers: exon 2-S 5′-GATGTTTGAGGGAGAGATGGC-3′ and exon 2-AS 5′-GGCTCTTCATCTTCAAGTGCT-3′; neo-S 5′-TCCCCTCAGAAGAACTCGTC-3′ and neo-AS 5′-AGCACGTACTCGGATGGAAG-3′.
Figure 1. Targeted deletion in the FN3K locus in mice.

(A) The hatched boxes represent the exons in the Fn3k gene, the grey boxes those in the Fn3k-RP gene [34] and the white boxes the neomycin-resistant cassette (Neo) and the β-galactosidase marker (β-Gal). Key restriction enzyme sites and localization of probes used during Southern blotting are shown under the disrupted allele. (B) Confirmation of the correct targeting of the 5′ and 3′ arms and excludes any additional insertion by Southern blotting. WT, wild-type; KO, knock-out. (C) Confirmation that transcription from the Fn3k locus has been ablated in various tissues in mice that are homozygous for the Fn3k deletion. RT-PCR analysis of the sequence spanning exons 1 (forward primer: 5′-CGTGTTTGTCAAGGTCAATCG-3′) to 3 (reverse primer: 5′-TGCCATCTGTTCCCCGAGC-3′) shows the absence of a detectable transcript in Fn3k−/− mice. (D) FN3K activity in erythrocytes, kidney and brain following the ablation of the Fn3k gene in mouse. The activity of FN3K is related to the amount of protein in the extract. In erythrocytes the activity was determined in extracts of two mice for each genotype. In kidney and brain extracts, the activity in each tissue was determined after pooling the extracts from two mice of each genotype prior to purification.
FN3K activity and total GlcHb
For the assay of mouse FN3K activity, the conversion of [14C]fructose-ϵ-lysine (labelled on its deoxyfructose moiety; [7]) to [14C]fructoselysine 3-phosphate was measured in erythrocyte cell extracts or after partial purification on Blue Sepharose (kidney and brain extracts) [10]. GlcHb was measured using phenylboronate affinity chromatography using the GLYCO-Tek Affinity column kit (Helena BioSciences Europe, Sunderland, U.K.), according to the manufacturer's protocol, except that erythrocytes were left in the lysis solution for 30 min before loading on to the column. According to Abraham et al. [11], the Schiff's bases are not retained on this type of column.
Detection of fructosamines in erythrocyte extracts by phosphorylation with fructoselysine 6-kinase after separation by SDS/PAGE
Cell extracts were resolved by SDS/PAGE (12%, w/v gels). The proteins were electrophoretically transferred on to a HybondTM-C Extra nitrocellulose membrane (GE Healthcare), which was washed three times in 20 mM Hepes, pH 7.1, for 10 min and blocked by incubation with 5% (w/v) non-fat dry milk in the same buffer for 60 min at 30 °C. The membrane was then incubated for 90 min at 30 °C with 0.7 units/ml fructoselysine 6-kinase in 20 mM Hepes, pH 7.1, 1 mM MgCl2, 25 mM KCl, 20 μM ATP-Mg and 6.5×106 c.p.m./ml [γ-32P]ATP. The phosphorylation was stopped and excess ATP eliminated by washing three times in 20 mM Hepes, pH 7.1, and 1 mM EDTA at 30 °C for 5 min. The membrane was dried and exposed to a PhosphoImager screen (Molecular Dynamics, Sunnyvale, CA, U.S.A.) for detection of radioactivity. The identity of the proteins corresponding to the increase in phosphorylation observed in the Fn3k−/− mice was confirmed by MS analysis of the corresponding bands in the SDS/PAGE gels.
Measurement of protein-bound fructoselysine and plasma fructosamines
To measure protein-bound fructoselysine, tissue extracts were extensively dialysed against water to eliminate free fructose-ϵ-lysine, and then 150 μg of protein was digested by pepsin, pronase E, aminopeptidase and prolidase, as described previously [12]. Digestion was stopped by extraction with perchloric acid and free fructose-ϵ-lysine was measured in the neutralized extract using E. coli fructoselysine 6-kinase and [γ-32P]ATP [13].
The concentration of extracellular fructosamines was measured using 100 μl of plasma isolated from mouse blood treated with EDTA. We used a colorimetric assay [14] that relies on the ability of fructosamines to act as reducing agents in alkaline solutions. 1-Deoxy-morpholinofructose was used as the standard fructosamine.
Assay of glycated enzymes
The extent of glycation of intracellular proteins in various tissues was determined by phenylboronate affinity chromatography using a GLYCO-Tek affinity column for haemolysates prepared from whole blood and in the supernatants of a 1/4 (w/v) tissue extracts made in 25 mM Hepes, 1 mM DTT (dithiothreitol), 1 μg/ml leupeptin and 1 μg/ml antipain. Whole blood (15 μl corresponding to approx. 2 mg of protein) and 1.4 units of TPI (triose-phosphate isomerase), or 0.14 unit of PGI (phosphoglucose isomerase) and 3-PGK (3-phosphoglycerate kinase) were added to 85 μl of the lysis buffer supplied with the kit and immediately applied on to the phenylboronate columns. For the tissues the volume of extract added to the lysis buffer (100 μl final volume) varied and corresponded to approx. 3 units of TPI, and 0.4 unit of PGI and 3-PGK. To standardize the amount of protein loaded on to the phenylboronate columns (2 mg), BSA was added to the diluted extract.
After loading the columns at room temperature (22 °C), the samples were left for 10 min before washing with 0.5 ml of solution A, followed by 4 ml of solution A (50 mM MgCl2, 100 mM glycine, pH 8.1 to 8.6; supplied with the kit), which was kept as Fraction 1. The retained glycated proteins were eluted by 3 ml of 20 mM Hepes, pH 8.5, 350 mM sorbitol and 0.01% sodium azide (Fraction 2). This solution was preferred to solution B (provided by the manufacturer), since the latter was found to cause inhibition of the enzymatic activity. To preserve the enzymatic activity we added 0.5 mg/ml BSA to Fraction 2, immediately after elution from the column. Fractions 1 and 2 were kept on ice and their enzymatic activities measured spectrophotometrically at 30 °C, which was used to determine the proportion of enzyme that was retained on the column. Some of the enzymatic activities detected in Fractions 1 and 2 did not withstand freezing and thawing and therefore had to be measured soon after elution from the column. When working as described, we recovered on average 100% (TPI and PGI) and 70% (3-PGK) of the activities from the extracts loaded onto the columns.
TPI was measured in 25 mM Hepes, pH 7.1, 25 mM KCl, 1 mM MgCl2, 0.17 mM NADH+H+, 1 unit/ml of desalted rabbit muscle glycerol 3-phosphate dehydrogenase and 40 μl or 15 μl of Fraction 1 or 450 μl or 100 μl of Fraction 2 for erythrocytes and other tissues respectively. The reaction was initiated by addition of 1 mM glyceraldehyde 3-phosphate. PGI was measured in 25 mM Hepes, pH 7.1, 2 mM MgCl2, 1 mM DTT, 0.25 mM NADP++H+, 1 unit/ml of desalted yeast glucose 6-phosphate dehydrogenase and 50 μl of Fraction 1 or 450 μl of Fraction 2. The reaction was initiated by addition of 2 mM fructose 6-phosphate. 3-PGK was measured in 85 mM triethanolamine buffer, pH 7.6, containing 2 mM EDTA, 1.5 mM MgSO4, 7 mM 3-phosphoglyceric acid, 1 unit/ml of rabbit muscle glyceraldehyde 3-phosphate dehydrogenase and 50 μl of Fraction 1 or 450 μl of Fraction 2. The reaction was initiated by addition of 1 mM ATP–Mg.
Protein concentration in tissue samples was measured using γ-globulin as a standard [15].
Assay of free fructose-ϵ-lysine
Free fructose-ϵ-lysine was assayed in neutralized perchloric acid extracts from erythrocytes, plasma, tissues and urine of Fn3k+/+ and Fn3k−/− mice. Freshly collected mouse blood treated with EDTA was centrifuged at 800 g for 10 min at 4 °C to recover the plasma. Erythrocytes were then washed three times with 0.15 M NaCl and the pellet was diluted with 1 vol. of water. Perchloric acid extracts (5%, v/v) were made by addition of 70% (v/v) HClO4 to the diluted erythrocytes or directly to the plasma or urine. For the tissues, the frozen material was directly homogenized in 2 vol. of 10% (v/v) HClO4. After centrifugation (16000 g for 15 min at 4 °C), the supernatant was neutralized with 3M KHCO3 and, with the exception of urine, diluted with 2 vol. of water and applied onto a 1 ml AG 1-X8 column (Cl− form) equilibrated with water in order to eliminate ATP. The flow-through and the wash fractions (4 ml) containing fructoselysine were collected, lyophilized and rediluted in 10 mM Hepes, pH 7.1, to the initial sample volume. Free fructose-ϵ-lysine was determined using a radiochemical assay where fructose-ϵ-lysine is phosphorylated to fructoselysine 6-phosphate in the presence of 0.5 units of fructoselysine 6-kinase in a 100 μl assay mixture containing 25 μl of extract, 25 mM Hepes, pH 7.1, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 20 mM NaF, 10 μM ATP–Mg and 10000 c.p.m. of [γ-32P]ATP/μl. After incubation for 10 and 15 min at 30 °C, the reaction was stopped by mixing 30 μl of sample with ice-cold HClO4 to a final volume of 200 μl and a final concentration of 5% (w/v). The samples were then neutralized with 3M KHCO3 and the insoluble precipitate was removed by centrifugation (16000 g for 15 min at 4 °C). The supernatant was diluted a further 3-fold in 0.15 M NaCl and applied on to a 1 ml AG 1-X8 column (Cl− form) equilibrated with 0.15 M NaCl. Fructoselysine 6-[32P]phosphate was eluted in the flow-through and in the washing fractions (4×2 ml of 0.15 M NaCl) and separated from the remaining [γ-32P]ATP. The sum of the radioactivity found in these fractions was used to calculate the concentration of fructose-ϵ-lysine in the extracts.
The fructose-ϵ-lysine in the neutralized perchloric acid extracts of urine was assayed spectrophotometrically using fructoselysine 6-kinase, pyruvate kinase and lactate dehydrogenase in an ATP-coupled assay described previously [8].
The concentration of fructose-ϵ-lysine measured in urine and erythrocytes was normalized to the concentration of creatinine [16] or haemoglobin [17] in the respective samples. Fructose-ϵ-lysine in tissues was calculated in relation to the weight of the samples.
Histological examinations
All tissue samples taken from Fn3k+/+ and Fn3k−/− mice were immediately fixed in formalin and routinely processed for paraffin embedding. Tissue sections (5 μm) were stained with haematoxylin/eosin, periodic acid Schiff and Masson's Blue Trichrome stain, and examined by two different pathologists (C.G. and I.T.).
RESULTS
Inactivation of the Fn3k gene
The construct that was used to inactivate the Fn3k gene through homologous recombination (Figure 1A) was designed to replace the first exon, the first intron and the second exon of the mouse Fn3k gene by an in-frame bacterial β-galactosidase gene and a neomycin-resistance gene. Fn3k−/− mice were viable and the genotyping of 736 pups born from heterozygous matings showed that their distribution was as expected for unbiased Mendelian inheritance. There was no difference in fertility or in litter size for Fn3k−/− mice. RT (reverse transcriptase)-PCR analysis of RNA from various tissues indicated the absence of Fn3k mRNA in Fn3k−/− mice (Figure 1C). Furthermore, FN3K activity was undetectable in erythrocytes, brain and kidney extracts (Figure 1D) of Fn3k−/− mice and in erythrocyte extracts from Fn3k+/− mice it amounted to 50% of the activity compared with the wild-type mice.
Since the β-galactosidase transgene was placed in frame with the initiator ATG codon of the Fn3k gene, we also tried to detect the presence of β-galactosidase in tissues from Fn3k−/− mice by activity staining and RT-PCR analysis. However, activity staining did not show the presence of β-galactosidase, and RT-PCR revealed only very low amounts of β-galactosidase mRNA in the adult brain, liver or kidney, which are tissues where FN3K is normally expressed [5,10]. These data indicated that the β-galactosidase transgene did not recapitulate the normal expression of the Fn3k gene, suggesting that the region from exon 1 to exon 2 of the Fn3k gene contains important regulatory elements.
General characterization of the Fn3k−/− mice
For both males and females, the ablation of the Fn3k gene did not affect growth, as indicated by the fact that there was no difference in body weight between Fn3k−/− and control mice at various ages (from 3 weeks to 10 months). For instance at 15 weeks, the body weight was 34.8±1.2 g and 34.2±1.0 g for male Fn3k+/+ and Fn3k−/− mice respectively and 27.1±1.0 g and 26.9±0.8 g for female Fn3k+/+ and Fn3k−/− mice respectively. No difference in the blood glucose concentration was observed for mice aged 1, 2, 4 and 6 months that were starved overnight (n=4 for each age and sex; results not shown). Furthermore, there was no difference in the concentration of extracellular plasma fructosamines between 18-month-old Fn3k−/− (1.59±0.16 mM; n=8) and wild-type (1.61±0.10 mM; n=8) mice. No glucosuria or increased urinary albumin excretion was observed (results not shown) in Fn3k−/− mice (males, aged 2–12 months) and their plasma urea or creatinine concentrations were normal. The Fn3k−/− mice looked healthy and there was no apparent difference in life span (approx. 2 years) compared with wild-type mice. Histological examinations of tissues from Fn3k−/− mice (brain, cerebellum, eyes, thymus, kidney, liver, heart, skeletal muscle, spleen, bone marrow, pancreas, stomach, intestine, testis, ovary, uterus and bone marrow) performed at birth and at 2, 4, 12 and 18 months did not disclose any particular finding compared with wild-type mice of the same age.
Effect of FN3K deficiency on haemoglobin glycation
The impact of FN3K ablation on protein glycation was determined by measuring GlcHb in blood from overnight-starved mice aged 1–6 months by affinity chromatography on phenylboronate columns. An approx. 2.5-fold increase in the amount of GlcHb was observed in Fn3k−/− mice, whereas Fn3k+/− mice had a GlcHb level similar to that observed in the wild-type control mice (Figure 2).
Figure 2. Glycation of haemoglobin in wild-type and Fn3k−/− mice.
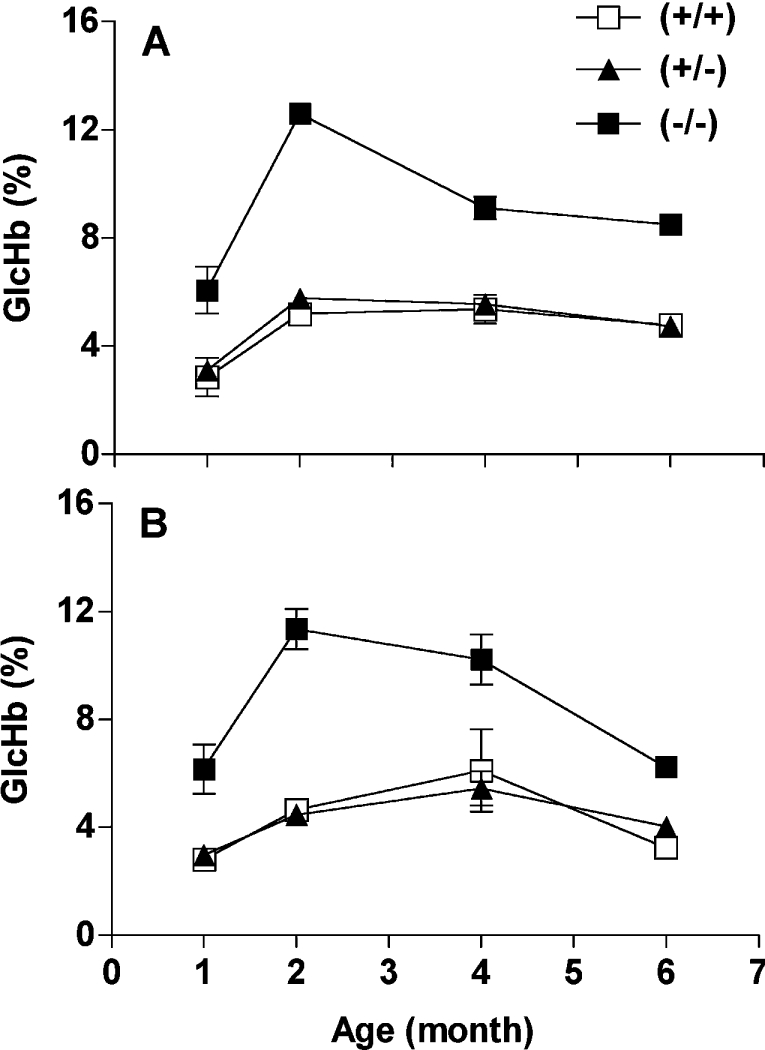
Total GlcHb that binds to phenylboronate affinity columns in erythrocytes from wild-type Fn3k+/+, Fn3k+/− and Fn3k−/− male (A) and female (B) mice aged 1, 2, 4 and 6 months. Each point represents the mean±S.E.M. from four mice.
To confirm that the increased glycation observed in Fn3k−/− mice corresponded to an increase in protein-bound fructosamines, erythrocyte proteins were resolved by SDS/PAGE and transferred on to a nitrocellulose membrane, which was then incubated with [γ-32P]ATP and E. coli fructoselysine 6-kinase. This enzyme acts best on free fructose-ϵ-lysine but also, more slowly, on proteinbound fructoselysine [8]. As shown in Figure 3, several fructosamine-containing bands were detected in this way and these were significantly more abundant in erythrocytes of Fn3k−/− versus Fn3k+/+ mice. MS analysis of tryptic peptides indicated that the lowest, most abundant band corresponded to haemoglobin monomers, the intermediate band also corresponded to haemoglobin (which is probably haemoglobin dimers), and the two upper bands corresponded to carbonic dehydratase II and haemoglobin (possibly tetramers). To compare the extent of glycation at various sites in haemoglobin between Fn3k−/− and Fn3k+/+ mice, we used a method described previously [13], in which the fructosamine residues are tagged by incubation with [γ-32P]ATP and FN3K. After reduction of the fructosamine 3-phosphates with borohydride, the protein was digested with trypsin and the resulting peptides were separated by HPLC, finally the radioactivity was counted in the fractions. Typical HPLC profiles, illustrated in Figure 4, show that the incorporated radioactivity was globally higher in haemoglobin from Fn3k−/− mice compared with wild-type mice, consistent with a higher degree of glycation in the FN3K deficient mice. Remarkably, some of the peaks found with haemoglobin from the Fn3k−/− mice were significantly reduced in the profiles from the wild-type mice, while other peaks were much less affected. These results indicate that in vivo deglycation takes place with efficiencies that differ largely from site to site.
Figure 3. Phosphorylation of glycated residues from proteins in erythrocyte extracts from wild-type and Fn3k−/− mice.

Whole blood extracts and erythrocyte lysates from Fn3k+/+ and Fn3k−/− mice (100 μg per lane) or control lysozyme or glycated (25 μg per lane) were separated by SDS/PAGE, the proteins were transferred on to a nitrocellulose membrane and phosphorylated with recombinant E. coli fructoselysine 6-kinase and [γ-32P]ATP. The membrane was overloaded to enhance the signal of the faintest bands. As a result of this, the phosphorylation of the lower band, corresponding to haemoglobin monomers, is underestimated compared with the other bands.
Figure 4. HPLC profile of radioactive peptides after tryptic digestion of haemoglobin from Fn3k+/+ and Fn3k−/− mice phosphorylated with FN3K and [γ-32P]ATP.

Haemoglobin from Fn3k+/+ and Fn3k−/− mice was partially purified on DEAE-Sepharose and incubated with recombinant mouse FN3K and [γ-32P]ATP for 2 h in vitro. After reduction of fructosamine 3-[32P]phosphate with NaBH4, protein was precipitated in acid, resuspended in urea and digested overnight with trypsin [13]. Peptides were separated by reverse-phase HPLC and radioactive peaks were detected by Cerenkov counting.
Glycation of proteins in tissues
Due to the wide tissue distribution of FN3K, we anticipated that FN3K ablation would cause an increase in the level of glycated proteins in many tissues. To investigate this point, we first determined the level of protein-bound fructoselysine with a specific enzymatic fructose-ϵ-lysine assay after extensive proteolytic digestion of total brain, kidney, liver and erythrocyte proteins from 1, 2 and 4 month-old mice. In the three age groups, protein-bound fructoselysine was consistently higher in erythrocytes and tissues in Fn3k−/− mice compared with wild-type mice from the same age groups. On average a 20% increase was measured in tissues from Fn3k−/− mice compared with wild-type mice (brain, 0.16±0.01 compared with 0.12±0.01, P=0.05; kidney, 0.18±0.002 compared with 0.16±0.005, P=0.035; liver, 0.17±0.02 compared with 0.13±0.01, P=0.2; results are presented as nmol of fructose-ϵ-lysine/mg of protein, n=3), whereas it was more than doubled in erythrocytes (Fn3k−/−, 0.78±0.22 compared with Fn3k+/+: 0.32±0.05 nmol of fructoselysine/mg of protein, P=0.1, n=3). As FN3K is a cytosolic protein, it was likely that the difference in total (i.e. both intracellular and extracellular) protein-bound fructoselysine values shown above underestimated the difference in the glycation levels of cytosolic proteins. We therefore measured the glycation extent of three cytosolic enzymes, TPI, PGI and 3-PGK by determining their degree of retention on phenylboronate affinity columns. The choice of these enzymes was directed by their high specific activity and their stability in cell extracts. Their glycation was found to be approx. 2-fold (TPI and 3-PGK) and 1.5-fold higher (PGI) in erythrocyte extracts from Fn3k−/− mice compared with wild-type mice (Figure 5). TPI and 3-PGK were also found to be glycated to a higher degree (by 40 to 60%) in liver, kidney, brain and skeletal muscle of FN3K-deficient mice (Figure 6). For PGI the increase in glycation was less important but still significant (with a mean of 23%). FN3K deficiency was not accompanied by any difference in the total activities of these enzymes (results not shown).
Figure 5. Glycation of cytosolic enzymes (TPI, PGI and 3-PGK) in erythrocytes from wild-type and Fn3k−/− mice.

The glycation in (A) TPI, (B) PGI and (C) 3-PGK was measured in extracts from erythrocytes from wild-type and Fn3k−/− mice aged 1, 2, 4 and 6 months after separation of the glycated and the non-glycated forms of the enzymes by phenylboronate affinity chromatography as described in the Experimental section. Error bars are calculated from measurements in two animals.
Figure 6. Increase in intracellular glycation of TPI, PGI and 3-PGK from liver, kidney, brain and muscle of FN3K-deficient mice.

Tissue extracts were diluted in the lysis solution and BSA was added to standardize the protein concentration to 2 mg in 100 μl, which was then loaded onto the phenylboronate affinity columns. Non-glycated and glycated proteins were separated as described in the legend for Figure 5. The results are means±S.E.M. of four measurements for extracts from mice aged 1, 2, 4 and 6 months. The difference between the values obtained for the samples from the wild-type and Fn3k−/− mice were compared using a Student's t test (***P<0.001; **0.001≤P<0.01; and, *0.01≤P<0.05).
Effect of FN3K deficiency on tissue and urinary fructose-ϵ-lysine
As shown in Table 1, free fructose-ϵ-lysine was present in the plasma of both wild-type Fn3k+/+ mice and FN3K-deficient fed mice. It was found at a concentration of nearly 3 μM in the control mice and this value was not significantly different in the FN3K-deficient mice. The tissue or cell concentration of free fructose-ϵ-lysine was lower than 1 nmol/g in brain and heart of Fn3k+/+ mice (its presence there is most likely contributed largely by the extracellular compartment), its concentration was intermediate in skeletal muscle and liver, and the highest in kidneys (probably reflecting contamination by small amounts of urine, which contains approx. 100-fold higher concentrations of this compound; see below). FN3K deficiency resulted in a marked increase (5–13-fold) in the cell or tissue content of fructose-ϵ-lysine except in kidneys, where the increase in the concentration did not reach statistical significance.
Table 1. Fructoselysine content of cells and tissues from Fn3k+/+ and Fn3k−/− mice.
Fructoselysine concentration was determined using a radiochemical assay (see Experimental section) in neutralized perchloric acid extracts prepared from washed erythrocytes or the indicated tissues taken from 12–14 month-old mice. Results are means±S.E.M. Figures in parentheses show the number of replicates (n) and the probability. ns, not significant.
| Fructoselysine (μmol/mg of creatinine) | |||
|---|---|---|---|
| Tissue/cell | Genotype… | Fn3k+/+ | Fn3k−/− |
| Red blood cells | 0.28±0.17 | 3.64±0.25 (n=5; P<0.0001) | |
| Serum | 2.87±0.81 | 2.65±0.76 (n=4; ns) | |
| Brain | 0.29±0.07 | 2.8±0.22 (n=5; P<0.0001) | |
| Kidney | 16.4±2.9 | 22.2±5.5 (n=5; ns) | |
| Heart | 0.37±0.13 | 3.7±1.3 (n=3; P=0.063) | |
| Liver | 4.67±1.09 | 18.8±4.84 (n=3; P<0.05) | |
| Skeletal muscle | 2.9±0.79 | 12.4±0.55 (n=3; P<0.001) | |
The urine of fed mice contained relatively high concentrations of fructose-ϵ-lysine (typically of the order of 1 mM) and the urinary excretion of fructose-ϵ-lysine, as estimated by the fructose-ϵ-lysine/creatinine ratio did not differ between Fn3k+/+ and Fn3k−/− mice. The urinary excretion of fructoselysine was, however, 10–20-fold lower in 36 h fasted mice, where a significant difference was observed: Fn3k−/− mice excreted 2.5-fold more fructose-ϵ-lysine compared with Fn3k+/+ or Fn3k+/− mice (Table 2).
Table 2. Fructoselysine in urine of Fn3k+/+ and Fn3k−/− mice.
Results are means±S.E.M. *Statistically different, P<0.0001 (Student's t test).
| Fructoselysine (μmol/mg of creatinine) | ||||
|---|---|---|---|---|
| Genotype… | Fn3k+/+ | Fn3k+/− | Fn3k−/− | |
| Urine from fed mice | 3.14±0.19 (n=7) | 3.41±0.90 (n=3) | 3.06±0.23 (n=6) | |
| Urine from 36 h starved mice | 0.117±0.006* (n=6) | 0.078±0.004 (n=3) | 0.272±0.011* (n=8) | |
DISCUSSION
Evidence that FN3K acts as a protein deglycating enzyme
The ability of FN3K to convert protein-bound fructosamines into unsTable 3-phospho derivatives suggested that it could act as a protein repair enzyme, responsible for deglycation of intracellular proteins [5–7]. The present work provides strong evidence for the operation of this mechanism in vivo. Targeted inactivation of the Fn3k gene leads to an increase in protein-bound fructosamines, as evidenced by a combination of biochemical analyses, i.e. (1) retention of proteins on phenylboronate affinity columns (which measures the proportion of proteins having cis-diol groups and is therefore not specific for fructosamines); (2) measurement of fructose-ϵ-lysine residues with a specific enzymatic assay; and, (3) 32P-tagging of protein-bound fructoselysine with human FN3K or E. coli fructoselysine 6-kinase. This increased glycation affects intracellular (haemoglobin, glycolytic enzymes) but not extracellular proteins, as indicated by the finding that the level of serum fructosamines is the same in Fn3k−/− and Fn3k+/+ mice. This last observation and the finding that the Fn3k−/− mice have a normal blood glucose level indicate that the difference in protein-bound intracellular fructosamines does not result from a change in glycaemia. Furthermore, the finding that increased glycation affects tissues where glucose entry is carried out by different types of glucose transporters (e.g. Glut1 in erythrocytes, Glut2 in liver and Glut4 in muscle; see [18] for a review) indicates that it is not the result from a change in intracellular glucose concentration consequent to a perturbed regulation of one of the glucose carriers.
The fact that protein-bound fructosamines are present on haemoglobin and on other cytosolic proteins in mice with a normal level of FN3K indicates that enzymatic deglycation does not proceed to completion. Conceptually, this could be either because FN3K activity is insufficient, i.e. that the rate of glycation largely exceeds the capacity of FN3K or, alternatively, because some of the glycation sites are inaccessible or poorly phosphorylated by FN3K. The findings that the GlcHb levels in Fn3k+/− and Fn3k+/+ mice are indistinguishable despite a 2-fold difference in FN3K activity is against the first possibility (as is also the observation that GlcHb as well as HbA1c (stable minor haemoglobin variant containing glycated N-terminal β chains) levels do not differ between humans with an approx. 4-fold difference in erythrocyte FN3K activity [19]). Accordingly the erythrocyte FN3K activity (3 nmol·min−1·ml−1 of packed cells) largely exceeds the rate of glycation that is calculated to take place at 5 mM glucose (approx. 0.01 nmol·min−1·ml−1 of packed erythrocytes). This conclusion most likely applies not only to erythrocytes but also to tissues, such as liver and brain, where the FN3K activity (expressed per g of tissue) represents at least 10% of the erythrocyte FN3K activity (Figure 1D) [10], although we cannot exclude that cell-to-cell differences in the expression of FN3K may make it rate-limiting for deglycation in some cell types.
Inaccessibility of FN3K to some glycation sites appears to be the major cause for deglycation being incomplete, as indicated by the large variability in the impact of FN3K ablation on the glycation extent at various sites in haemoglobin. Some of the glycation sites were barely affected by the absence of FN3K, whereas a nearly all-or-none situation was found for others (as e.g. peak 3, Figure 4), in agreement with results obtained with haemoglobin derived from human erythrocytes incubated with a competitive inhibitor of FN3K or without [13]. The conclusion from the latter study was that well-accessible fructoselysine residues (e.g. those corresponding to α-Lys16, β-Lys17, α-Lys139 in human haemoglobin) were extensively deglycated in intact erythrocytes (in the absence of the FN3K inhibitor), but that fructosamine removal was negligible at other positions (β-Val1 and β-Lys66), either for accessibility reasons (β-Lys66) or because fructosamines bound to α amines are much poorer substrates for FN3K than those bound to the side-chain of lysines [13]. The action of FN3K provides therefore an explanation for the long-standing observation that some of the lysines that are preferentially glycated following incubation of purified haemoglobin with glucose are undetectably glycated in intact erythrocytes [20].
An important question with respect to the monitoring of diabetes is whether variations in FN3K activity lead to changes in the HbA1c value, independent of changes in blood glucose level. The finding that the large variation in FN3K activity (4-fold range) in human erythrocytes does not detectably affect HbA1c levels, suggests that the glycated valine residue present in human haemoglobin is not a substrate for FN3K in intact cells [19]. Our attempts to confirm these findings in mice (results not shown) were met with difficulties in obtaining a reproducible assay for HbA1c for this species. For instance, mouse HbA1c was poorly separated from other forms of haemoglobin when an HPLC method was used.
Action of FN3K on free fructose-ϵ-lysine
As FN3K phosphorylates free fructose-ϵ-lysine with high affinity (Km≈10 μM), leading to a compound that spontaneously degrades to lysine, 3-deoxyglucosone and inorganic phosphate [6], it can participate in the recovery of lysine from its glycated derivative. Our findings indicate that this recovery mechanism applies differently to endogenously produced and alimentary fructoselysine.
That FN3K participates in the metabolism of intracellular-generated free fructose-ϵ-lysine is indicated by the finding that the free fructose-ϵ-lysine concentration was about 5–13-fold higher in tissues (with the apparent exception of kidneys; see Table 1) from Fn3k−/− mice than from Fn3k+/+ mice, whereas no difference was observed for the plasma concentration of this amino acid derivative. Since the increase in intracellular protein glycation amounts to only about 1.5–2.5-fold, the 5–13-fold increase in tissue free fructose-ϵ-lysine in Fn3k−/− mice is probably best explained by the observation that FN3K also acts on free fructose-ϵ-lysine.
The greater than 10-fold higher urinary excretion of free fructose-ϵ-lysine observed in fed compared with starved mice implies an alimentary origin for most of the urinary fructose-ϵ-lysine excreted by fed animals. The finding that FN3K deficiency does not affect this excretion in fed mice indicates, therefore, that alimentary fructose-ϵ-lysine escapes the ‘salvage’ mechanism initiated by FN3K. This agrees with the observation made in mice and other species that parenterally administered fructose-ϵ-lysine is largely recovered intact in urine after a few hours (J. Delplanque, unpublished work and [21,22]). It should be noted that orally administered fructose-ϵ-lysine appears to be largely metabolized by bacteria present in the hindgut of species such as pig, rat and human [23–25], explaining that little alimentary fructose-ϵ-lysine is recovered in urine in these species. The finding that substantial amounts of fructose-ϵ-lysine were recovered in urine from fed mice indicates therefore that their intestinal bacterial flora does not comprise fructose-ϵ-lysine-metabolizing microorganisms.
In contrast with fed mice, starved Fn3k−/− mice had a significantly higher excretion of fructose-ϵ-lysine compared with Fn3k+/+ mice. This observation results from the fact that in starved mice, all of the fructose-ϵ-lysine found in urine comes from the degradation of endogenous proteins (more glycated in Fn3k−/− mice compared with Fn3k+/+ mice) and that free fructose-ϵ-lysine is no longer phosphorylated by FN3K in Fn3k−/− mice.
Potential physiological importance of FN3K
The results presented here, indicate that FN3K can catalyse the repair of proteins in vivo. Protein deglycation by FN3K is therefore a newly recognized protein repair mechanism, alongside the reduction of methionine sulfoxide residues by peptide methionyl sulfoxide reductase [26,27], the repair of isoaspartate residues by isoaspartyl methyltransferase [28], and the isomerization of cis-proline residues by proline cis-trans-isomerase [29]. Mice deficient in either of the first two protein repair mechanisms show a phenotype, which in the case of protein L-isoaspartyl/(D-aspartyl) O-methyltransferase deficiency is quite dramatic [30,31], and is much milder and dependent on the O2 concentration in the case of methionine sulfoxide reductase deficiency [32].
Although data obtained with siRNA (small interfering RNA) indicate that decreased FN3K expression is toxic to cultured human fibroblasts [33], the Fn3k−/− mice that we have generated looked healthy, indicating either that the observed effects of siRNA were unrelated to depletion of FN3K, or that compensatory mechanisms for FN3K deficiency exist in vivo. The conservation of FN3K in mammals and birds [10] indicates that it must play an important role and that animals with this enzyme have a selective advantage that can be linked either to the protein-repair activity of FN3K or to its action on free fructose-ϵ-lysine. It is likely that this selective advantage, which was not apparent in animals reared in the laboratory, would only become detectable under ‘stressed’ conditions. Identifying the reason why protein repair is important is a difficult task, because a multitude of proteins are potential targets for protein repair mechanisms. Furthermore, the repair to damage occurs at a number of different sites on each protein and with a low stoichiometry at each site. Conceivably, repairing lysine residues may be important to restore enzymatic activity, protein–protein interaction or recognition sites for phosphorylation (which often comprise basic residues) or ubiquitinylation. The availability of an animal model will allow for a better understanding of the physiological importance of this new repair mechanism.
Acknowledgments
We thank Kate Peel and Jean-François Cornut for their expert technical help. This work was supported by grants from the European Foundation for the Study of Diabetes, the Juvenile Diabetes Foundation, the Interuniversity Attraction Poles Programme-Belgian Science Policy and the Actions de Recherches Concertées of the French Community of Belgium. M.V.D.C. and P.J. are Chercheur Qualifié of the Belgian Fonds National de la Recherche Scientifique.
References
- 1.Hodge J. E. The Amadori rearrangement. Adv. Carbohydr. Chem. 1955;10:169–205. doi: 10.1016/s0096-5332(08)60392-6. [DOI] [PubMed] [Google Scholar]
- 2.Baynes J. W., Watkins N. G., Fisher C. I., Hull C. J., Patrick J. S., Ahmed M. U., Dunn J. A., Thorpe S. R. The Amadori product on protein: structure and reactions. Prog. Clin. Biol. Res. 1989;304:43–67. [PubMed] [Google Scholar]
- 3.Brownlee M. Glycation and diabetic complications. Diabetes. 1994;43:836–841. doi: 10.2337/diab.43.6.836. [DOI] [PubMed] [Google Scholar]
- 4.Brownlee M., Vlassara H., Cerami A. Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann. Intern. Med. 1984;101:527–537. doi: 10.7326/0003-4819-101-4-527. [DOI] [PubMed] [Google Scholar]
- 5.Delpierre G., Rider M. H., Collard F., Stroobant V., Vanstapel F., Santos H., Van Schaftingen E. Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes. 2000;49:1627–1634. doi: 10.2337/diabetes.49.10.1627. [DOI] [PubMed] [Google Scholar]
- 6.Szwergold B. S., Howell S., Beisswenger P. J. Human fructosamine-3-kinase: purification, sequencing, substrate specificity, and evidence of activity in vivo. Diabetes. 2001;50:2139–2147. doi: 10.2337/diabetes.50.9.2139. [DOI] [PubMed] [Google Scholar]
- 7.Delpierre G., Collard F., Fortpied J., Van Schaftingen E. Fructosamine3-kinase is involved in an intracellular deglycation pathway. Biochem. J. 2002;365:801–808. doi: 10.1042/BJ20020325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiame E., Delpierre G., Collard F., Van Schaftingen E. Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J. Biol. Chem. 2002;277:42523–42529. doi: 10.1074/jbc.M200863200. [DOI] [PubMed] [Google Scholar]
- 9.Tybulewicz V. L., Crawford C. E., Jackson P. K., Bronson R. T., Mulligan R. C. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 10.Delplanque J., Delpierre G., Opperdoes F. R., Van Schaftingen E. Tissue distribution and evolution of fructosamine 3-kinase and fructosamine 3-kinase-related protein. J. Biol. Chem. 2004;279:46606–46613. doi: 10.1074/jbc.M407678200. [DOI] [PubMed] [Google Scholar]
- 11.Abraham E. C., Perry R. E., Stallings M. Application of affinity chromatography for separation and quantitation of glycosylated hemoglobins. J. Lab. Clin. Med. 1983;102:187–197. [PubMed] [Google Scholar]
- 12.Ahmed N., Argirov O. K., Minhas H. S., Cordeiro C. A., Thornalley P. J. Assay of advanced glycation endproducts (AGEs): surveying AGEs by chromatographic assay with derivatization by 6- aminoquinolyl-N-hydroxysuccinimidyl-carbamate and application to Nϵ-carboxymethyl-lysine and Nϵ-(1-carboxyethyl)lysine-modified albumin. Biochem J. 2002;364:1–14. doi: 10.1042/bj3640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delpierre G., Vertommen D., Communi D., Rider M. H., Van Schaftingen E. Identification of fructosamine residues deglycated by fructosamine-3-kinase in human hemoglobin. J. Biol. Chem. 2004;279:27613–27620. doi: 10.1074/jbc.M402091200. [DOI] [PubMed] [Google Scholar]
- 14.Baker J. R., Metcalf P. A., Johnson R. N., Newman D., Rietz P. Use of protein-based standards in automated colorimetric determinations of fructosamine in serum. Clin. Chem. 1985;31:1550–1554. [PubMed] [Google Scholar]
- 15.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye-binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 16.Jaffé M. Uber den Niederschlag, welchen pikrinsäure in normalen Harn erzeugt und uber eine neue Reaktion des Kreatinins. Z. Phys. Chem. 1886;10:391–400. [Google Scholar]
- 17.Drabkin D. L., Austie J. H. Spectrophotometric studies. Preparations from washed blood cells, nitric oxide hemoglobin and sulfhemoglobin. J. Biol. Chem. 1935;112:51–65. [Google Scholar]
- 18.Gould G. W., Holman G. D. The glucose transporter family: structure, function and tissue-specific expression. Biochem. J. 1993;295:329–341. doi: 10.1042/bj2950329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delpierre G., Veiga-da-Cunha M., Vertommen D., Buysschaert M., Van Schaftingen E. Variability in erythrocyte fructosamine-3-kinase activity in humans correlates with polymorphisms in the FN3K gene and impacts on haemoglobin glycation at specific sites. Diabetes Metab. 2006;32:31–39. doi: 10.1016/s1262-3636(07)70244-6. [DOI] [PubMed] [Google Scholar]
- 20.Shapiro R., McManus M. J., Zalut C., Bunn H. F. Sites of nonenzymatic glycosylation of human hemoglobin A. J. Biol. Chem. 1980;255:3120–3127. [PubMed] [Google Scholar]
- 21.Erbersdobler H. F., Brandt A., Scharrer E., Von Wangenheim B. Transport and metabolism studies with fructose amino acids. Prog. Food Nutr. Sci. 1981;5:257–263. [PubMed] [Google Scholar]
- 22.Finot P. A., Magnenat E. Metabolic transit of early and advanced Maillard products. Prog. Food Nutr. Sci. 1981;5:193–207. [PubMed] [Google Scholar]
- 23.Mori B., Nakatsuji H. Utilization in rats of 14C-L-lysine labeled casein browned by amino-carbonyl reaction. Agric. Biol. Chem. Tokyo. 1977;41:345–350. [Google Scholar]
- 24.Rérat A., Calmes R., Vaissade P., Finot P. A. Nutritional and metabolic consequences of the early Maillard reaction of heat treated milk in the pig. Significance for man. Eur. J. Nutr. 2002;41:1–11. doi: 10.1007/s003940200000. [DOI] [PubMed] [Google Scholar]
- 25.Lee K., Erbersdobler H. F. Balance experiments on human volunteers with ϵ-fructose lysine (FL) and lysinoalanine (LASL) In: Labuza T. P., Reineccius G. A., Monnier V. M., O'Brien J., Baynes J. W., editors. Maillard Reactions in Chemistry Food and Health. London: Royal Society of Chemistry; 1994. pp. 358–363. [Google Scholar]
- 26.Grimaud R., Ezraty B., Mitchell J. K., Lafitte D., Briand C., Derrick P. J., Barras F. Repair of oxidized proteins: identification of a new methionine sulfoxide reductase. J. Biol. Chem. 2001;276:48915–48920. doi: 10.1074/jbc.M105509200. [DOI] [PubMed] [Google Scholar]
- 27.Sun H., Gao J., Ferrington D. A., Biesiada H., Williams T. D., Squier T. C. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry. 1999;38:105–112. doi: 10.1021/bi981295k. [DOI] [PubMed] [Google Scholar]
- 28.Clarke S. Aging as war between chemical and biochemical processes: protein methylation and the recognition of age-damaged proteins for repair. Ageing Res. Rev. 2003;2:263–285. doi: 10.1016/s1568-1637(03)00011-4. [DOI] [PubMed] [Google Scholar]
- 29.Schiene C., Fischer G. Enzymes that catalyze the restructuring of proteins. Curr. Opin. Struc. Biol. 2000;10:40–45. doi: 10.1016/s0959-440x(99)00046-9. [DOI] [PubMed] [Google Scholar]
- 30.Kim E., Lowenson J. D., MacLaren D. C., Clarke S., Young S. G. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6132–6137. doi: 10.1073/pnas.94.12.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim E., Lowenson J. D., Clarke S., Young S. G. Phenotypic analysis of seizure-prone mice lacking L-isoaspartate (D-aspartate) O-methyltransferase. J. Biol. Chem. 1997;274:20671–20678. doi: 10.1074/jbc.274.29.20671. [DOI] [PubMed] [Google Scholar]
- 32.Moskovitz J., Bar-Noy S., Williams W. M., Requena J., Berlett B. S., Stadtman E. R. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conner J. R., Beisswenger P. J., Szwergold B. S. Some clues as to the regulation, expression, function, and distribution of fructosamine-3-kinase and fructosamine-3-kinase-related protein. Ann. N.Y. Acad. Sci. 2005;1043:824–836. doi: 10.1196/annals.1333.095. [DOI] [PubMed] [Google Scholar]
- 34.Collard F., Delpierre G., Stroobant V., Matthijs G., Van Schaftingen E. A mammalian protein homologous to fructosamine-3-kinase is a ketosamine-3-kinase acting on psicosamines and ribulosamines but not on fructosamines. Diabetes. 2003;52:2888–2895. doi: 10.2337/diabetes.52.12.2888. [DOI] [PubMed] [Google Scholar]