

Abstract
Various bacterial pathogens secrete toxins, which are not only responsible for fatal pathogenesis of disease, but also facilitate evasion of host defences. One of the best-known bacterial toxin groups is the mono-ADP-ribosyltransferase family. In the present study, we demonstrate that human neutrophil α-defensins are potent inhibitors of the bacterial enzymes, particularly against DT (diphtheria toxin) and ETA (Pseudomonas exotoxin A). HNP1 (human neutrophil protein 1) inhibited DT- or ETA-mediated ADP-ribosylation of eEF2 (eukaryotic elongation factor 2) and protected HeLa cells against DT- or ETA-induced cell death. Kinetic analysis revealed that inhibition of DT and ETA by HNP1 was competitive with respect to eEF2 and uncompetitive against NAD+ substrates. Our results reveal that toxin neutralization represents a novel biological function of HNPs in host defence.
Keywords: α-defensin, diphtheria toxin, human neutrophil protein 1 (HNP1), mono-ADP-ribosyltransferase, Pseudomonas exotoxin A
Abbreviations: ART, mono-ADP-ribosyltransferase; CHO cell, Chinese-hamster ovary cell; CT, cholera toxin; DT, diphtheria toxin; DTA, catalytic fragment of DT with C-terminal His6 tag; DTT, dithiothreitol; E, free enzyme; eEF2, eukaryotic elongation factor 2; ϵ-NAD+, etheno-β-NAD+; ES complex, enzyme–substrate complex; ETA, Pseudomonas exotoxin A; ETAc, catalytic fragment of ETA with C-terminal His6 tag; FCS, fetal calf serum; HNP1, human neutrophil protein 1; LB plot, Lineweaver–Burk plot; LF, lethal factor; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide; PT, pertussis toxin
INTRODUCTION
Mono-ADP-ribosylation, a covalent chemical modification process, is not only a component of host cell intracellular signalling but is also a widely used mechanism of bacterial pathogenesis [1]. This reaction is catalysed by ARTs (mono-ADP-ribosyltransferases), which transfer the ADP-ribose moiety of NAD+ to a target protein with nicotinamide release [2]. This enzyme family is structurally classified into two groups: the CT (cholera toxin) group and the DT (diphtheria toxin) group [3,4]. Although these two groups do not share any significant sequence homology, they show conserved secondary and tertiary structural elements. The CT group bears an NAD+-binding pocket characterized by the catalytic glutamic acid residue, a β/α structural element and an arginine residue. Most of the bacterial ARTs and mammalian mono-ARTs belong to this group. On the other hand, the DT group including DT and ETA (Pseudomonas exotoxin A) comprises a conserved glutamic acid residue, the β/α motif and a histidine residue instead of arginine. It is well established that DT and ETA cause cell death by inactivating eEF2 (eukaryotic elongation factor 2) via ADP-ribosylation [5].
A recent study showed that mammalian ART-1 modifies Arg14 of HNP1 (human neutrophil protein 1) [6]. HNPs are small cationic peptides, which belong to the α-defensin family. They are structurally characterized by their β-sheet dominant structure and intramolecular disulfide bridges [7]. HNP1–3 are identical in sequence in all but their N-terminal residues [8]. In vitro, these peptides display microbicidal activity against a broad spectrum of bacteria, fungi and viruses [7]. Recently, we showed that HNP1–3 inhibit the activity of LF (lethal factor), a metalloprotease produced by Bacillus anthracis [9]. In the present paper, we report another example of neutralization of bacterial toxins by HNPs: HNP1–3 neutralize bacterial ARTs, particularly DT and ETA of the DT group but not members of the CT group. Together with our previous findings that HNP1–3 neutralize LF of B. anthracis [9], our results reveal that toxin neutralization represents a novel biological function of HNP1–3 in host defence.
EXPERIMENTAL
Bacterial toxins and peptides
Murine ART-1 was purified from HEK-293T cells [human embryonic kidney-293 cells expressing the large T-antigen of SV40 (simian virus 40)] transfected with pCDM8-mART1.Fc (a gift from Dr Friedrich Nolte, University of Hamburg, Hamburg, Germany) as described in [10]. All bacterial toxins were purchased from Quadratech (U.K.). For kinetic experiments, ETAc (catalytic fragment of ETA with C-terminal His6 tag) and DTA (catalytic fragment of DT with C-terminal His6 tag), and Saccharomyces cereviseae eEF2 were purified as described previously [11–13]. Synthetic HNP1 was obtained from Bachem (Weil am Rhein, Germany). Natural HNP1–3 were purified from human buffy coats (German Red Cross) as described in [14]. Synthetic LL-37 was generously provided by Dr Hubert Kalbacher (University of Tübingen, Tübingen, Germany).
ART assay
ADP-ribosylation of HNP1 was analysed as described in [6]. In brief, 10 μg of HNP1 was incubated overnight in 200 μl of 50 mM potassium phosphate buffer (pH 7.5), with 100 ng of each ART (activated) and 3 mM NAD+ at 30 °C. After overnight incubation, the reactions were applied to a XTerra® C18 reverse-phase HPLC column (Waters, Milford, MA, U.S.A.) and analysed. Activation of each toxin was performed as previously described [15–19]. In order to remove DTT (dithiothreitol), which will cause linearization of HNP1, the buffers for toxin activation were modified by using a NAP-10 desalting column (Amersham, U.K.) before each experiment. PT (pertussis toxin) A promoter was obtained from List Biological Laboratories (Quadratech, U.K.).
ADP-ribosylation of eEF2 was examined as follows. HeLa cell lysates (3 μg), [32P]NAD+ and 1 ng of activated DT or ETA were incubated in 20 mM Tris/HCl (pH 8.0) and 1 mM EDTA in the presence of HNP1 or LL-37 at room temperature for 30 min. The reactions were terminated by adding SDS-loading buffer [50 mM Tris/HCl (pH 6.8), 2% (w/v) SDS, 10% (v/v) glycerol, 1% (v/v) 2-mercaptoethanol, 12.5 mM EDTA and 0.02% Bromophenol Blue] and analysed by SDS/PAGE.
Toxin protection assay
One day before the assay, HeLa cells were seeded in 96-well plates at a density of 1.5×104 cells per well in 100 μl of RPMI 1640 medium containing 10% (v/v) FCS (fetal calf serum). For this assay, 20 ng/ml DT or 100 ng/ml ETA and the indicated amounts of HNP1 or LL-37 were added simultaneously to cells in serum-free RPMI 1640. Cell viability was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide] assay 24 h after treatment.
Binding assay and kinetic analysis
The quenching of the intrinsic tryptophan fluorescence of toxins was used to determine the binding constant (Kd) for HNP1 with both DTA and ETAc as described elsewhere [20].
In order to determine IC50 values, the substrates, ϵ-NAD+ (etheno-β-NAD+) and eEF2, at saturating doses of 500 and 14 μM respectively, and various concentrations of HNP1 (0–20 μM) were added to the assay medium (20 mM Tris/HCl, pH 7.9) in a 70 μl total volume. The reaction mixture was incubated for 5 min at 25 °C and initiated by adding 10 nM (final concentration) ETAc or DTA. The reaction was monitored as previously described [21].
The NAD+-dependent and eEF2-dependent ART activities of the toxins were measured as reported previously [21,22]. In brief, the NAD+-dependent ART assay was performed with the eEF2 concentration in the assay medium held to 14 μM, while the concentration of ϵ-NAD+ varied between 0 and 500 μM in the presence of HNP1 at 0, 5, 10, 15 and 20 μM. The reactions were initiated with 20 nM (final concentration) ETAc or DTA. The eEF2-dependent ART assay was performed at 500 μM ϵ-NAD+ while varying the eEF2 substrate (0–15 μM) in the presence of HNP1 at 0, 5, 7.5 and 10 μM. The results were analysed by linear regression analysis of both the Hanes–Woolf and the LB (Lineweaver–Burk) plots. The Ki values were determined using Dixon plots, and also from secondary plots of the slope of the LB plots versus inhibitor concentration.
Infection assay
One day before the assay, HeLa cells were seeded in 96-well plates at a density of 2×104 cells per well in 100 μl of RPMI 1640 medium containing 10% FCS. For the assay, 4×105 Coryne-bacterium diphtheriae cells (A.T.C.C. no. 13812) were added per well and the indicated amounts of HNP1 were then added to the cells in serum-free RPMI 1640. Bacteria were killed by 100 units/ml penicillin and 100 μg/ml streptomycin 6 h after infection, and cells were incubated for a further 24 h. Cytotoxicity was determined by the CytoTox 96® cytotoxicity assay (Promega).
RESULTS AND DISCUSSION
HNP1 is not a substrate of bacterial ARTs
It has been shown that HNP1 is a substrate of mammalian ART-1 [6]. Therefore we first examined whether bacterial ARTs were also able to transfer ADP-ribose to HNP1. CT and PT were tested as representatives of the CT group, and DT and ETA as those of the DT group. Consistent with previous findings [6], we found distinct absorbance peaks in HNP1 incubated with mouse ART-1 corresponding to the ADP-ribose HNP1 product(s) (Figure 1, denoted by arrows). In contrast, HNP1 treated with bacterial ARTs showed the same profile as the control (HNP1 without treatment), i.e. no ADP-ribose product was observed (Figure 1). We conclude that HNP1 does not serve as a substrate for these bacterial ARTs.
Figure 1. Separation of reaction products of HNP1 after incubation with mono-ARTs.

HNP1 was incubated with 50 mM potassium phosphate buffer (pH 7.5) alone, murine ART-1 (mART-1), DT, ETA, CT or PT and then analysed by reverse-phase HPLC. The arrows indicate the peaks corresponding to the ADP-ribose-HNP1.
HNPs protect cells from DT- or ETA-mediated cell death
Next, we determined whether HNPs had any effect on bacterial ART-treated cells. HeLa cells treated with DT or ETA succumbed to these toxins within 24 h. In marked contrast, the addition of HNP1 completely abolished cytotoxicity in a dose-dependent manner (Figures 2A and 2B). Purified natural HNP1–3 mixtures from human leucocytes showed similar protective capabilities (results not shown), whereas LL-37, another neutrophil cationic peptide with similar size and net charge as HNP1–3, did not display any significant effect (Figures 2A and 2B).
Figure 2. Effect of HNP1 on DT- or ETA-treated cells.
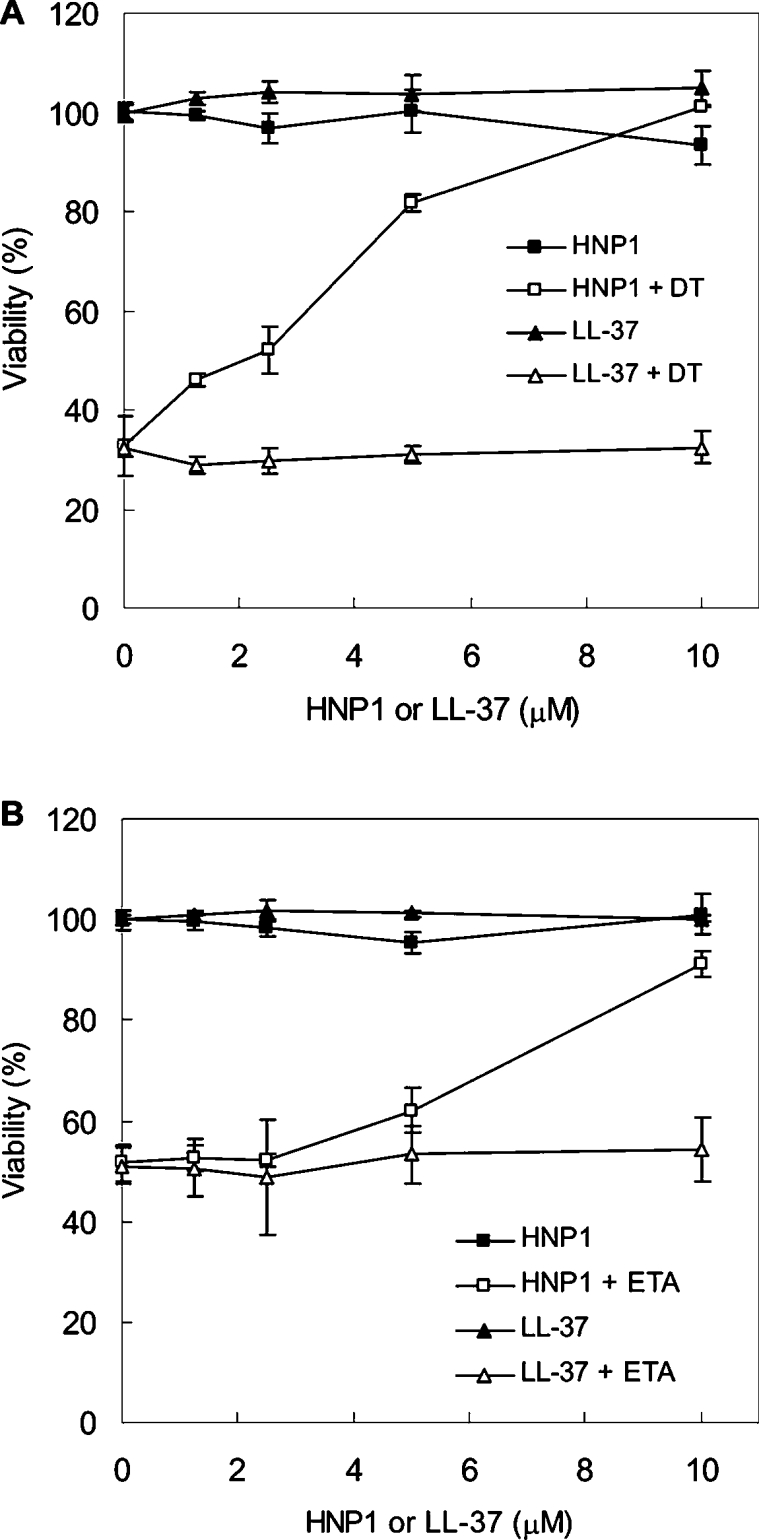
HeLa cells were treated with DT (A) or ETA (B) in the presence of the indicated amounts of HNP1 or LL-37. Bacterial toxins and peptides were treated at the same time and incubated for 24 h. Cell viability was determined by the MTT assay.
We also examined the effect of HNPs on CT- or PT-treated CHO (Chinese-hamster ovary) cells. However, we did not detect any significant difference in cAMP levels between toxin-only-treated groups and toxin- and HNPs-treated groups, indicating that HNPs do not prevent CT- or PT-mediated intoxication in CHO cells (results not shown).
HNP1 inhibits ADP-ribosylation of eEF2
To determine whether HNP1 directly inhibits ART activity of DT and ETA, we examined the transfer of radiolabelled ADP-ribose from NAD+ to eEF2 in vitro. The eEF2 in HeLa cell extracts was efficiently ADP-ribosylated after incubation with activated DT or ETA. In contrast, ADP-ribosylation of eEF2 was inhibited in the presence of HNP1 in a dose-dependent manner. LL-37 had no significant effect on this reaction, suggesting that the inhibition by HNP1 was specific (Figure 3). Surprisingly, linearized HNP1 also showed comparable inhibition activity with intact HNP1. HNP1 contains three intramolecular disulfide linkages to maintain its stable and compact conformation. Our previous study suggested that the unique structure of HNP1 determined by disulfide bonds is a crucial requirement for inhibition against anthrax LF activity [9]. In contrast with LF, DT and ETA were efficiently inhibited by DTT-treated HNP1, indicating that DT and ETA recognize the primary sequence of HNP1 rather than a structural motif in HNP1.
Figure 3. Effect of HNP1 on DT or ETA-mediated ADP-ribosylation of eEF2.

HeLa cell lysates were incubated with DT (A) or ETA (B) in the presence of the indicated amounts of HNP1 or LL-37. The samples were analysed by SDS/PAGE followed by autoradiography. In the presence of HNP1 at 0.1, 0.5 or 2.5 μM, DT activity decreased to 25, 8 and 0.5% of control DT activity and ETA activity declined to 37, 12 and 1% of control ETA activity respectively.
HNP1 competitively inhibits eEF2 and uncompetitively inhibits NAD+
Figures 4(A) and 4(B) show the fluorescence quench curves for the intrinsic tryptophan fluorescence of ETAc and DTA on titration with HNP1 as the ligand. In this assay, HNP1 appears to bind to DTA more tightly (Kd=6.8±1.2 μM) than to ETAc (Kd=21.5±2.1 μM).
Figure 4. Binding properties of HNP1 to DT and ETA.
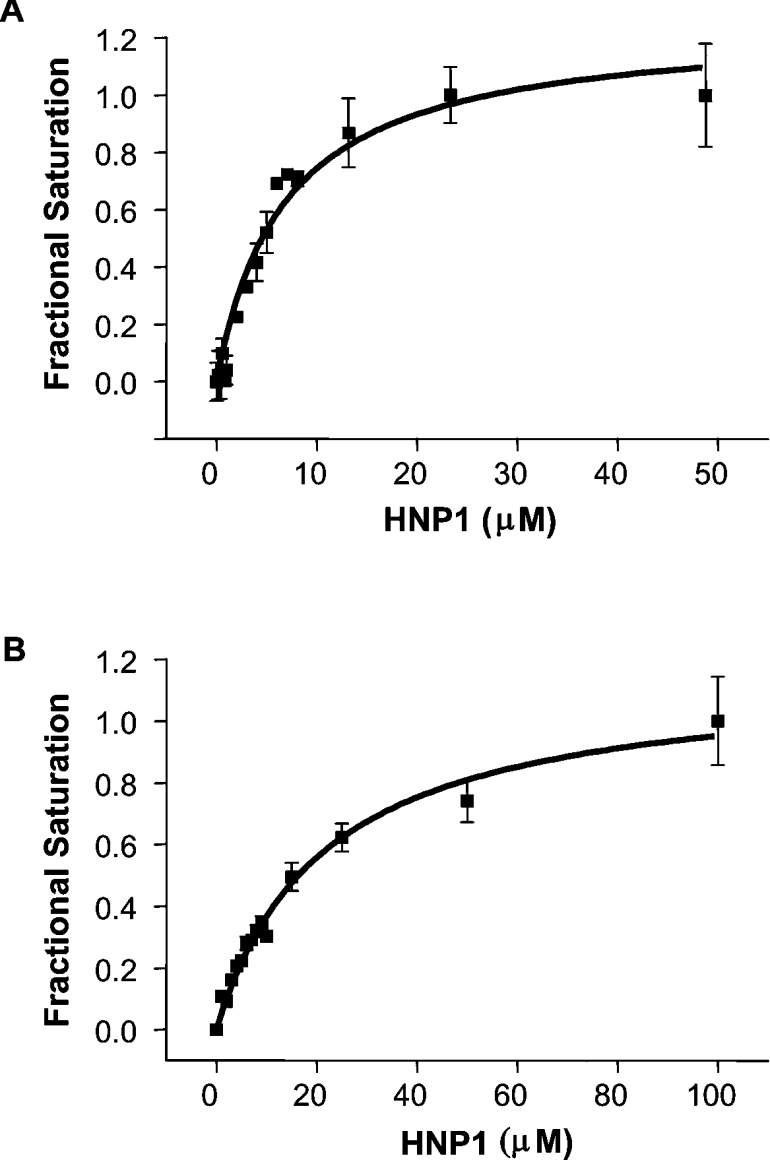
The binding curve for HNP1 with DTA (A) or ETAc (B) was determined from the quenching of the intrinsic protein fluorescence. The raw fluorescence quenching results were converted into fractional saturation values [20] and plotted against the HNP1 concentration.
To elucidate the mode of action, we characterized the kinetics of inhibition of HNP1 against both DTA and ETAc. The initial rates of enzyme reaction in the presence of various concentrations of HNP1 provided an IC50 value of 11.1±2.7 μM against DTA and 19.2±6.5 μM against ETAc (Figures 5A and 5B). In competition assays, when the concentration of eEF2 was held constant and that of NAD+ was varied, HNP1 was identified as an uncompetitive inhibitor against both toxins, invoking a change in both Km and Vmax (Figures 6A and 6B). On the other hand, when the concentration of NAD+ was fixed and that of eEF2 was varied, HNP1 acted as a competitive inhibitor (Figure 6C; inhibition results against DTA not shown). Kinetic and thermodynamic parameters for the interaction of HNP1 with both DTA and ETAc in the present study are summarized in Table 1. These results show that HNP1 is a more effective inhibitor against DTA than ETAc (IC50 values: 11.1±2.6 and 19.2±6.5 μM respectively; Ki values with variable NAD+: 7.3±1.7 and 13.3±0.9 μM respectively), which correlates well with the results from the HeLa cell protection assay (Figure 2).
Figure 5. Inhibitory properties of HNP1 against DTA and ETAc.
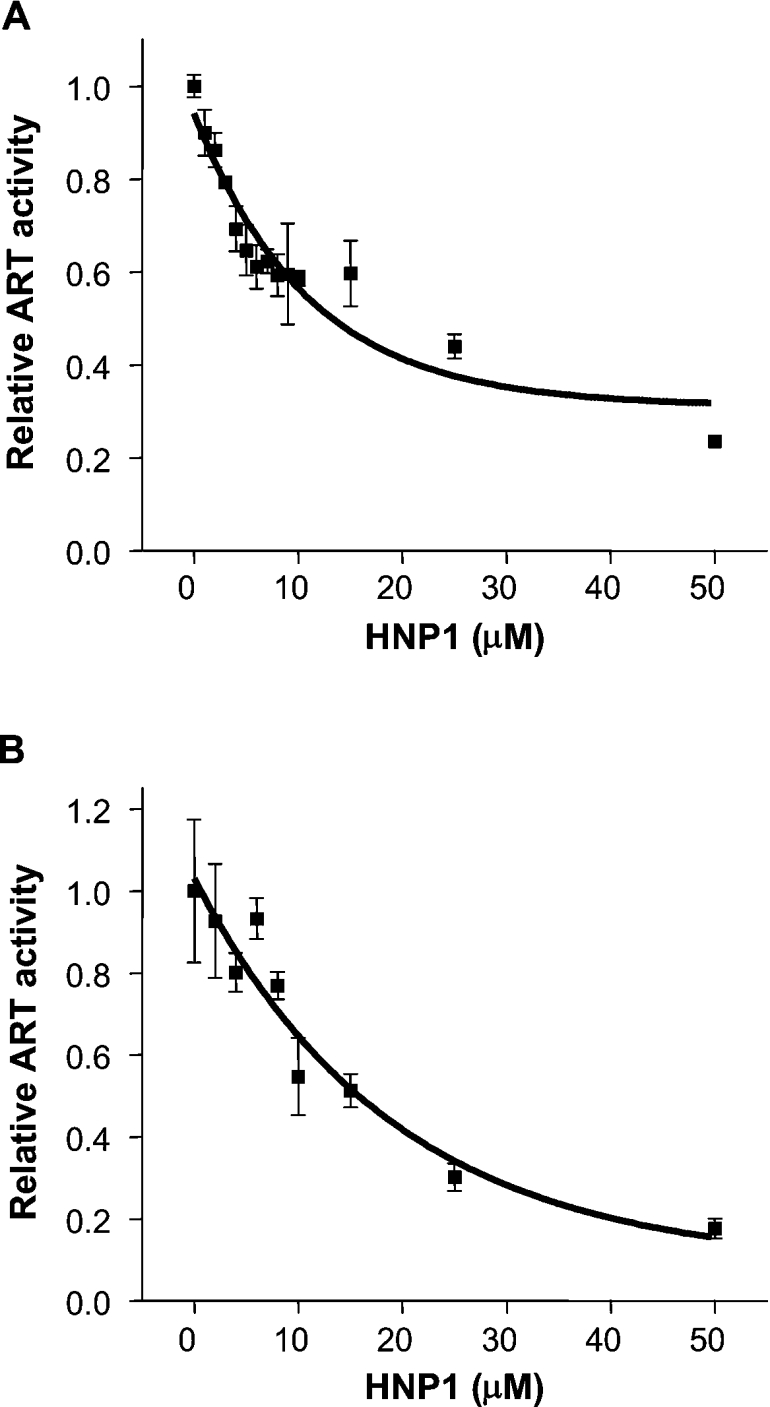
The substrates, ϵ-NAD+ and eEF2, in the fluorescence-based ART assay (see the Experimental section) were used for examining the inhibitory properties of HNP1 against DTA (A) and ETAc (B). The IC50 value was determined by non-linear regression curve fitting using Origin 6.1 (OriginLab, Northampton, MA, U.S.A.). The data were fitted to the exponential first-order decay function.
Figure 6. Mode of inhibition of HNP1 against DTA and ETAc.
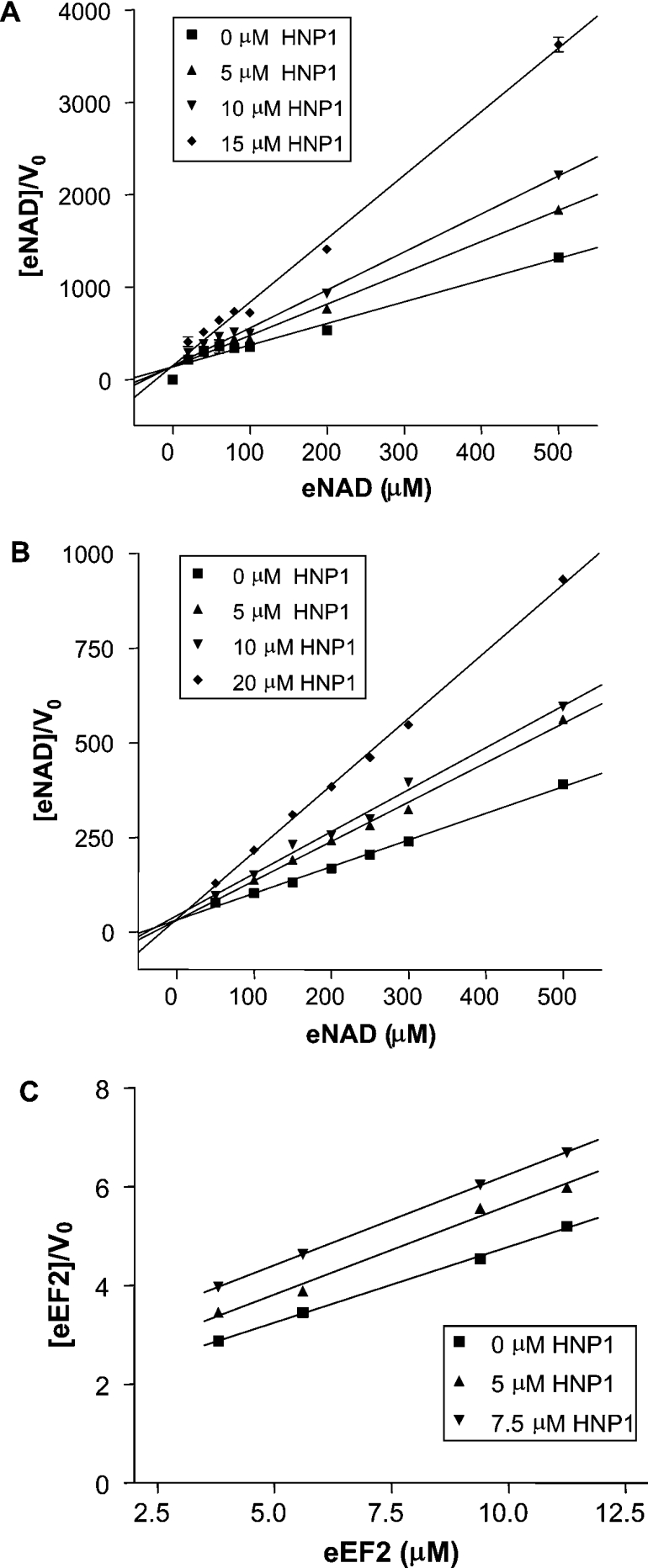
The NAD+-dependent DTA (A) and ETAc (B) assays were performed with the fixed amount of eEF2 and variable concentrations of HNP1 and ϵ-NAD+. The eEF2-dependent ETAc assay (C) was performed at a fixed concentration of ϵ-NAD+ while varying the amounts of eEF2 in the presence of HNP1. The data were analysed by linear regression analysis of the Hanes–Woolf plot. V0, initial velocity of the progress curves for the ADPRT reaction in units of μM/s.
Table 1. Kinetic and thermodynamic parameters for the binding and inhibition of bacterial ART activity by HNP1.
| Variable NAD+ substrate: uncompetitive inhibition | Variable eEF2 substrate: competitive inhibition | |||
|---|---|---|---|---|
| Toxin | IC50 (μM) | Ki (μM) | Kd (μM) | Ki (μM) |
| DT | 11.1±2.6 | 7.3±1.7 | 6.8±1.2 | 6.7±1.1 |
| ETA | 19.2±6.5 | 13.3±0.9 | 21.5±2.1 | 5.3±0.4 |
Our kinetic studies indicate that HNP1 is a competitive inhibitor against both DT and ETA with respect to eEF2. Mammalian ART-1 has been shown to transfer ADP-ribose to HNP1 [6]. As a corollary, HNP1 would be expected to recognize and bind to the eEF2 docking site within both DT and ETA. It is, therefore, not surprising that HNP1 is a competitive inhibitor against the eEF2 substrate for the toxins. On the other hand, HNP1 acted as an uncompetitive inhibitor for NAD+ substrates for the toxins. An uncompetitive inhibitor is defined as an inhibitor binding to the ES (enzyme–substrate) complex but not to the E (free enzyme). Although HNP1 bound to DT and ETA in the absence of NAD+ (Figures 4A and 4B), it was identified as an uncompetitive inhibitor with respect to NAD+ in our assays. The following explanation is a plausible rationalization of this observation. It is possible that HNP1 binds to the E in a unique manner without affecting the binding of NAD+. Yet, once NAD+ is accommodated in the E-binding pocket, HNP1 may bind to the ES complex in an inhibitory mode, which results in a kinetically impaired E (lower Vmax and higher Km). It remains to be elucidated whether unique HNP1–3-binding pockets exist in such functionally distinct bacterial toxins as LF [9], DT and ETA.
HNP1 rescues HeLa cells from C. diphtheriae-induced cell death
Having identified DT neutralization as a biological function of HNP1, we decided to examine whether HNP1 neutralizes DT that has been secreted by bacteria. HeLa cells were infected with C. diphtheriae and were treated immediately with the indicated amounts of HNP1. As shown in Figure 7, we confirmed that HNP1 protects cells from C. diphtheriae-induced cell death in a dose-dependent manner.
Figure 7. Protection of infected cells by HNP1.
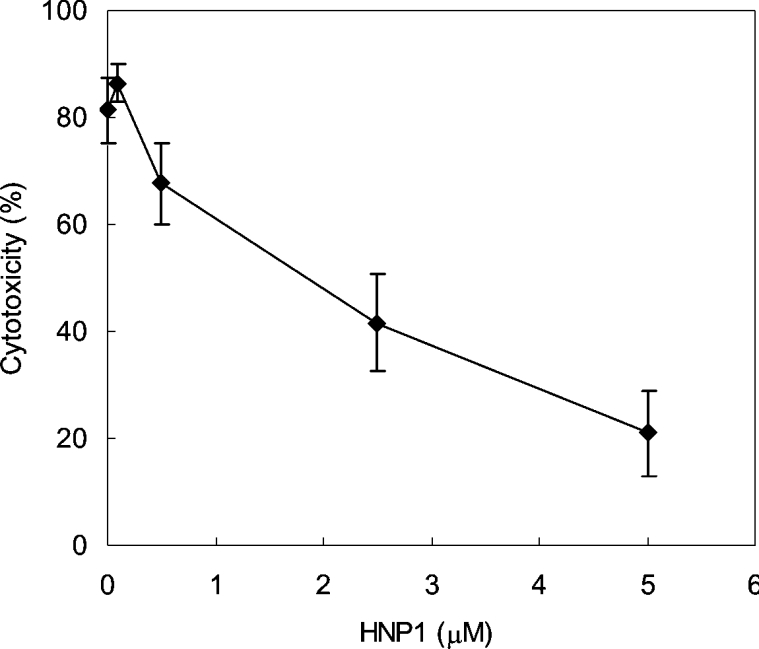
HeLa cells were infected with C. diphtheriae and then treated with the indicated amounts of HNP1. Cytotoxicity was determined by measuring lactate dehydrogenase levels released from HeLa cells.
Recently, we identified a new biological function of HNPs, namely neutralization of a secreted bacterial enzyme, the LF of B. anthracis [9]. Consistent with, and extending of our previous findings [9], in the present study we show that HNP1–3 act as inhibitors of two additional bacterial toxins, DT and ETA. It is well known that these toxins are critical for the pathogenesis caused by C. diphtheriae and Pseudomonas aeruginosa [23]. These two toxins use molecular mimicry of an essential part of the eukaryotic host cell's rRNA in order to minimize the risk that the host could develop resistance to them. This is because mutations that might confer resistance to the host cell would also most likely interfere with translation efficiency and fidelity in the host [24]. As an alternative defence strategy, the host has developed the capacity to neutralize these toxins by binding their enzyme domains with the α-defensins, HNP1–3. The HNP1–3 can be internalized into host cells [25] and the Kd of HNP1 for DT and ETA indicate that it can compete efficiently with eEF2 for association with these toxins inside host cells.
HNP1–3 have several advantages over known NAD+ analogue inhibitors. First, HNP1–3 can prevent toxin binding to ribosomes by blocking the toxin's association with eEF2. It has been suggested that ART toxins can complex with eEF2 on the ribosomes [24,26]. Since NAD+ analogues fail to prevent toxins from binding to eEF2, the toxin may still be able to interfere with eEF2 on translational complexes. Secondly, HNP1–3 are more specific for the toxins because NAD+ analogues will potentially inhibit various host metabolic enzymes such as dehydrogenases, eventually becoming toxic to host cells.
Although HNPs are well known as antimicrobial peptides [27], emerging evidence suggests additional biological functions [28]. Many bacterial pathogens secrete enzymes into the host environment to avoid destruction by the immune system. Because recruited human neutrophils release abundant HNP1–3 locally in the inflammatory regions, we assume that HNP1–3 achieve efficient neutralization of bacterial enzymes in vivo. Hence we propose that neutralization of a group of bacterial toxins constitutes a major function of HNP1–3 in antimicrobial host defence.
Acknowledgments
We thank Dr Friedrich Nolte for murine ART-1 expression vector and helpful recommendations for purification. We also thank Dr Hubert Kalbacher for providing LL-37. We are grateful to Robert Hurwitz and Gerry Prentice for expert technical support. S.H.E.K. acknowledges financial support from EU-FP6 (Anthrax Euronet). A.R.M. received support from the Canadian Institutes of Health Research and the Canadian Cystic Fibrosis Research Foundation.
References
- 1.Corda D., Di Girolamo M. Mono-ADP-ribosylation: a tool for modulating immune response and cell signaling. Science STKE 2002. 2002:PE53. doi: 10.1126/stke.2002.163.pe53. [DOI] [PubMed] [Google Scholar]
- 2.Okazaki I. J., Moss J. Mono-ADP-ribosylation: a reversible posttranslational modification of proteins. Adv. Pharmacol. 1996;35:247–280. doi: 10.1016/s1054-3589(08)60277-x. [DOI] [PubMed] [Google Scholar]
- 3.Domenighini M., Rappuoli R. Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Mol. Microbiol. 1996;21:667–674. doi: 10.1046/j.1365-2958.1996.321396.x. [DOI] [PubMed] [Google Scholar]
- 4.Han S., Tainer J. A. The ARTT motif and a unified structural understanding of substrate recognition in ADP-ribosylating bacterial toxins and eukaryotic ADP-ribosyltransferases. Int. J. Med. Microbiol. 2002;291:523–529. doi: 10.1078/1438-4221-00162. [DOI] [PubMed] [Google Scholar]
- 5.Collier R. J. Understanding the mode of action of diphtheria toxin: a perspective on progress during the 20th century. Toxicon. 2001;39:1793–1803. doi: 10.1016/s0041-0101(01)00165-9. [DOI] [PubMed] [Google Scholar]
- 6.Paone G., Wada A., Stevens L. A., Matin A., Hirayama T., Levine R. L., Moss J. ADP ribosylation of human neutrophil peptide-1 regulates its biological properties. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8231–8235. doi: 10.1073/pnas.122238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehrer R. I., Lichtenstein A. K., Ganz T. Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu. Rev. Immunol. 1993;11:105–128. doi: 10.1146/annurev.iy.11.040193.000541. [DOI] [PubMed] [Google Scholar]
- 8.Selsted M. E., Harwig S. S., Ganz T., Schilling J. W., Lehrer R. I. Primary structures of three human neutrophil defensins. J. Clin. Invest. 1985;76:1436–1439. doi: 10.1172/JCI112121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim C., Gajendran N., Mittrucker H. W., Weiwad M., Song Y. H., Hurwitz R., Wilmanns M., Fischer G., Kaufmann S. H. Human α-defensins neutralize anthrax lethal toxin and protect against its fatal consequences. Proc. Natl. Acad. Sci. U.S.A. 2005;102:4830–4835. doi: 10.1073/pnas.0500508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braren R., Glowacki G., Nissen M., Haag F., Koch-Nolte F. Molecular characterization and expression of the gene for mouse NAD+:arginine ecto-mono(ADP-ribosyl)transferase, Art1. Biochem. J. 1998;336:561–568. doi: 10.1042/bj3360561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yates S. P., Merrill A. R. Characterization of oxidized nicotinamide adenine dinucleotide (NAD(+)) analogues using a high-pressure-liquid-chromatography-based NAD(+)-glycohydrolase assay and comparison with fluorescence-based measurements. Anal. Biochem. 2005;340:41–51. doi: 10.1016/j.ab.2005.01.051. [DOI] [PubMed] [Google Scholar]
- 12.Weiss M. S., Blanke S. R., Collier R. J., Eisenberg D. Structure of the isolated catalytic domain of diphtheria toxin. Biochemistry. 1995;34:773–781. doi: 10.1021/bi00003a010. [DOI] [PubMed] [Google Scholar]
- 13.Andersen C. F., Anand M., Boesen T., Van L. B., Kinzy T. G., Andersen G. R. Purification and crystallization of the yeast translation elongation factor eEF3. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1304–1307. doi: 10.1107/S0907444904010716. [DOI] [PubMed] [Google Scholar]
- 14.Harwig S. S., Ganz T., Lehrer R. I. Neutrophil defensins: purification, characterization, and antimicrobial testing. Methods Enzymol. 1994;236:160–172. doi: 10.1016/0076-6879(94)36015-4. [DOI] [PubMed] [Google Scholar]
- 15.Mekalanos J. J., Collier R. J., Romig W. R. Enzymic activity of cholera toxin. I. New method of assay and the mechanism of ADP-ribosyl transfer. J. Biol. Chem. 1979;254:5849–5854. [PubMed] [Google Scholar]
- 16.Mekalanos J. J., Collier R. J., Romig W. R. Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J. Biol. Chem. 1979;254:5855–5861. [PubMed] [Google Scholar]
- 17.Collier R. J., Kandel J. Structure and activity of diphtheria toxin. I. Thiol-dependent dissociation of a fraction of toxin into enzymically active and inactive fragments. J. Biol. Chem. 1971;246:1496–1503. [PubMed] [Google Scholar]
- 18.Drazin R., Kandel J., Collier R. J. Structure and activity of diphtheria toxin. II. Attack by trypsin at a specific site within the intact toxin molecule. J. Biol. Chem. 1971;246:1504–1510. [PubMed] [Google Scholar]
- 19.Leppla S. H., Martin O. C., Muehl L. A. The exotoxin. P. aeruginosa: a proenzyme having an unusual mode of activation. Biochem. Biophys. Res. Commun. 1978;81:532–538. doi: 10.1016/0006-291x(78)91567-x. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong S., Li J. H., Zhang J., Merrill A. R. Characterization of competitive inhibitors for the transferase activity of Pseudomonas aeruginosa exotoxin A. J. Enzyme Inhib. Med. Chem. 2002;17:235–246. doi: 10.1080/1475636021000010914. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong S., Merrill A. R. Application of a fluorometric assay for characterization of the catalytic competency of a domain III fragment of Pseudomonas aeruginosa exotoxin A. Anal. Biochem. 2001;292:26–33. doi: 10.1006/abio.2001.5052. [DOI] [PubMed] [Google Scholar]
- 22.Yates S. P., Merrill A. R. Elucidation of eukaryotic elongation factor-2 contact sites within the catalytic domain of Pseudomonas aeruginosa exotoxin A. Biochem. J. 2004;379:563–572. doi: 10.1042/BJ20031731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yates S. P., Jorgensen R., Andersen G. R., Merrill A. R. Stealth and mimicry by deadly bacterial toxins. Trends Biochem. Sci. 2006;31:123–133. doi: 10.1016/j.tibs.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Jorgensen R., Merrill A. R., Yates S. P., Marquez V. E., Schwan A. L., Boesen T., Andersen G. R. Exotoxin A-eEF2 complex structure indicates ADP ribosylation by ribosome mimicry. Nature. 2005;436:979–984. doi: 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- 25.Nassar T., Akkawi S., Bar-Shavit R., Haj-Yehia A., Bdeir K., Al-Mehdi A. B., Tarshis M., Higazi A. A. Human alpha-defensin regulates smooth muscle cell contraction: a role for low-density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. Blood. 2002;100:4026–4032. doi: 10.1182/blood-2002-04-1080. [DOI] [PubMed] [Google Scholar]
- 26.Spahn C. M., Gomez-Lorenzo M. G., Grassucci R. A., Jorgensen R., Andersen G. R., Beckmann R., Penczek P. A., Ballesta J. P., Frank J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004;23:1008–1019. doi: 10.1038/sj.emboj.7600102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganz T. Defensins: antimicrobial peptides of vertebrates. C. R. Biol. 2004;327:539–549. doi: 10.1016/j.crvi.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Yang D., Biragyn A., Kwak L. W., Oppenheim J. J. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 2002;23:291–296. doi: 10.1016/s1471-4906(02)02246-9. [DOI] [PubMed] [Google Scholar]