

Abstract
The Pgp (P-glycoprotein) multidrug transporter couples ATP hydrolysis at two cytoplasmic NBDs (nucleotide-binding domains) to the transport of hydrophobic compounds. Orthovanadate (Vi) and fluoroaluminate (AlFx) trap nucleotide in one NBD by forming stable catalytically inactive complexes (Pgp–M2+–ADP–X), which are proposed to resemble the catalytic transition state, whereas the complex formed by beryllium fluoride (BeFx) is proposed to resemble the ground state. We studied the trapped complexes formed via incubation of Pgp with ATP (catalytically forward) or ADP (reverse) and Vi, BeFx or AlFx using Mg2+ or Co2+ as the bivalent cation. Quenching of intrinsic Pgp tryptophan fluorescence by acrylamide, iodide and caesium indicated that conformational changes took place upon formation of the trapped complexes. Trapping with Vi and ATP led to a 6-fold increase in the acrylamide quenching constant, KSV, suggesting that large conformational changes take place in the Pgp transmembrane regions on trapping in the forward direction. Trapping with Vi and ADP gave only a small change in quenching, indicating that the forward- and reverse-trapped complexes are different. TNP (trinitrophenyl)–ATP/TNP–ADP interacted with all of the trapped complexes, however, the fluorescence enhancement differed for the trapped states, suggesting a change in polarity in the nucleotide-binding sites. The nucleotide-binding site of the BeFx-trapped complex was much more polar than that of the Vi and AlFx complexes. Functionally, all the trapped complexes were able to bind drugs and TNP–nucleotides with unchanged affinity compared with native Pgp.
Keywords: ATP-binding cassette superfamily (ABC superfamily), fluorescence quenching, multidrug resistance, nucleotide-binding domain, P-glycoprotein (Pgp), transition state
Abbreviations: ABC, ATP-binding cassette; FRET, fluorescence resonance energy transfer; H33342, Hoechst 33342; NATA, N-acetyltryptophanamide; NBD, nucleotide-binding domain; Pgp, P-glycoprotein; PMPC, palmitoylmyristoyl phosphatidylcholine; R123, rhodamine 123; TM, transmembrane; TNP, trinitrophenyl
INTRODUCTION
The ABC (ATP-binding cassette) transporter superfamily is one of the largest groups of proteins in the proteome, and many of them play an important role in human health and disease [1,2]. The Pgp (P-glycoprotein) multidrug transporter (also called ABCB1) can export hundreds of structurally unrelated hydrophobic natural products, drugs and peptides out of the cell, powered by coupled ATP hydrolysis. Pgp plays an important role in the resistance to multiple chemotherapeutic drugs displayed by many human cancers [3,4]. The location of Pgp at the apical surface of epithelial cells, in the placenta and in the endothelial cells of the blood–brain barrier suggests that its physiological role is protection of tissues and organs from ingested toxic compounds. The ‘vacuum cleaner’ model proposes that substrates gain access to the transporter after partitioning into the lipid bilayer [5]. The drug-binding region is located within the cytoplasmic leaflet [6], probably formed by the TM (transmembrane) helices. Substrates are likely to interact with Pgp via a flexible binding pocket, using multiple van der Waals forces and surface complementarity, in a similar fashion to soluble multidrug-binding proteins [7].
Drug transport by Pgp is powered by ATP hydrolysis at two cytoplasmic NBDs (nucleotide-binding domains). Each NBD contains highly conserved Walker A, B and C signature motifs. Elucidation of the mechanism of drug transport will require a detailed understanding of the various steps in the catalytic cycle of these domains. The addition of the phosphate analogue, orthovanadate (Vi), to membrane-bound Pgp in the presence of ATP resulted in stable trapping of MgADP·Vi in one of the catalytic sites [8], and beryllium fluoride (BeFx) [9] and fluoroaluminate (AlFx) [10] also formed stable trapped complexes. These observations led to the proposal that Pgp operates by an alternating catalytic sites mechanism, whereby only one active site can enter the transition state at any point in time, and the NBDs alternate in hydrolysing ATP [11,12]. The two NBDS of Pgp are likely to be interdigitated with one another [13], as seen in the X-ray crystal structures of the ABC proteins BtuCD, Rad50cd and the E171Q mutant of MJ0796 [14]. The two Walker A motifs of Pgp can be chemically cross-linked [15], and FRET (fluorescence resonance energy transfer) studies indicate that the active sites are close to each other [16]. This physical proximity is presumably necessary for tight functional co-operativity between the two NBDs. In recent years, biophysical/spectroscopic studies and protease-susceptibility approaches have been used to explore the conformational changes taking place in Pgp on substrate binding, and during the catalytic cycle of ATP hydrolysis and drug transport [17]. Fluorescence approaches have allowed quantification of the binding affinity of nucleotides and various substrates to the protein at several points in the catalytic cycle [18–20].
Previous studies with myosin established that Vi trapping of ADP and a bivalent cation at the active site produced a stable complex with no ATPase activity. The trigonal bipyramidal geometry around the penta-co-ordinate vanadium atom in the myosin·MgADP·Vi complex suggests that this species structurally resembles the transient catalytic transition state formed during ATP hydrolysis [21]. In contrast, the geometry around the Be atom in myosin·MgADP·BeFx is tetrahedral, and it occupies the position normally taken up by the γ-phosphorus atom of ATP [22]. Thus it resembles an MgATP complex immediately prior to hydrolysis. The crystal structures of the F1-ATP synthase with bound MgADP [23], and F1 trapped using MgADP with AlFx [24], suggested that the AlFx-trapped complex resembles the catalytic transition state. AlFx can form different complexes with proteins, so that either AlF4− or AlF3 may be bound [25]; the F co-ordination number appears to be regulated by pH. AlFx complexes appear to mimic the transition state in other ATP- and GTP-utilizing enzymes [26,27]. In myosin and G-proteins, the structure of the AlF4− complex is thought to represent a transition state analogue, but it has octahedral square planar geometry in crystal structures [22,27] and is clearly different from the Vi complex [21].
Vanadate-trapped complexes of the bacterial ABC protein, MsbA (a lipid A flippase), have recently been examined using X-ray crystallography and site-directed spin labelling to explore structural changes taking place during the catalytic cycle [28,29]. In the present study, we have used purified Pgp to generate 12 trapped complexes, and have isolated them from unbound ligands. These complexes have been characterized both structurally and functionally using various fluorescence spectroscopic approaches. We were interested in any conformational differences between the trapped states, and how these might relate to the catalytic cycle of the transporter. We confirm that Vi, BeFx and AlFx can all form stable trapped complexes with Pgp, starting from either ATP or ADP, and show that the complexes trapped with Co2+ are exceptionally stable. We used collisional quenching of intrinsic tryptophan fluorescence to assess conformational changes within the TM regions of Pgp that take place on trapping. In the case of vanadate, trapping from ATP leads to a substantially different conformation from trapping with ADP. All the trapped states bound fluorescent TNP (trinitrophenyl)-labelled nucleotides with similar affinity, although the properties of the complexes varied, indicating differences in the polarity of the nucleotide-binding site. Functionally, the trapped complexes did not differ from Pgp in their ability to bind various drug substrates and TNP-labelled nucleotides.
EXPERIMENTAL
Materials
Acrylamide was obtained from Bio-Rad Laboratories (Mississauga, ON, Canada). KI and CsCl were purchased from Fisher Scientific (Unionville, ON, Canada). TNP–ATP, TNP–ADP, H33342 (Hoechst 33342) and R123 (rhodamine 123) were supplied by Molecular Probes (Eugene, OR, U.S.A.). CHAPS, disodium ATP, sodium ADP, sodium orthovanadate, NaF and NATA (N-acetyltryptophanamide) were purchased from Sigma (St Louis, MO, U.S.A.). A stock solution of 100 mM sodium orthovanadate was prepared at pH 10, and samples were boiled for 4 min before use. Beryllium sulfate tetrahydrate (BeSO4·4H2O) and AlCl3 were obtained from Aldrich Chemicals (Milwaukee, WI, U.S.A.). PMPC (palmitoylmyristoyl phosphatidylcholine) was supplied by Avanti Polar Lipids (Alabaster, AL, U.S.A.) and asolectin was purchased from Fluka (Ronkonkoma, NY, U.S.A.).
Plasma membrane preparation and Pgp purification
Pgp was isolated from multidrug-resistant CHRB30 CHO (Chinese-hamster ovary) cell plasma membrane vesicles [30] using a modification of a previously reported two-step selective extraction procedure with the detergent CHAPS [31]. After treatment of the membrane vesicles with 25 mM CHAPS buffer and centrifugation, the resulting S1 pellet was solubilized in 15 mM CHAPS buffer. The soluble S2 fraction was subjected to affinity chromatography on concanavalin A–Sepharose to give purified Pgp, which was kept on ice and used within 24 h. The final Pgp preparation was 90–95% pure in 50 mM Tris/HCl/0.15 M NaCl/5 mM MgCl2 buffer (pH 7.5) containing 2 mM CHAPS. Protein was quantified by the methods of either Peterson [32] or Bradford [33] using BSA (crystallized and lyophilized; Sigma) as a standard.
Measurement of Pgp ATPase activity
The release of Pi from ATP was used to measure the Mg2+-dependent ATPase activity of purified Pgp [34]. Loss of ATPase activity during formation of the trapped complexes was monitored by assaying samples removed at various times after initiation of the reaction. Recovery of ATPase activity at 22 °C, resulting from breakdown of the trapped complexes, was followed over time by comparison with a sample that was treated identically, but in the absence of trapping agent. For breakdown of trapped complexes formed with Mg2+, the data were fitted to a first-order rate equation (SigmaPlot; Systat Software, Point Richmond, CA, U.S.A.), and values of t1/2 were calculated. For breakdown of trapped complexes formed with Co2+, t1/2 values were estimated directly from plots of ATPase activity versus time.
Formation and isolation of trapped complexes of Pgp
For preparation of the Mg2+- and Co2+-trapped complexes of Pgp with Vi, 200 μg of purified protein in 2 mM CHAPS/50 mM Tris/HCl (pH 7.5) was incubated with 1 mM ATP or 1.5 mM ADP, 1 mM MgCl2 or 5 mM CoCl2, and 0.3 mM sodium orthovanadate in a final volume of 2.7 ml for 20 min at 37 °C. The BeFx-trapped complexes of Pgp were formed by incubating 200 μg of purified Pgp with 0.2 mM BeSO4·4H2O, 1 mM NaF, 1 mM ATP or 1.5 mM ADP, and 1 mM MgCl2 or 5 mM CoCl2, for 20 min at 37 °C. AlFx-trapped complexes were formed by incubating 200 μg of purified Pgp with 1 mM AlCl3, 5 mM NaF, 1 mM ATP or 1.5 mM ADP, and 1 mM MgCl2 or 5 mM CoCl2, for 20 min at 37 °C. Trapping was initiated by adding nucleotide to the sample. After incubation, samples were eluted through a gel-filtration column of Bio-Gel-P6 equilibrated with 2 mM CHAPS buffer to remove excess reagents. All Co2+-trapped Pgp samples were subjected to an ATPase assay after completion of the fluorescence experiment to confirm that no catalytic activity was present.
The stated level of ATP in the ADP preparation was less than 1%, and there was no measurable release of Pi from ADP over a 20 min period at 37 °C when 1 mM ADP was substituted for 1 mM ATP in the ATPase assay (see above). Thus formation of trapped complexes in the presence of ADP is not due to catalytic turnover of contaminating ATP.
Fluorescence measurements
Fluorescence spectra were recorded on a PTI Alphascan-2 spectrofluorimeter (Photon Technology International, London, ON, Canada) thermostated at 22 °C, using a 2 nm excitation and emission band pass. Emission spectra were corrected using a built-in automatic correction system. The measured fluorescence intensity was corrected for light scattering using controls containing no Pgp. In the fluorescence-quenching titrations, the inner filter effect was corrected as described previously [31,35]. Quenching data with iodide and caesium ions were corrected for ionic strength effects using parallel titrations with KCl.
Quenching of the intrinsic tryptophan fluorescence of Pgp and its trapped complexes
Stock solutions of 5 M acrylamide, 5 M KI or 5 M CsCl were added as 5 μl aliquots in buffer to 0.5 ml of 100 μg/ml Pgp in 2 mM CHAPS buffer containing 0.5 mg/ml PMPC as large unilamellar vesicles, which had been prepared by extrusion through 100 nm polycarbonate filters [31]. All quencher solutions were freshly prepared, and 0.1 mM Na2S2O3 was added to the KI stock solution to prevent I3− formation. Fluorescence excitation was at 290 nm, with emission measured at 330 nm. Parallel quenching experiments were carried out using NATA in aqueous solution. Quenching data were analysed using the Stern–Volmer equation [35]:
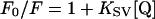 |
where F0 and F are the fluorescence intensities in the absence and presence of quencher respectively, [Q] is the quencher concentration, and KSV is the Stern–Volmer quenching constant.
Determination of TNP-labelled nucleotide-binding affinity to Pgp and its trapped complexes
A fixed concentration of native Pgp (50 μg in 0.5 ml), or Pgp trapped complexes formed from either ATP or ADP, was titrated at 22 °C with increasing concentrations of TNP–ATP or TNP–ADP. A 0.5 ml sample of native Pgp or trapped complex (50 μg of protein) was mixed with 0.5 mg/ml of 100 nm asolectin vesicles in a 0.5 cm quartz cuvette, and titrated with 5 μl samples of TNP-labelled nucleotide in 2 mM CHAPS/50 mM Tris/HCl (pH 7.5) containing 0.5 mg/ml asolectin vesicles. The fluorescence emission was measured at 535 nm after excitation at 408 nm. A solution of 2 mM CHAPS/50 mM Tris/HCl (pH 7.5) containing 0.5 mg/ml asolectin vesicles was titrated as a control in all experiments. Titration data were fitted to the following equation, describing interaction with a single type of binding site:
 |
where ΔF is the change in fluorescence intensity on addition of TNP-labelled nucleotide at a concentration [S], ΔFmax is the maximum fluorescence intensity change, and Kd is the dissociation constant. Fitting was performed using SigmaPlot, and values for Kd and ΔFmax were obtained.
Quenching of Pgp intrinsic tryptophan fluorescence by binding of TNP-ATP
Tryptophan fluorescence quenching upon TNP–ATP binding was used to monitor its binding to native or trapped Pgp as described previously [36]. A fixed concentration of native Pgp (50 μg in 0.5 ml), or trapped complexes formed from either ATP or ADP, was titrated at 22 °C with increasing concentrations of TNP–ATP. Tryptophan residues were excited at 290 nm and emission was monitored at 330 nm.
Determination of the affinity of substrate binding to Pgp and its trapped complexes
Tryptophan fluorescence quenching was used to determine the affinity of native or trapped Pgp for R123 and H33342 [19,36]. Pgp (50 μg in 0.5 ml) was titrated in 2 mM CHAPS buffer containing 0.5 mg/ml PMPC vesicles, with increasing concentrations of drug, and the quenching of tryptophan fluorescence was monitored at 330 nm with excitation at 290 nm. Kd and ΔFmax values were obtained following fitting of the data to an equation describing binding to a single affinity site:
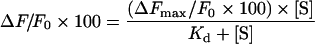 |
where ΔF/F0×100 represents the percentage change in fluorescence intensity relative to the initial value after addition of drug at a concentration [S], and ΔFmax/F0×100 is the maximum percentage quenching of the fluorescence intensity that occurs upon saturation of the drug-binding site. H33342 binding affinity was also assessed by monitoring FRET from Pgp tryptophan residues to the bound drug [19]. A fixed concentration of native or trapped-state Pgp was titrated with increasing concentrations of H33342. Fluorescence emission from H33342 at 460 nm (the emission λmax for the drug) was measured following excitation of tryptophan residues at 290 nm. Kd and ΔFmax values were obtained after fitting of the data to a binding equation. Finally, the binding affinity for H33342 was determined by measuring enhancement of drug fluorescence intensity on titration with native or trapped Pgp (emission at 460 nm following excitation at 350 nm) [19].
RESULTS AND DISCUSSION
Formation of trapped complexes of Pgp
The phosphate analogues Vi, BeFx and AlFx have proved very useful in investigating the ground-state and transition-state-like structures formed at the active sites of proteins that hydrolyse or transfer phosphate groups [37]. Stable complexes of Pgp were formed with three different trapping agents, Vi, BeFx and AlFx. The trapped complexes were formed by reaction in both the forward direction (after one round of ATP hydrolysis) and the reverse direction (from ADP). Trapping in the forward direction was carried out by incubation of purified Pgp with ATP and a bivalent cation in the presence of trapping agent. Following one round of catalysis, the trapped complex consists of Pgp·ADP·M2+·X, where M2+ represents a bivalent cation, and X is Vi, BeFx or AlFx. Trapping in the reverse direction requires no catalytic turnover, and was accomplished by incubating Pgp with ADP and a bivalent cation in the presence of trapping agent. The formation of the trapped state can be followed by measuring the loss of Pgp ATPase activity [8,16]. Previous work in our laboratory [16] and by others [8] pointed out the usefulness of using Co2+ as the bivalent cation in forming the trapped complex of Pgp with Vi. Trapped complexes of Pgp were formed from ATP at 37 °C in the presence of Mg2+ or Co2+ for all three trapping agents. The loss of ATPase activity approached 100% when trapping was complete. Trapping occurred very rapidly, with 80% of the ATPase activity lost after 2–5 min, and was essentially complete after 6–8 min. The rate of formation of the trapped state was slightly lower when ADP was used, as reported previously [8]. Trapped complexes formed in the presence of Mg2+ all showed similar kinetics of formation, as did those formed from Co2+. Trapping in the presence of Co2+ was somewhat slower than with Mg2+, and again, trapping was slower when using ADP.
The 12 trapped complexes were separated from unbound ligands, and the time-course of their dissociation was examined. We determined the kinetics of the return of ATPase activity after prolonged incubation, which corresponds to dissociation of the trapping agent and ADP to re-form active Pgp. Trapped complexes formed using Mg2+ as the bivalent cation all showed similar kinetics for the reappearance of ATPase activity. The kinetic data fitted well to a first-order rate equation, and t1/2 values for dissociation were obtained by the fitting process (see Table 1). The t1/2 values for dissociation were relatively rapid (approx. 30 min) for the BeFx-and AlFx-trapped states, and almost twice as slow for the Vi-trapped state. In general, the Mg2+-trapped complexes dissociated and regained full catalytic activity when compared with a control sample after approx. 5 h. Our results agree with those of Urbatsch et al. [8] who reported virtually identical dissociation kinetics for the Vi-trapped states of plasma membrane Pgp formed from MgATP and MgADP. In contrast, the six Co2+-trapped complexes were much more stable, with t1/2 values for dissociation of between 6 and 11 h (Table 1), and required 24 h to regain activity. For both Mg2+ and Co2+, the dissociation rates of the trapped complexes formed with ATP were almost identical with those formed with ADP, irrespective of which of the three trapping agents was used (Table 1). The remarkable stability of the six Co2+-trapped complexes makes them very useful tools for spectroscopic analysis.
Table 1. Half-times (t1/2) for dissociation at 22 °C of trapped complexes of Pgp formed from ATP or ADP in the presence of Mg2+ or Co2+ ions.
Dissociation of trapped complexes of Pgp was indicated by recovery of catalytic activity. Trapped complexes were formed with ATP/ADP, and either Mg2+ or Co2+, in the presence of Vi, BeFx or AlFx. After isolation by gel-filtration chromatography, the complexes were incubated at 22 °C for various times, and their ATPase activity was determined, using a 10 min assay time, relative to a sample of native Pgp subjected to a mock treatment without trapping agent. Two independent experiments were carried out for determination of each value of t1/2, with different preparations of Pgp and the three trapped complexes in each case. For the breakdown of trapped complexes formed in the presence of Co2+, t1/2 values were estimated directly from kinetic plots of ATPase activity versus time. For breakdown of trapped complexes formed in the presence of Mg2+, the kinetic plots were fitted to a first-order rate equation, and values of t1/2 were obtained by the fitting process.
| t1/2 for dissociation of trapped complexes | |||
|---|---|---|---|
| Trapping agent | Mg2+ (min) | Co2+ (h) | |
| ATP | Vi | 61 | 11 |
| BeFx | 35 | 8.5 | |
| AlFx | 32 | 5.7 | |
| ADP | Vi | 68 | 11 |
| BeFx | 35 | 8.6 | |
| AlFx | 32 | 6.0 | |
Quenching of intrinsic tryptophan fluorescence of Pgp trapped complexes by acrylamide
Tryptophan residues are widely used as reporters for protein conformational changes. Pgp contains 11 tryptophan residues, three in putative TM segments, two in extracellular loops and six in intracellular loops and domains [36]. The fluorescence emission spectrum of Pgp shows a low λmax of 333 nm, indicating that the emitting tryptophan residues are located in a very hydrophobic environment, and drug binding can quench tryptophan fluorescence very efficiently. For these reasons, Pgp intrinsic fluorescence is believed to arise from tryptophan residues in the TM regions of the protein [36]. Dynamic quenchers are also able to quench intrinsic tryptophan fluorescence, the extent of quenching depending on the solvent exposure of the tryptophan residues. Changes in the conformation or arrangement of tryptophan residues in the TM domains of Pgp in the trapped complexes would be reflected in alterations in quenching characteristics. Native Pgp and the six Co2+-trapped complexes were subjected to acrylamide quenching to detect conformational differences as a change in the Stern–Volmer quenching constant, KSV. As noted previously [36], the emitting tryptophan residues formed a single class of fluorophores, as indicated by linear Stern–Volmer plots up to an acrylamide concentration of 0.5 M. The tryptophan residues in native Pgp were highly inaccessible to acrylamide (Figure 1), with a 22-fold lower value for KSV compared with the soluble Trp analogue NATA (Table 2). Thus native Pgp is tightly folded so that the emitting tryptophan residues are buried and relatively inaccessible to acrylamide in the aqueous solvent.
Figure 1. Stern–Volmer plots for quenching of trapped complexes of Pgp by acrylamide.

Complexes were formed using the trapping agents Vi (A), BeFx (B) and AlFx (C), in the presence of Co2+, with either ATP (▼) or ADP (▽), and isolated by gel-filtration chromatography. Samples (5 μl) of a 5 M solution of acrylamide were added at 22 °C to 0.5 ml of trapped complex (50 μg of Pgp) in 0.5 ml of 2 mM CHAPS buffer containing 0.5 mg/ml PMPC vesicles, and the fluorescence emission intensity was determined at 330 nm after each addition following excitation at 290 nm. Parallel experiments were carried out using native Pgp (○; 50 μg in 0.5 ml), and the soluble Trp analogue, NATA (●; 30 μM). Fluorescence intensities were corrected for dilution and scattering as described in the Experimental section.
Table 2. Stern–Volmer constants for acrylamide, KI and CsCl quenching of trapped complexes of Pgp formed from ATP or ADP in the presence of Co2+ ions.
Two independent experiments were carried out for determination of each value of KSV, with different preparations of Pgp and the three trapped complexes in each case. The results are expressed as the means±range. NL, non-linear.
| KSV | |||
|---|---|---|---|
| Acrylamide | KI | CsCl | |
| Native Pgp | 1.40±0.012 | 1.17±0.036 | 0.343±0.019 |
| Pgp+Vi+ATP | 8.85±0.142 | 0.82±0.026 | 0.616±0.023 |
| Pgp+Vi+ADP | 2.49±0.019 | 0.71±0.030 | 0.355±0.015 |
| Pgp+BeFx+ATP | 2.88±0.032 | 0.96±0.038 | 0.275±0.010 |
| Pgp+BeFx+ADP | 2.05±0.025 | 0.99±0.013 | 0.101±0.008 |
| Pgp+AlFx+ATP | 1.37±0.066 | 1.09±0.080 | 0.262±0.014 |
| Pgp+AlFx+ADP | 2.34±0.079 | 0.83±0.032 | NL |
| NATA | 31.0±1.17 | 11.2±0.26 | 2.468±0.041 |
The Vi-trapped complex formed from ATP clearly possesses a conformation in which the emitting tryptophan residues are much more accessible to quencher (Figure 1A and Table 2). Thus the TM regions of Pgp appear to undergo considerable conformational rearrangement upon formation of the Vi-trapped complex from ATP, so that tryptophan residues are more exposed to the aqueous surroundings. Cryo-electron microscopy at 10 Å resolution showed that Pgp undergoes a dramatic structural change on Vi trapping [38], suggestive of an extensive re-organization of the TM helices. Proteolysis data also indicate that the Vi-trapped complex has an altered conformation when compared with the native protein. The Vi-trapped state was reported to show a peptide profile intermediate between those of the ATP- and ADP-bound states in trypsin-sensitivity experiments [39]. In contrast, Julien and Gros [40] found that Vi-trapped Pgp displayed a unique peptide pattern on proteolysis and was highly resistant to trypsin digestion.
Interestingly, the Vi-trapped complex formed from ADP behaved quite differently, and its quenching behaviour was more similar to that of native Pgp, with a KSV value elevated only 1.8-fold (Figure 1A and Table 2). This dramatic difference in quenching properties strongly suggests that the TM domains are arranged differently in the forward- and reverse-trapped Pgp species formed with Vi, which thus do not appear to be identical.
In the case of the other trapping agents, BeFx (Figure 1B) and AlFx (Figure 1C), the trapped complexes formed from ADP displayed properties similar to that of the Vi-trapped state, with KSV values slightly higher than those of the native Pgp (Table 2). The trapped complexes formed using ATP, however, did not show the high quenching level of the Vi-trapped state. The complex formed from ATP showed slightly higher quenching than that of the ADP complex in the case of BeFx, and for AlFx, the complex formed from ATP behaved identically with native Pgp. Thus, for BeFx and AlFx, the conformation of the TM regions of the protein in the ATP-trapped state is probably not dramatically different from that of the ADP-trapped state. Nevertheless, reproducible differences in KSV values can be seen for the complexes formed from ATP and ADP, indicating that even for these trapped states, there is a small difference in conformation of the membrane-bound regions which is reflected in the accessibility of tryptophan residues to acrylamide.
Quenching of intrinsic fluorescence of Pgp-trapped complexes by iodide and caesium ions
The use of charged quenchers can shed light on the local electrostatic environment of the fluorophore. The Stern–Volmer plots with iodide ion as the quencher were linear for native Pgp and all six trapped states (Figure 2). As was observed with acrylamide, the KSV value for native Pgp was much lower than for NATA (approx. 10-fold; Table 2), again implying that the emitting tryptophan residues are buried, without contact with the aqueous solvent. In general, the six trapped complexes showed lower quenching and smaller KSV values than native Pgp. Only in the case of the AlFx complex formed with ATP was quenching slightly enhanced over that of untrapped protein. For Vi and AlFx, the trapped complexes formed from ATP and ADP showed somewhat different quenching properties, reflected in differences in the KSV values for iodide ion (Table 2), again suggesting that they adopt slightly different conformations. Quenching with caesium ion was carried out (Table 2), and also showed differential quenching between the trapped states formed from ATP and ADP, especially for complexes trapped with Vi. In addition, the KSV values for quenching of the Vi-trapped complex by caesium ion were larger than those for native Pgp, indicating that this quencher has enhanced access to the Trp residues after complex formation.
Figure 2. Stern–Volmer plots for quenching of trapped complexes of Pgp by iodide ion.

Complexes were formed using the trapping agents Vi (A), BeFx (B) and AlFx (C), in the presence of Co2+ with either ATP (▼) or ADP (▽), and isolated by gel-filtration chromatography. Samples (5 μl) of a 5 M solution of KI were added at 22 °C to 0.5 ml of trapped complex (50 μg of Pgp) in 0.5 ml of 2 mM CHAPS buffer containing 0.5 mg/ml PMPC vesicles, and the fluorescence emission intensity was determined at 330 nm after each addition following excitation at 290 nm. A parallel experiment was carried out using native Pgp (○; 50 μg in 0.5 ml). Fluorescence intensities were corrected for dilution, scattering and ionic strength effects as described in the Experimental section.
Interaction of native Pgp and its trapped complexes with TNP-labelled nucleotides
The fluorescent nucleotides TNP–ATP and TNP–ADP can interact with the NBDs of Pgp. TNP–ATP is a poor Pgp substrate, and behaves as a classical competitive inhibitor of ATPase activity [41]. TNP-labelled nucleotides are only weakly fluorescent in aqueous solution, but display greatly enhanced emission upon transfer to the hydrophobic nucleotide-binding site of Pgp [41], allowing estimation of their binding affinity. Figure 3 shows the emission spectra of TNP–ATP bound to various Pgp-trapped complexes. Native Pgp displays the highest fluorescence enhancement, with the Vi-trapped complexes displaying roughly half the enhancement, consistent with binding of only one nucleotide molecule per protein at the unoccupied NBD [20].
Figure 3. Fluorescence emission scans at 22 °C for TNP–ATP bound to the various complexes of Pgp trapped with Co2+ and either ATP or ADP.

Samples of native Pgp (a), or complexes trapped with Vi and ATP (b), Vi and ADP (c), BeFx and ATP (d), and BeFx and ADP (e), contained 50 μg of protein in 0.5 ml of 2 mM CHAPS buffer with 0.5 mg/ml asolectin vesicles. The TNP–ATP concentration was 100 μM. The fluorescence emission spectrum was recorded over the range 480–650 nm after excitation at 408 nm.
Spectral properties of the bound nucleotides can provide information on the local polarity of the binding site. The complexes formed from ATP and ADP in the presence of Vi show similar emission spectra, with a blue-shifted emission maximum of 539 nm compared with aqueous TNP–ATP, reflecting the hydrophobic environment of the NBD. In contrast, the emission spectra of TNP–ATP bound to the BeFx-trapped complexes showed a very small fluorescence enhancement, and a barely detectable blue shift (Figure 3). This suggests that the local environment of the TNP–nucleotide is substantially more hydrophilic in these complexes. This difference is not unexpected, since BeFx-trapped complexes are thought to resemble the ground-state structurally, whereas the other two complexes likely mimic the transition state.
Titration of native or trapped Pgp with TNP-labelled nucleotide results in a saturable increase in fluorescence for all six trapped states (Figure 4). Fitting of the data to a binding equation allows estimation of the Kd, and the maximum enhancement, ΔFmax [20,41]. Figure 4(A) and Table 3 indicate that ΔFmax for the Vi-trapped complexes is approximately half that of native Pgp. Previous work showed that native Pgp binds two TNP–nucleotide molecules, whereas the Vi-trapped complex binds only one [20]. The fluorescence enhancement of this single TNP–nucleotide bound to the trapped state is similar to that of TNP–nucleotides bound to native Pgp, probably reflecting an environment with a similar non-polar nature. Binding of TNP–ATP and TNP–ADP to the AlFx-trapped complex also gave a similar ΔFmax when trapping used ADP, but a lower value when ATP was used (Figure 4C and Table 3), suggesting a difference between the molecular properties of the nucleotide-binding site in the two trapped states. TNP–ATP also bound more tightly to the AlFx-trapped complexes (Kd of 17–19 μM) compared with those trapped with Vi (Kd of 54–58 μM) (Table 3).
Figure 4. Binding of TNP–ATP at 22 °C to the various complexes of Pgp trapped with Co2+ and either ATP (●) or ADP (○), assessed by enhancement of TNP–ATP fluorescence.

Binding to Pgp trapped with (A) Vi (binding of TNP–ATP to native untrapped Pgp is also shown for comparison, ♦), (B) BeFx and (C) AlFx. Samples contained 50 μg of protein in 0.5 ml of 2 mM CHAPS buffer with 0.5 mg/ml asolectin vesicles, and were titrated successively with 12×5 μl aliquots of TNP–ATP working solutions as follows; three aliquots of 0.25 mM TNP–ATP, followed by three aliquots each of 0.5 mM, 1.0 mM and 2.0 mM TNP-ATP. All working solutions were made up from a 6.4 mM stock TNP–ATP solution. The fluorescent nucleotide was excited at 408 nm, and fluorescence emission was measured at 535 nm. A control titration was carried out using buffer and asolectin vesicles in the absence of Pgp, and the fluorescence intensities were subtracted from those determined in the presence of Pgp.
Table 3. Affinity for TNP-labelled nucleotide binding of trapped Pgp complexes formed from ATP or ADP in the presence of Co2+ ions and determined by enhancement of TNP–nucleotide fluorescence.
Two independent experiments were carried out for determination of each value of Kd, with different preparations of Pgp and the three trapped complexes in each case. The results are expressed as the means±range.
| TNP–ATP | TNP–ADP | |||
|---|---|---|---|---|
| Kd (μM) | ΔFmax | Kd (μM) | ΔFmax | |
| Native Pgp | 50.3±4.7 | 17.8±1.4 | 48.0±4.8 | 18.6±1.0 |
| Pgp+Vi+ATP | 53.8±1.6 | 7.1±0.4 | 31.6±1.5 | 7.3±0.2 |
| Pgp+Vi+ADP | 58.0±3.0 | 8.6±0.5 | 38.0±5.5 | 10.5±0.5 |
| Pgp+BeFx+ATP | 54.2±1.4 | 1.5±0.2 | 42.6±2.1 | 1.8±0.1 |
| Pgp+BeFx+ADP | 59.3±1.4 | 1.7±0.2 | 47.1±5.5 | 2.6±0.1 |
| Pgp+AlFx+ATP | 19.3±2.7 | 4.6±0.2 | 35.7±2.9 | 4.0±0.2 |
| Pgp+AlFx+ADP | 16.9±3.8 | 7.2±0.6 | 31.2±4.1 | 6.5±0.3 |
The BeFx-trapped complexes showed very low fluorescence enhancement, and correspondingly small ΔFmax values of 1.5–2.6, compared with Vi and AlFx (Figure 4B and Table 3). Thus the local environment of the bound TNP–nucleotide must be considerably more polar in these complexes, in agreement with the observation that there is a barely detectable blue shift in the emission spectrum. We used another approach to show that the BeFx-trapped complex does indeed bind TNP–nucleotide with similar affinity to the other complexes. We previously showed that TNP–ATP/ADP binding to Pgp results in quenching of the tryptophan fluorescence by an energy transfer mechanism [36]. As shown in Figure 5, all three trapped complexes of Pgp showed saturable quenching of tryptophan fluorescence over the same TNP–ATP concentration range, with comparable estimated Kd values, indicating that the BeFx-trapped complex has a similar intrinsic ability to bind TNP–ATP.
Figure 5. Binding of TNP-ATP at 22 °C to the various complexes of Pgp trapped with Co2+ and either ATP (●) or ADP (○), assessed by quenching of intrinsic tryptophan fluorescence.

Binding to Pgp trapped with (A) Vi (binding of TNP-ATP to native untrapped Pgp is also shown for comparison, ♦), (B) BeFx and (C) AlFx. A 0.5 ml sample (in 2 mM CHAPS buffer with 0.5 mg/ml asolectin vesicles) containing 50 μg of native Pgp, or Pgp-trapped complexes formed from either ATP or ADP, was titrated with increasing concentrations of TNP–ATP. Tryptophan residues were excited at 290 nm and emission was monitored at 330 nm. A control titration was carried out using buffer and asolectin vesicles in the absence of Pgp, and the fluorescence intensities were subtracted from those determined in the presence of Pgp.
Thus the nucleotide-binding pockets of the three trapped species display different characteristics that are reflected in the binding affinity and the fluorescence properties of bound TNP–nucleotides. Forward- and reverse-trapped complexes again displayed differences in ΔFmax values, suggesting that the local environment around the bound nucleotide is not identical, likely as a result of differences in local structure at the nucleotide-binding site.
Interaction of drug substrates with native Pgp and its trapped complexes
Drugs are believed to gain access to Pgp from the lipid bilayer, and both the H33342 and R123 drug-binding sites have been mapped to the cytoplasmic leaflet of the membrane [6,42]. Tryptophan residues in the TM helices may play an important role in binding drugs, especially those with aromatic rings [43]. The tryptophan fluorescence quenching that arises on drug binding allows estimation of the binding affinity [36]. Substantial tryptophan quenching was observed on addition of the transport substrate R123 to the various trapped complexes of Pgp (Figures 6A–6C), confirming that they can bind this compound. The estimated Kd values were very similar for native Pgp and all six trapped complexes, whereas the maximal quenching was significantly lower in the trapped complexes. R123 is proposed to quench Pgp tryptophan residues within the membrane by a FRET-based mechanism [36]. Therefore, a reduction in the maximal quenching reflects the fact that a conformational difference exists between the trapped complexes and native Pgp.
Figure 6. Binding of R123 to various complexes of Pgp trapped with Co2+ and either ATP (●) or ADP (○).

Vi (A), BeFx (B) and AlFx (C) trapping. Binding was measured by quenching of tryptophan residues within Pgp (excitation at 290 nm, emission at 330 nm). Samples contained 50 μg of protein in 500 μl of 2 mM CHAPS buffer with 0.5 mg/ml PMPC vesicles, and were titrated successively with 10×5 μl aliquots of three different R123 working solutions. R123 binding to native untrapped Pgp (♦) is shown in each panel for comparison. A control titration was carried out using buffer and PMPC vesicles in the absence of Pgp, and the fluorescence intensities were subtracted from those determined in the presence of Pgp.
Binding affinity was also determined for H33342, a substrate that displays enhanced fluorescence on interaction with Pgp, and also quenches its tryptophan fluorescence by a FRET mechanism [6,19,42]. For each of the six trapped complexes, enhancement of H33342 fluorescence, quenching of intrinsic tryptophan fluorescence by drug binding and FRET emission from bound drug following tryptophan excitation were measured. Once again, the binding affinities were comparable between all of the trapped complexes and native Pgp, whereas there were differences between the values of ΔFmax, tryptophan quenching and FRET emission (results not shown). We previously showed that Vi-trapped Pgp formed from ATP displays high-affinity binding for several drugs [19], in contrast with an earlier report that its affinity for a photo-active drug was very low [44]. The results obtained with R123 and H33342 indicate that all six trapped complexes of Pgp bind drug substrates with high affinity, and appear functionally indistinguishable. However, the observed differences in the maximal values of Trp quenching, FRET emission, and fluorescence enhancement for the trapped complexes, relative to native Pgp, suggest that protein conformation changes altered the local environment around the site where the drugs bind.
Based on photo-affinity labelling studies, Vi-trapped Pgp was proposed to show 30-fold decreased affinity for [125I]iodoarylazidoprazosine [44]. This led to a proposed transport mechanism in which the drug binding site changed from facing the cytoplasmic leaflet to facing the extracellular medium, coupled with a large fall in binding affinity, thus resulting in drug release to the outside [45]. This proposed mechanism may indeed be correct, but the underlying assumption that the stable trapped states should display similar binding properties to the true catalytic transition state is not. The trapped complexes are only structural mimics of the transition state that forms during ATP hydrolysis. Based on the low affinity of Pi association with Pgp, it was proposed that a large decrease in free energy occurs on release of Pi after ATP hydrolysis [11,12]. The trapping agents enter the active site of the NBD after Pi dissociation (Scheme 1), thus these complexes are very stable and of low energy, whereas the true transition state is transient and of high energy. Drug transport is most likely coupled to the large release of free energy resulting from loss of Pi from the active site [11], and is thus complete before formation of the trapped states (Scheme 1). The switch to low-affinity drug binding and back to high-affinity drug binding is thus expected to be complete before the trapped states are formed. This provides an explanation for the high-affinity drug binding properties observed for the BeFx- and AlFx-trapped states in the present study, and for the Vi-trapped state in previous work [19].
Scheme 1. Kinetics of the formation of forward- and reverse-trapped complexes of Pgp.

Binding to native Pgp of substrate (S) and ATP, together with a bivalent cation (M2+), results in formation of a ternary complex. Hydrolysis of ATP to ADP and Pi proceeds via a transient high-energy catalytic transition state (ts*). Loss of Pi from the post-hydrolysis complex leads to a large fall in free energy, with concerted release of substrate on the other side of the membrane (i.e. transport). This release is likely to take place via a shift in the location of the drug from the high-affinity binding site in the cytoplasmic leaflet to a low-affinity site with access to the extracellular leaflet of the medium. Vi (or BeFx/AlFx) can enter the active site of either the post-hydrolysis complex (forward trapping) or the ADP-bound form of Pgp (reverse trapping). The two complexes appear to have different conformations, as indicated by dynamic quenching experiments. The post-hydrolysis conformation may retain some of the free energy released on Pi dissociation (indicated by *).
The use of photolabelling to estimate binding affinity is subject to serious technical difficulties [19], including changes in cross-linking efficiency in different protein conformations and low site occupancy. The results of such experiments are difficult to interpret; in fact, altered labelling efficiency alone may account for the observed changes in the amount of drug bound. Indeed, the Vi-trapped complex of TAP1/2 (transporter associated with antigen processing 1/2) showed normal peptide-binding affinity [46]. MRP1 (multidrug resistance-associated protein 1) mutants also did not display the ‘expected’ reduction in LTC4 (leukotriene C4) affinity when trapped with Vi [47]. These reports are consistent with our observations that the trapped states of Pgp show normal high-affinity drug binding, and with the mechanistic proposal presented above.
Conformational inequivalence of the trapped complexes formed from ATP and ADP
Our results suggest that the trapped complexes formed from ATP are not conformationally equivalent to those formed from ADP. A similar observation was made recently when spin labelling was used to examine Vi-trapped complexes of the ABC lipid A flippase, MsbA [29]. The conformation of the trapped complex formed using ADP was different from that formed using ATP. The ATP-trapped complex displayed a conformation consistent with occlusion of the substrate binding chamber, whereas the ADP-trapped form closely resembled the ligand-free protein, where the chamber is open and accessible to water. The chemical composition of the forward- and reverse-trapped complexes of Pgp appears to be identical; ADP was trapped at a stoichiometry of approx. 1 in both cases [8,48]. The trapped Pgp complexes formed using 8-azido-ATP and 8-azido-ADP were also functionally indistinguishable [49]. For some ABC proteins, the trapped state cannot be reached equally readily in the forward and reverse directions; for example, MalK can form the Vi-trapped state only from ATP, not ADP [50], implying that energy input from ATP hydrolysis is required for its formation. The trapped complex formed from ATP may retain some of the energy released during ATP hydrolysis, resulting in a conformation that is different from that of the complex formed from ADP. A proposed kinetic scheme for formation and dissociation of the forward- and reverse-trapped states is shown in Scheme 1. As yet, it is not known whether it is possible to interconvert these two species. Clearly, future work should take into account the inequivalence of the two trapped species.
The trapped complexes of Vi, AlFx and BeFx with ATPases and GTPases have proved very useful as mimics of the transition state and the ground state, and have helped to delineate the structural changes taking place at the active site during catalysis. The highly stable Co2+ complexes of Pgp with these three agents allow insight into various features of ATP hydrolysis at different stages along the reaction pathway. Trapping approaches to generate stable complexes will likely also prove useful for exploring structure and function of other transporters in the ABC superfamily.
Acknowledgments
This work was supported by a research grant from the Canadian Cancer Society. F.J.S. holds a Tier 1 Canada Research Chair in Membrane Protein Biology.
References
- 1.Dassa E. Phylogenetic and functional classification of ABC (ATP-binding cassette) systems. In: Holland I. B., Cole S. P. C., Kuchler K., Higgins C. F., editors. ABC Proteins: from Bacteria to Man. London: Academic Press; 2003. pp. 3–35. [Google Scholar]
- 2.Borst P., Oude Elferink R. P. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 3.Gottesman M. M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 4.Schinkel A. H., Jonker J. W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 5.Higgins C. F., Gottesman M. M. Is the multidrug transporter a flippase? Trends Biochem. Sci. 1992;17:18–21. doi: 10.1016/0968-0004(92)90419-a. [DOI] [PubMed] [Google Scholar]
- 6.Qu Q., Sharom F. J. Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry. 2002;41:4744–4752. doi: 10.1021/bi0120897. [DOI] [PubMed] [Google Scholar]
- 7.Schumacher M. A., Brennan R. G. Structural mechanisms of multidrug recognition and regulation by bacterial multidrug transcription factors. Mol. Microbiol. 2002;45:885–893. doi: 10.1046/j.1365-2958.2002.03039.x. [DOI] [PubMed] [Google Scholar]
- 8.Urbatsch I. L., Sankaran B., Weber J., Senior A. E. P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 1995;270:19383–19390. doi: 10.1074/jbc.270.33.19383. [DOI] [PubMed] [Google Scholar]
- 9.Sankaran B., Bhagat S., Senior A. E. Inhibition of P-glycoprotein ATPase activity by beryllium fluoride. Biochemistry. 1997;36:6847–6853. doi: 10.1021/bi970034s. [DOI] [PubMed] [Google Scholar]
- 10.Sankaran B., Bhagat S., Senior A. E. Inhibition of P-glycoprotein ATPase activity by procedures involving trapping of nucleotide in catalytic sites. Arch. Biochem. Biophys. 1997;341:160–169. doi: 10.1006/abbi.1997.9944. [DOI] [PubMed] [Google Scholar]
- 11.Urbatsch I. L., Sankaran B., Bhagat S., Senior A. E. Both P-glycoprotein nucleotide-binding sites are catalytically active. J. Biol. Chem. 1995;270:26956–26961. doi: 10.1074/jbc.270.45.26956. [DOI] [PubMed] [Google Scholar]
- 12.Senior A. E., al-Shawi M. K., Urbatsch I. L. The catalytic cycle of P-glycoprotein. FEBS Lett. 1995;377:285–289. doi: 10.1016/0014-5793(95)01345-8. [DOI] [PubMed] [Google Scholar]
- 13.Jones P. M., George A. M. Subunit interactions in ABC transporters: towards a functional architecture. FEMS Microbiol. Lett. 1999;179:187–202. doi: 10.1111/j.1574-6968.1999.tb08727.x. [DOI] [PubMed] [Google Scholar]
- 14.Moody J. E., Thomas P. J. Nucleotide binding domain interactions during the mechanochemical reaction cycle of ATP-binding cassette transporters. J. Bioenerg. Biomembr. 2005;37:475–479. doi: 10.1007/s10863-005-9494-8. [DOI] [PubMed] [Google Scholar]
- 15.Loo T. W., Clarke D. M. Drug-stimulated ATPase activity of human P-glycoprotein is blocked by disulfide cross-linking between the nucleotide-binding sites. J. Biol. Chem. 2000;275:19435–19438. doi: 10.1074/jbc.C000222200. [DOI] [PubMed] [Google Scholar]
- 16.Qu Q., Sharom F. J. FRET analysis indicates that the two ATPase active sites of the P-glycoprotein multidrug transporter are closely associated. Biochemistry. 2001;40:1413–1422. doi: 10.1021/bi002035h. [DOI] [PubMed] [Google Scholar]
- 17.Sharom F. J. Probing of conformational changes, catalytic cycle and ABC transporter function. In: Holland I. B., Kuchler K., Higgins C., Cole S. P., editors. ABC Proteins: from Bacteria to Man. London: Academic Press; 2003. pp. 107–133. [Google Scholar]
- 18.Sharom F. J., Liu R., Qu Q., Romsicki Y. Exploring the structure and function of the P-glycoprotein multidrug transporter using fluorescence spectroscopic tools. Semin. Cell Dev. Biol. 2001;12:257–266. doi: 10.1006/scdb.2000.0251. [DOI] [PubMed] [Google Scholar]
- 19.Qu Q., Chu J. W., Sharom F. J. Transition state P-glycoprotein binds drugs and modulators with unchanged affinity, suggesting a concerted transport mechanism. Biochemistry. 2003;42:1345–1353. doi: 10.1021/bi0267745. [DOI] [PubMed] [Google Scholar]
- 20.Qu Q., Russell P. L., Sharom F. J. Stoichiometry and affinity of nucleotide binding to P-glycoprotein during the catalytic cycle. Biochemistry. 2003;42:1170–1177. doi: 10.1021/bi026555j. [DOI] [PubMed] [Google Scholar]
- 21.Smith C. A., Rayment I. X-ray structure of the magnesium(II)·ADP·vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry. 1996;35:5404–5417. doi: 10.1021/bi952633+. [DOI] [PubMed] [Google Scholar]
- 22.Fisher A. J., Smith C. A., Thoden J. B., Smith R., Sutoh K., Holden H. M., Rayment I. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP·BeFx and MgADP·AlF4−. Biochemistry. 1995;34:8960–8972. doi: 10.1021/bi00028a004. [DOI] [PubMed] [Google Scholar]
- 23.Abrahams J. P., Leslie A. G., Lutter R., Walker J. E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature (London) 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 24.Braig K., Menz R. I., Montgomery M. G., Leslie A. G., Walker J. E. Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ ADP and aluminium fluoride. Structure Fold. Des. 2000;8:567–573. doi: 10.1016/s0969-2126(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 25.Schlichting I., Reinstein J. pH influences fluoride coordination number of the AlFx phosphoryl transfer transition state analog. Nat. Struct. Biol. 1999;6:721–723. doi: 10.1038/11485. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y. W., Morera S., Janin J., Cherfils J. AlF3 mimics the transition state of protein phosphorylation in the crystal structure of nucleoside diphosphate kinase and MgADP. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3579–3583. doi: 10.1073/pnas.94.8.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sondek J., Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. GTPase mechanism of G proteins from the 1.7-Å crystal structure of transducin a.GDP·AlF4−. Nature (London) 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 28.Reyes C. L., Chang G. Structure of the ABC transporter MsbA in complex with ADP·vanadate and lipopolysaccharide. Science (Washington, D.C.) 2005;308:1028–1031. doi: 10.1126/science.1107733. [DOI] [PubMed] [Google Scholar]
- 29.Dong J., Yang G., McHaourab H. S. Structural basis of energy transduction in the transport cycle of MsbA. Science (Washington, D.C.) 2005;308:1023–1028. doi: 10.1126/science.1106592. [DOI] [PubMed] [Google Scholar]
- 30.Doige C. A., Sharom F. J. Strategies for the purification of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Protein Expr. Purif. 1991;2:256–265. doi: 10.1016/1046-5928(91)90081-s. [DOI] [PubMed] [Google Scholar]
- 31.Liu R., Sharom F. J. Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry. 1996;35:11865–11873. doi: 10.1021/bi960823u. [DOI] [PubMed] [Google Scholar]
- 32.Peterson G. L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 33.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Sharom F. J., Yu X., Chu J. W. K., Doige C. A. Characterization of the ATPase activity of P-glycoprotein from multidrug-resistant Chinese hamster ovary cells. Biochem. J. 1995;308:381–390. doi: 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakowicz J. R. Principles of Fluorescence Spectroscopy. New York: Kluwer Academic Publishers; 1999. [Google Scholar]
- 36.Liu R., Siemiarczuk A., Sharom F. J. Intrinsic fluorescence of the P-glycoprotein multidrug transporter: sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry. 2000;39:14927–14938. doi: 10.1021/bi0018786. [DOI] [PubMed] [Google Scholar]
- 37.Petsko G. A. Chemistry and biology. Proc. Natl. Acad. Sci. U.S.A. 2000;97:538–540. doi: 10.1073/pnas.97.2.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenberg M. F., Velarde G., Ford R. C., Martin C., Berridge G., Kerr I. D., Callaghan R., Schmidlin A., Wooding C., Linton K. J., Higgins C. F. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 2001;20:5615–5625. doi: 10.1093/emboj/20.20.5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G., Pincheira R., Zhang M., Zhang J. T. Conformational changes of P-glycoprotein by nucleotide binding. Biochem. J. 1997;328:897–904. doi: 10.1042/bj3280897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Julien M., Gros P. Nucleotide-induced conformational changes in P-glycoprotein and in nucleotide binding site mutants monitored by trypsin sensitivity. Biochemistry. 2000;39:4559–4568. doi: 10.1021/bi992744z. [DOI] [PubMed] [Google Scholar]
- 41.Liu R., Sharom F. J. Fluorescence studies on the nucleotide binding domains of the P-glycoprotein multidrug transporter. Biochemistry. 1997;36:2836–2843. doi: 10.1021/bi9627119. [DOI] [PubMed] [Google Scholar]
- 42.Lugo M. R., Sharom F. J. Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry. 2005;44:643–655. doi: 10.1021/bi0485326. [DOI] [PubMed] [Google Scholar]
- 43.Pawagi A. B., Wang J., Silverman M., Reithmeier R. A., Deber C. M. Transmembrane aromatic amino acid distribution in P-glycoprotein. A functional role in broad substrate specificity. J. Mol. Biol. 1994;235:554–564. doi: 10.1006/jmbi.1994.1013. [DOI] [PubMed] [Google Scholar]
- 44.Ramachandra M., Ambudkar S. V., Chen D., Hrycyna C. A., Dey S., Gottesman M. M., Pastan I. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry. 1998;37:5010–5019. doi: 10.1021/bi973045u. [DOI] [PubMed] [Google Scholar]
- 45.Sauna Z. E., Smith M. M., Müller M., Kerr K. M., Ambudkar S. V. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J. Bioenerg. Biomembr. 2001;33:481–491. doi: 10.1023/a:1012875105006. [DOI] [PubMed] [Google Scholar]
- 46.Chen M., Abele R., Tampé R. Peptides induce ATP hydrolysis at both subunits of the transporter associated with antigen processing. J. Biol. Chem. 2003;278:29686–29692. doi: 10.1074/jbc.M302757200. [DOI] [PubMed] [Google Scholar]
- 47.Payen L., Gao M., Westlake C., Theis A., Cole S. P. C., Deeley R. G. Functional interactions between nucleotide binding domains and leukotriene C4 binding sites of multidrug resistance protein 1 (ABCC1) Mol. Pharmacol. 2005;67:1944–1953. doi: 10.1124/mol.104.007708. [DOI] [PubMed] [Google Scholar]
- 48.Urbatsch I. L., Tyndall G. A., Tombline G., Senior A. E. P-glycoprotein catalytic mechanism – studies of the ADP-vanadate inhibited state. J. Biol. Chem. 2003;278:23171–23179. doi: 10.1074/jbc.M301957200. [DOI] [PubMed] [Google Scholar]
- 49.Sauna Z. E., Smith M. M., Müller M., Ambudkar S. V. Functionally similar vanadate-induced 8-azidoadenosine 5′-[α-32P]diphosphate-trapped transition state intermediates of human P-glycoprotein are generated in the absence and presence of ATP hydrolysis. J. Biol. Chem. 2001;276:21199–21208. doi: 10.1074/jbc.M100886200. [DOI] [PubMed] [Google Scholar]
- 50.Sharma S., Davidson A. L. Vanadate-induced trapping of nucleotides by purified maltose transport complex requires ATP hydrolysis. J. Bacteriol. 2000;182:6570–6576. doi: 10.1128/jb.182.23.6570-6576.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]