

Abstract
In addition to genetic effects on disease risk, age-at-onset (AAO) of Alzheimer’s disease (AD) is also genetically controlled. Using AAO as a covariate, a linkage signal for AD has been detected on chromosome 14q32 near the a1-antichymotrypsin (ACT) gene. Previously, a signal peptide polymorphism (codon -17A>T) in the ACT gene has been suggested to affect AD risk, but with inconsistent findings. Given that a linkage signal for AAO has been detected near ACT, we hypothesized that ACT genetic variation affects AAO rather than disease risk and this may explain the previous inconsistent findings between ACT genetic variation and AD risk. We examined the impact of the ACT signal peptide polymorphism on mean AAO in 909 AD cases. The ACT polymorphism was significantly associated with AAO and this effect was independent of the APOE polymorphism. Mean AAO among ACT/AA homozygotes was significantly lower than that in the combined AT+TT genotype group (p=0.019) and this difference was confined to male AD patients (p=0.002). Among male AD patients, the ACT/AA genotype was also associated with shorter disease duration before death as compared to the ACT/AT + TT genotypes (p=0.012). These data suggest that the ACT gene may affect AAO and disease duration of AD.
1. Introduction
Alpha-1-antichymotrypsin (ACT), also known as serine proteinase inhibitor A3 (SERPINA3), is an acute phase reactant protein that is believed to be involved in the pathogenesis of Alzheimer’s disease (AD). ACT is a major component of neuritic plaques [1] and it promotes the assembly of amyloid-ß peptide (Aß) into filaments [13] and its deposition in the brain [16, 18] and it also affects cognitive impairment [17]. Compared to controls, ACT levels are elevated in the cerebrospinal fluid of AD patients [3, 12] and elevated levels are also associated with AD severity [3]. Originally our group reported that the ACT/codon -17*A allele was associated with AD risk in an APOE-dependent fashion [7], however, subsequent studies showed inconsistent results [4, 25]. Additional evidence that the ACT/-17*A allele may be functional comes from data implicating this allele in deleterious cerebral glucose metabolism [5], severity of cerebral amyloid angiopathy [24] and its enhanced secretion from astroglial cell lines [19]. Despite these data, the role of ACT genetic variation in AD risk has not gained special attention mainly because of the inconsistent association data and the apparent lack of linkage signal on chromosome 14q32, where the ACT gene resides. Recently linkage analysis methods have been developed that allow covariates to be included in the analysis, thus enabling the detection of additional linkage signals that might be undetectable using conventional linkage methods. Age-at-onset (AAO) is an important covariate for AD and is also highly heritable [2, 9]. Using AAO as a covariate for risk of AD, a linkage signal with lod score of 1.89 was detected on chromosome 14q32 near D14S1015 [20], which is within 2 Mb of the ACT gene.
To test the hypothesis that ACT genetic variation affects AAO of AD, we examined the impact of the ACT signal peptide polymorphism at codon -17 (A>T) on mean AAO in 909 AD cases. We also examined if this polymorphism is associated with disease duration.
2. Materials and Methods
2.1. Subjects
The study sample comprised 909 AD cases (67% women; 33% men) with AAO ranging from 60-91 years and mean AAO of 72.19 ± [SE] 0.21 years. All AD cases were recruited by the University of Pittsburgh Alzheimer’s Disease Research Center and their use for genetic studies was approved by the University of Pittsburgh Institutional Review Board. All cases were clinically evaluated and satisfied NINCDS/ADRDA criteria for probable AD [14].
2.2. Genotyping
ACT and apolipoprotein E (APOE) genotyping were performed as described elsewhere [6, 7].
2.3. Statistical Analyses
The mean AAO between different genotype groups was compared using a general linear models approach including the effects of ACT genotype (AA vs. AT + TT), APOE genotype (APOE*4 carriers vs. non-APOE*4 carriers), APOE by ACT genotype interaction, sex and sex by ACT genotype interaction. A priori, we decided to analyze ACT genotype data as two groups (AA vs. AA + TT) based on results of our previous studies (7). Statistical analyses were performed using the R software environment (http://www.maths.lth.se/help/R/.R/doc/html/index.html) which is similar to S-plus. Kaplan-Maier survival analysis was also used to compare the AAO between genotypes. The disease duration was estimated by subtracting the AAO from the age at death and then mean disease duration was compared among ACT genotype after adjusting for the effects of sex and APOE genotype. Data on disease duration were available on 157 AD patients.
3. Results
A regression analysis including ACT genotype, APOE genotype, sex, sex by ACT genotype interaction and ACT by APOE genotype interaction revealed that ACT (p=0.033), APOE (p<0.0001) and sex by ACT genotype (p=0.059) were significantly associated with AAO of AD. There was no statistically significant interaction between ACT and APOE (p=0.54).
Table 1 presents mean AAO among APOE genotypes. As expected, the lowest AAO was in the 4/4 genotype and the highest AAO in the 2/2 genotype. The sex-adjusted mean AAO was about three years lower among APOE*4 carriers than non-APOE*4 carriers (70.96 ± 0.28 vs. 73.96; p<0.0001). However, this difference between APOE*4 carriers and non-APOE*4 carriers was more pronounced among females (70.97 ± 0.32 vs. 74.57 ±0.38; p<0.0001) than males (70.92 ± 0.42 vs. 72.66 ± 0.62; p=0.017). The APOE genotype explained 5.7% of the phenotypic variation in AAO. Kaplan-Maier survival analysis revealed a gene-dosage effect of the APOE*4 allele on AAO (Fig. 1A).
Table 1.
Sex-adjusted (± SE) mean age-at-onset (AAO) among APOE genotypes
| APOE Genotype | N | AAO (year) | |
|---|---|---|---|
| 2/2 | 3 | 79.37 ± 2.91 | |
| 2/3 | 33 | 72.57 ± 0.96 | |
| 33 | 336 | 74.05 ± 0.34 | |
| 2/4 | 24 | 71.68 ± 1.11 | |
| 34 | 433 | 71.36 ± 0.29 | |
| 44 | 80 | 68.59 ± 0.59 | |
| p<0.0001 |
Figure 1.
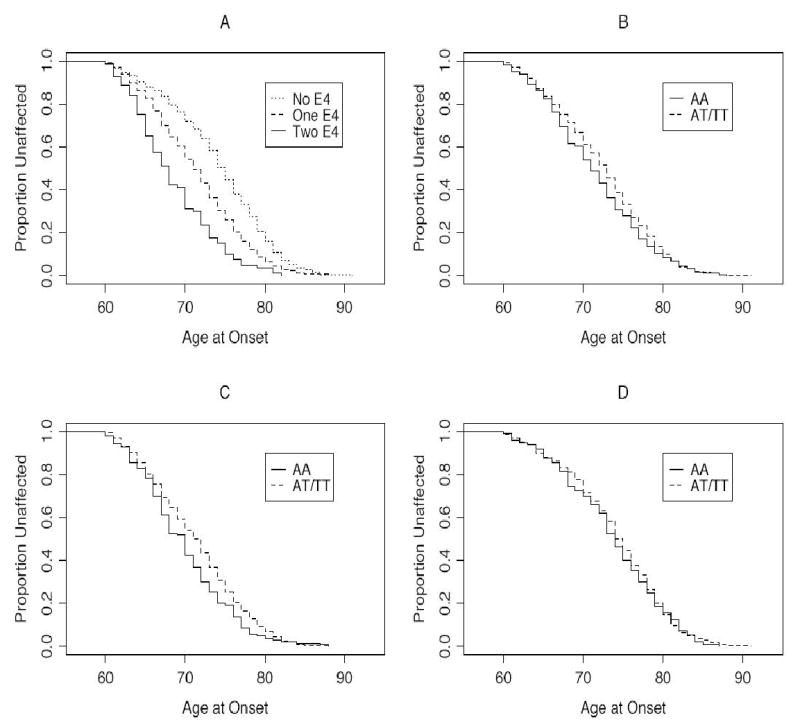
Kaplan-Maier survival analysis for age-at-onset (AAO). The AAO is plotted against the proportion unaffected by AD. (A) = Comparison of no copy of APOE*4 allele (2/2, 2/3, 3/3 genotypes), one copy of APOE*4 allele (2/4, 3/4 genotype) and two copies of APOE*4 allele (4/4 genotype) on AAO. At age 70, 72.1% subjects with no APOE*4, 54.8% with one APOE*4 and 31.7% with two APOE*4 are unaffected by AD (log rank 75.2, df = 5, p<0.0001). (B) = Comparison of ACT/AA and AT + TT genotypes on AAO. At age 70, 61.2% subjects with AT + TT genotype and 53.8% with AA genotype are unaffected by AD (log rank 3.4, df = 1, p=0.066). (C) = Comparison of ACT genotype on AAO among APOE*4 carriers. At age 70, 54% subjects with ACT/AT + TT genotype and 42.4% with ACT/AA genotype are unaffected by AD (log rank = 5.8, df = 1, p=0.016). (D) = Comparison of ACT genotype on AAO among non-APOE*4 carriers (log rank 0.3, df = 1, p=0.61).
APOE-adjusted mean AAO among ACT genotypes is presented in Table 2. Because there was a suggestive sex by ACT genotype interaction effect in the regression model, mean AAO is also presented separately for males and females. Mean AAO was significantly lower in the AA genotype than the combined AT and TT genotypes in the total sample (71.41 ± 0.38 vs. 72.48 ± 0.23; p=0.019) and furthermore, this difference was significant in males (70.01 ± 0.64 vs. 72.42 ± 0.41; p=0.009) but not females (72.14 ± 0.47 vs. 72.49 ± 0.28; p=0.131). Although there was no statistically significant interaction between ACT and APOE, we also examined the impact of ACT genotype on AAO based on the presence or absence of the APOE*4 allele in the combined sample (Table 2). The mean AAO was significantly lower between ACT/AA homozygotes and ACT/AT+TT genotypes among APOE*4 carriers only (p=0.015). As noted above, the mean AAO difference between APOE*4 carriers and non-APOE*4 carriers is about 3 years. However, this difference is influenced by the ACT genotype such that within the APOE*4 carriers the ACT/AA, AT and TT genotypes have 3.75, 2.81 and 2.62 years lower AAO, respectively, than the corresponding ACT genotypes within non-APOE*4 carriers. Kaplan-Maier survival analysis (Fig. 2B–D) also showed that ACT/AA genotype had lower AAO than ACT/AT + TT genotypes (p=0.066) and this difference was significant among APOE*4 carriers (p=0.016) but not among non-APOE*4 carriers (p=0.61).
Table 2.
APOE-adjusted mean age-at-onset (AAO) among ACT genotypes stratified by gender
| N | AA | AT | TT | p-Value | |
|---|---|---|---|---|---|
| Male | 298 | 70.01 ± 0.64 (n = 81) | 72.38 ± 0.52 (n = 144) | 72.51 ± 0.68 (n = 73) | 0.009 (0.002)* |
| Female | 611 | 72.14 ± 0.47 (n = 155) | 72.89 ± 0.34 (n = 292) | 71.78 ± 0.49 (n = 164) | 0.131 (0.522)* |
| Combined | 909 | 71.41 ± 0.38 (n = 236) | 72.73 ± 0.29 (n = 436) | 72.01 ± 0.40 (n = 237) | 0.021 (0.019)*† |
| Combined APOE*4 carriers | 537 | 69.92 ± 0.48 (n = 139) | 71.56 ± 0.37 (n = 260) | 70.90 ± 0.65 (n = 138) | 0.029 (0.015)* |
| Combined non-APOE*4 carriers | 372 | 73.67 ± 0.63 (n = 97) | 74.37 ± 0.46 (n = 176) | 73.52 ± 0.65 (n = 99) | 0.480 (0.595)* |
p-value in parenthesis is based on comparison between AA vs. AT + TT groups
p-values in the combined sample are adjusted for sex and APOE genotype
We also examined the impact of ACT genotypes on the disease duration (age at death – AAO) after adjusting for the effect of APOE. While there was no difference among females, the disease duration was significantly shorter (p=0.021) among male ACT/AA patients (disease duration 7.69 ± 0.81 years) than AT (10.13 ± 0.61 years) and TT (11.61 ± 1.26 years). Comparison of ACT/AA homozygote with combined ACT/AT + TT genotypes showed about a 3 year difference in disease duration (7.69 ± 0.81 vs. 10.67 ± 0.61; p=0.012). While there was no impact of the APOE polymorphism on disease duration in the total sample (p=0.20), disease duration among females was shorter in APOE*4 carriers than non-APOE*4 carriers (8.74 ± 0.52 vs. 10.56 ± 0.55; p=0.028). These gender-specific effects of ACT and APOE on disease duration are analogous to those seen for AAO.
4. Discussion
New emerging data indicate that in addition to disease risk, genetic factors also affect AAO of AD [2, 9] with an estimate of heritability of AAO >40% [9]. Identification of genes for AAO may help to delay the onset of disease, which could save billions of dollars in healthcare cost. In addition to disease risk, APOE also affects variation in AAO. However, the APOE genetic variation explains only 4 to 9% of the variation in AAO [2, 21]. A simulation study has suggested that in addition to APOE, seven additional genes with effect size ranging from <1% to 57% may influence the AAO of AD [2], but their identity is unknown. Using AAO as a covariate, Olsen et al. [20] reported a linkage signal within 2 Mb of the ACT gene on chromosome 14q32. In their baseline model without AAO as a covariate, they obtained no linkage signal (lod score = 0.0), but they did obtain a suggestive linkage signal (lod score = 1.89) when AAO was used as a covariate. Several studies have examined the role of the ACT signal peptide polymorphism (−17 A>T) with AD risk, but the results have been inconsistent. In the current investigation, we tested the hypothesis that the ACT signal peptide polymorphism affects AAO rather than the risk of AD, and that this hypothesized relationship might explain the previous inconsistent association data between ACT genotypes and AD risk. Indeed, we found that the ACT signal peptide polymorphism has moderate, but significant impact on AAO. The ACT/AA homozygotes were associated with lower mean AAO than the other two ACT genotypes and this effect was independent of the APOE polymorphism. Overall, the ACT polymorphism explained 0.7% of the phenotypic variation in AAO, which is less than the contribution of the APOE polymorphism on AAO (5.7%) in this sample. However, this 0.7% contribution of the ACT polymorphism to AAO is consistent with the simulation results in which two of the seven postulated genes have <1% effect each on AAO [2].
Previously, Talbot et al. [22] reported a similar lowering, but non-significant (p=0.06), effect of he AA genotype of the ACT signal peptide polymorphism on AAO in 202 AD cases, however, this association was significant among APOE*4 carriers (p=0.012). On the other hand, Licastro et al. [11] reported that the TT genotype of the ACT signal peptide polymorphisms was associated with lower AAO among an unspecified number of late-onset AD cases (>65 years) of the total 281 AD cases examined. Using data on 909 late-onset AD cases, we observed a significant association between ACT genotypes and AAO (p=0.019). Similar to the report by Talbot et al. [22], the ACT/AA genotype was associated with a lower AAO of AD. Furthermore, when the ACT genotypes were stratified by APOE genotype, the significant association was confined to APOE*4 carriers, which was also confirmed by Kaplan-Maier survival analysis. Our data revealed that the significant impact of the ACT polymorphism on AAO was confined to male AD patients, in which the AA homozygotes had AAO more than two years earlier than the other two genotypes (70.01 ± 0.64 vs. 72.42 ± 0.41; p=0.002). In addition to having earlier AAO, male AD patients with the AA genotype also had about 3 years shorter disease duration before death than male AD patients with the other two genotypes (7.69 ± 0.81 vs. 10.67 ± 0.61; p=0.012). These data suggest that the ACT polymorphism is not only associated with AAO but also with disease duration in a gender-specific manner.
Recently, the ACT signal peptide polymorphism has also been reported to be associated with AAO of Parkinson’s disease [23]. This association between ACT polymorphism and AAO of both AD and Parkinson’s disease is similar to that observed for APOE, because APOE also affects AAO for both AD and Parkinson’s disease [8]. Our data suggest that the ACT gene confers AD risk by affecting AAO of AD and this may explain the previous inconsistent reports of an association between the ACT signal peptide polymorphism and AD risk. An alternative explanation for the current observation is that the ACT signal peptide polymorphism may be in linkage disequilibrium with a functional variant either within the ACT gene or in a nearby gene. Recently, a polymorphisms in the ACT promoter (−51G>T) has been described that affects ACT expression in transfected glial cell lines [15] and AD risk [10]. We found that although this polymorphism is in strong linkage disequilibrium with the signal peptide polymorphism, it was not associated with either the risk or AAO of AD (data not shown). Because a linkage signal has been observed for AAO in the vicinity of the ACT gene [20], a renewed interest in this region may help to determine if the ACT gene or a nearby gene affects AAO for AD, and perhaps Parkinson’s disease.
Acknowledgments
This work was supported by National Institute on Aging grants, AG 13672 and AG 05133.
References
- 1.Abraham CR, Selkoe DJ, Potter H. Immunochemical identification of the serine protease inhibitor a1-antichymotrypsin in the brain amyloid deposits of Alzheimer’s disease. Cell. 1988;52:487–451. doi: 10.1016/0092-8674(88)90462-x. [DOI] [PubMed] [Google Scholar]
- 2.Daw EW, Heath SC, Wijsman EM. Multipoint oligogenic analysis of age-at-onset data with application to Alzheimer disease pedigrees. Am J Hum Genet. 1999;64:839–851. doi: 10.1086/302276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeKosky ST, Ikonomovic MD, Wang X, Farlow M, Wisniewski S, Lopez OL, Becker JT, Saxton J, Klunk WE, Sweet R, Kaufer DI, Kamboh MI. Plasma and cerebrospinal fluid a1-antichymotrypsin levels in Alzheimer’s disease: Correlation with Cognitive impairment. Ann Neurol. 2003;53:81–90. doi: 10.1002/ana.10414. [DOI] [PubMed] [Google Scholar]
- 4.Haines JL, Pritchard ML, Saunders AM, Schildkraut JM, Growdon JH, Gaskell PC, Farrer LA, Auerbach SA, Gusella JF, Locke PA, Rosi BL, Yamaoka L, Small GW, Conneally PM, Roses AD, Pericak-Vance MA. No genetic effect of a1-antichymotrypsin in Alzheimer disease. Genomics. 1996;33:53–56. doi: 10.1006/geno.1996.0158. [DOI] [PubMed] [Google Scholar]
- 5.Higuchi M, Arai H, Nakagawa T, Higuchi S, Muramatsu T, Matsushita S, Kosaka Y, Itoh M, Sasaki H. Regional cerebral glucose utilization is modulated by the dosage of apolipoprotein E type 4 allele and a1-antichymotrypsin type A allele in Alzheimer disease. Neuroreport. 1997;8:2639–2643. doi: 10.1097/00001756-199708180-00001. [DOI] [PubMed] [Google Scholar]
- 6.Kamboh MI, Aston CE, Hamman RF. The relationship of APOE polymorphism and cholesterol levels in normoglycemic and diabetic subjects in a biethnic population from the San Luis Valley, Colorado. Atherosclerosis. 1995;112:145–159. doi: 10.1016/0021-9150(94)05409-c. [DOI] [PubMed] [Google Scholar]
- 7.Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST. APOE*4-associated Alzheimer’s disease risk is modified by a1-antichymotrypsin polymorphism. Nat Genet. 1995;10:486–488. doi: 10.1038/ng0895-486. [DOI] [PubMed] [Google Scholar]
- 8.Li YJ, Hauser MA, Scott WK, Martin ER, Booze MW, Qin XJ, Walter JW, Nance MA, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Small GW, Mastaglia F, Haines JL, Pericak-Vance MA, Vance JM. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology. 2004;62:2005–2009. doi: 10.1212/01.wnl.0000128089.53030.ac. [DOI] [PubMed] [Google Scholar]
- 9.Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Allen FA, Jr, Goetz CG, Mastaglia F, Stajich JM, Gibson RA, Middleton LT, Saunders AM, Scott BL, Small GW, Nicodemus KK, Reed AD, Schmechel DE, Welsh-Bohmer KA, Conneally PM, Roses AD, Gilbert JR, Vance JM, Haines JL, Pericak-Vance MA. Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet. 2002;70:985–993. doi: 10.1086/339815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Licastro F, Chiappelli M, Grimaldi LME, Morgan K, Kalsheker N, Calabrese E, Ritchie A, Porcellini E, Salani G, Franceschi M, Canal N. A new promoter polymorphism in the alpha-1-antichymotrypsin gene is a disease modifier of Alzheimer's disease. Neurobiol Aging. 2005;26:449–453. doi: 10.1016/j.neurobiolaging.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Licastro F, Pedrini S, Ferri C, Casadei V, Govoni M, Pession A, Sciacca FL, Veglia F, Annoni G, Bonafe M, Olivieri F, Franceschi C, Grimaldi LM. Gene polymorphism affecting alpha1-antichymotrypsin and interleukin-1 plasma levels increases Alzheimer's disease risk. Ann Neurol. 2000;48:388–391. [PubMed] [Google Scholar]
- 12.Licastro F, Parnetti L, Morini MC, Davis LJ, Cucinotta D, Gaiti A, Senin U. Acute phase reactant a1-antichymotrypsin is increased in cerebrospinal fluid and serum of patients with probable Alzheimer’s disease. Alzheimer Dis Assoc Disord. 1995;9:112–118. doi: 10.1097/00002093-199509020-00009. [DOI] [PubMed] [Google Scholar]
- 13.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins a1-antitrypsin and apolipoprotein E promote assembly of Alzheimer ß-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 15.Morgan K, Licastro F, Tilley L, Ritchie A, Morgan L, Pedrini S, Kalsheker N. Polymorphism in the alpha(1)-antichymotrypsin (ACT) gene promoter: effect on expression in transfected glial and liver cell lines and plasma ACT concentrations. Hum Genet. 2001;109:303–310. doi: 10.1007/s004390100575. [DOI] [PubMed] [Google Scholar]
- 16.Mucke L, Yu GQ, McConlogue L, Rockenstein EM, Abraham CR, Masliah E. Astroglial expression of human a1-antichymotrypsi enhances Alzheimer’s –like pathology in amyloid protein precursor transgenic mice. Am J Pathol. 2000;157:2003–2010. doi: 10.1016/s0002-9440(10)64839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsson LN, Arendash GW, Leighty RE, Costa DA, Low MA, Garcia MF, Cracciolo JR, Rojiani A, Wu X, Bales KR, Paul SM, Potter H. Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol Aging. 2004;25:1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson LN, Bales KR, DiCarlo G, Gordon MN, Morgan D, Paul SM, Potter H. a1-antichymotrypsin promotes ß-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nilsson LN, Das S, Potter H. Effect of cytokines,dexamethazone and the A/T-signal peptide polymorphism on the expression of alpha1-antichymotrypsin in astrocytes: significance for Alzheimer’s disease. Neurochem Int. 2000;39:361–370. doi: 10.1016/s0197-0186(01)00043-2. [DOI] [PubMed] [Google Scholar]
- 20.Olson JM, Goddard KA, Dudek DM. A second locus for very-late-onset Alzheimer disease: a genome scan reveals linkage to 20p and epistasis between 20p and the amyloid precursor protein region. Am, J Hum Genet. 2002;71:154–161. doi: 10.1086/341034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slooter AJC, Cruts M, Kalmijn S, Hofman A, Breteler MMB, van Broeckhoven C, van Duijn CM. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: The Rotterdam Study. Arch Neurol. 1998;55:964–968. doi: 10.1001/archneur.55.7.964. [DOI] [PubMed] [Google Scholar]
- 22.Talbot T, Houlden H, Craddock N, Crook R, Hutton M, Lendon C, Prihar G, Morris JC, Hardy J, Goate A. Polymorphism in the AACT gene may lower age of onset of Alzheimer’s disease. NeuroReport. 1996;7:534–536. doi: 10.1097/00001756-199601310-00038. [DOI] [PubMed] [Google Scholar]
- 23.Wang YC, Liu HC, Liu TY, Hong CJ, Tsai SJ. Genetic association analysis of alpha-1-antichymotrypsin polymorphism in Parkinson’s disease. Eur Neurol. 2001;45:254–256. doi: 10.1159/000052138. [DOI] [PubMed] [Google Scholar]
- 24.Yamada M, Sodeyama N, Itoh Y, Suematsu N, Otomo E, Matsushita M, Mizusawa H. Association of alpha1-antichymotrypsin polymorphism with cerebral amyloid angiopathy. Ann Neurol. 1998;44:129–131. doi: 10.1002/ana.410440120. [DOI] [PubMed] [Google Scholar]
- 25.Yoshiiwa A, Kamino K, Yamamoto H, Kobayashi T, Imagawa M, Nonomura Y, Yoneda H, Sakai T, Nishiwaki Y, Sato N, Rakugi H, Miki T, Ogihara T. a1-antichymotrypsin as a risk modifier for late-onset Alzheimer’s disease in Japanese apolipoprotein E epsilon 4 carriers. Ann Neurol. 1997;42:115–117. doi: 10.1002/ana.410420118. [DOI] [PubMed] [Google Scholar]