

Abstract
Amphipathic antimicrobial peptides can destroy bacteria cells by inducing membrane permeabilization, forming one strategy for innate defense by various organisms. However, although the antimicrobial peptides are considered a promising alternative for use against multidrug-resistant bacteria, large-scale screening of potential candidate antimicrobial peptides will require a simple, rapid assay for antimicrobial activity. Here, we describe a novel fluorescence resonance energy transfer (FRET)-based assay system for antimicrobial peptides which takes advantage of pH-related changes in FRET efficiency due to the instability of enhanced yellow fluorescent protein versus the stability of enhanced cyan fluorescent protein in a reduced-pH environment. We successfully showed that quantification of antimicrobial activity is possible through a difference of FRET efficiency between ECFP-EYFP fusion molecules released from disrupted Escherichia coli in an extracellular environment (pH 6) and those retained in an intracellular environment (pH ∼7). Thus, we herein suggest a new simple, effective, and efficient pH-controlled FRET-based antimicrobial peptide screening method applicable to high-throughput screening of candidate peptide libraries.
Conventional chemical-based antibiotics, such as penicillin, usually destroy bacteria by blocking synthesis of the bacterial cell wall or by disrupting the translational machinery (3, 4). However, due to the wholesale use of chemical antibiotics over the past decades, many bacteria have become antibiotic resistant. Amphipathic peptides, which have both hydrophilic (cationic) and hydrophobic properties, have been found in organisms ranging from bacteria to humans; many of these peptides have antibiotic properties, including effects against multidrug-resistant bacteria. Although the mechanism of antimicrobial peptides (AMPs) action is not clearly understood, the AMPs are generally believed to bind lipopolysaccharide on gram-negative bacteria or lipoteichoic acid on gram-positive bacteria. This charge-based binding perturbs the fundamental structure of bacterial membranes, forming transient pores that eventually lead to bacterial lysis (5, 17).
Owing to the potential superiority of AMPs as antibiotics, many researchers have sought to screen potential candidate peptides from various types of organisms. However, the traditional screening methods used to test AMP candidates have proven time consuming and laborious, limiting the effectiveness of these studies. To overcome these limitations, we here introduce a novel fluorescence resonance energy transfer (FRET)-based assay for rapid and high-throughput screening of AMPs. In general, FRET is the distance-dependent transfer of energy from a donor fluorophore to an acceptor fluorophore (12). For this process to occur, the distance between two fluorophores should be 2 to 6 nm, and the emission spectrum of the donor must overlap with the excitation spectrum of the acceptor by more than 30% (15). These requirements have been used to develop FRET-based fluorescent biosensors, in which ionic distributions or kinetics are assessed with regard to the alteration of the distance between two fluorescent proteins by specific ions, chemicals, or other compounds (2, 10). However, for employment of distance-based FRET as an AMP screening system, its major limitation will be a difficulty in obtaining a sufficient concentration gradient of ions or chemicals between the areas inside and outside of cells.
The novel technique presented herein takes advantage of a previous study showing that the stabilities of enhanced green fluorescent protein mutants, such as enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP), were pH dependent and that while ECFP and EYFP maintained their native fluorescence signatures at a physiologically normal pH of 7.0, the stability of EYFP, but not of ECFP, was significantly decreased at a reduced pH (9). In particular, we based our method on the hypothesis that the FRET efficiency of an ECFP-EYFP fusion protein expressed in Escherichia coli would differ based on pH and that providing a reduced-pH extracellular environment would allow us to distinguish between extracellular molecules released by disrupted E. coli cells and molecules retained in the intracellular environment (pH ∼7).
MATERIALS AND METHODS
Plasmid construction and strain.
Recombinant plasmid pCPY-S was constructed to encode an ECFP-EYFP fusion protein (Fig. 1). The ecfp and eyfp genes were PCR amplified from pECFP and pEYFP (Clontech) with the primers (ecfp gene, 5′-GCATATGGTGAGCAAGGGCGAG-3′ and 5′-GGAATTCCTTGTACAGCTCGTCCAT-3′; eyfp gene, 5′-GGCGGCCGCATGGTGAGCAAGGG-3′ and 5′-CCTCGAGTTACTTGTACAGCTC-3′) and inserted into the NdeI and EcoRI or NotI and XhoI sites of pET22b(+) (Novagen), respectively. This vector, containing the ecfp::eyfp fusion gene with a 33-bp spacer sequence, was used to transform E. coli BL21(DE3) (Novagen). Recombinant E. coli cells were induced with 1 mM isopropyl-β-d-thiogalactopyranoside, cultured in LB medium containing 50 μg/ml ampicillin for 1 h at 37°C, and harvested for experiments.
FIG. 1.
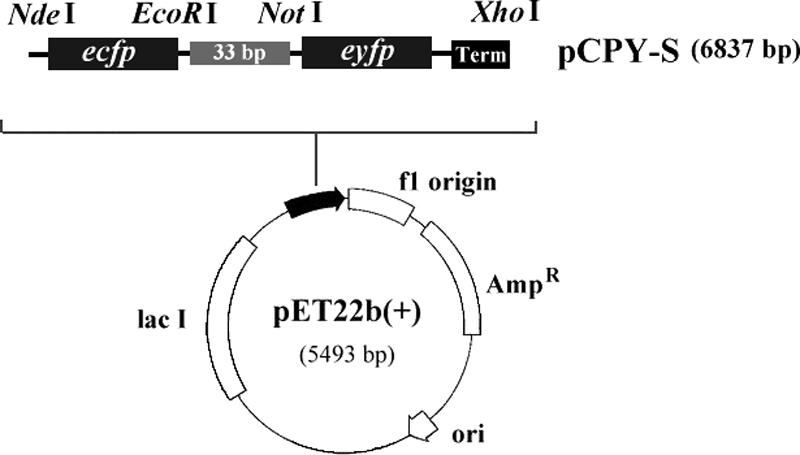
Gene map of recombinant plasmid pCPY-S. Plasmid pET22b(+) was used as a parent vector. Abbreviations: ecfp, enhanced cyan fluorescent protein gene; eyfp, enhanced yellow fluorescent protein gene; Term, termination sequence; Ampr, ampicillin resistance gene; lacI, overexpressed lac repressor gene; ori, replication origin.
Assessing the efficiency of pH-controlled FRET.
To investigate the efficiency of pH-based changes in FRET (Fig. 1B), ∼1 × 109 recombinant E. coli cells expressing intracellular ECFP-EYFP were completely disrupted by sonication in phosphate-buffered saline (PBS) buffers of various pH values (8.5, 8.0, 7.5, 7.0, 6.5, 6.0, 5.5, and 5.0). The cell lysates were centrifuged, and the emission spectra of the supernatant were scanned using a fluorescence spectrophotometer (Shimadzu), with excitation at 433 nm.
Antimicrobial assay using pH-controlled FRET.
The procedure consisted of two steps, an antimicrobial reaction at pH 7.0 followed by equilibration at pH 6.0. Equilibration should be allowed to proceed for no more than 20 min; longer equilibration times yielded huge deviations in the results, due to additional ECFP-EYFP expression by living cells during that time (data not shown). For experiments, ∼1 × 109 ECFP-EYFP-expressing cells were suspended in 20 μl of PBS buffer containing 0, 0.25, 0.50, 0.75, or 1 nM bovine serum albumin (BSA) (Sigma), Hal 18 (a gift from I.-H. Lee of Hoseo University), or magainin II (Sigma). The antimicrobial reaction was allowed to proceed for 30 min. After that time, each sample was equilibrated in 2 ml of PBS buffer (pH 6.0) for 20 min, and the emission spectrum and fluorescence intensity of ECFP at 475 nm and EYFP at 527 nm were measured with excitation at 433 nm using a fluorescence spectrophotometer. pH-controlled FRET assays were also performed for other nonpeptide compounds (lysozyme [Sigma], sodium dodecyl sulfate [SDS] [Kanto chemical], and kanamycin [Sigma]). For experiments involving the fluorescence microplate reader system, the cell number was reduced to ∼1 × 108, but antimicrobial reactions were performed as described above. For equilibration, 180 μl of PBS buffer (pH 5.84) was added to each sample, adjusting the final pH to 6.0. The fluorescence intensities at 475 and 527 nm were measured with excitation at 433 nm, using a fluorescence microplate reader (Molecular Devices).
Antimicrobial assays using colony counting and MIC.
For the colony counting assay, noninduced recombinant E. coli cells (6 × 106) were subjected to antimicrobial reactions as described above. The samples were then serially diluted, and the dilutions were incubated on agar plates at 37°C for 12 h. Antimicrobial activity was calculated with regard to the number of CFU remaining in each sample. For the MIC assay, noninduced recombinant E. coli cells were diluted to approximately 1 × 105 CFU/ml. Cells (90 μl) and peptide (10 μl) were mixed and incubated overnight at 37°C. The MIC was determined as the lowest concentration which inhibited bacterial growth completely.
RESULTS AND DISCUSSION
Control of FRET efficiency using different pH dependencies of stabilities between ECFP and EYFP.
We designed our recombinant ECFP-EYFP fusion protein such that at a physiological pH of 7.0, the EYFP will accept emitted energy from an ECFP molecule excited at 433 nm, leading to emission of yellow fluorescence at 527 nm (Fig. 2A, left). At a reduced pH, however, the EYFP will be significantly unstable and unable to accept the emitted energy from ECFP; in this case, fluorescence at 527 nm will decrease, leaving only the ECFP fluorescence at 475 nm (Fig. 2A, right). Thus, we should be able to evaluate antimicrobial activity by examining the difference of fluorescence intensity (FI) between ECFP-EYFP fusion molecules exposed to a physiologically normal intracellular pH (pHi) and those released into a reduced extracellular pH (pHex) environment. Although one might expect that the cyan FI signal from ECFP might increase as the pH decreased, because its emitted energy would no longer be transferred to EYFP, this effect was not observed (Fig. 2B). This might be due to a slight instability of ECFP at a reduced pH, which could lead to a decrease in the absolute emission energy at 475 nm.
FIG. 2.
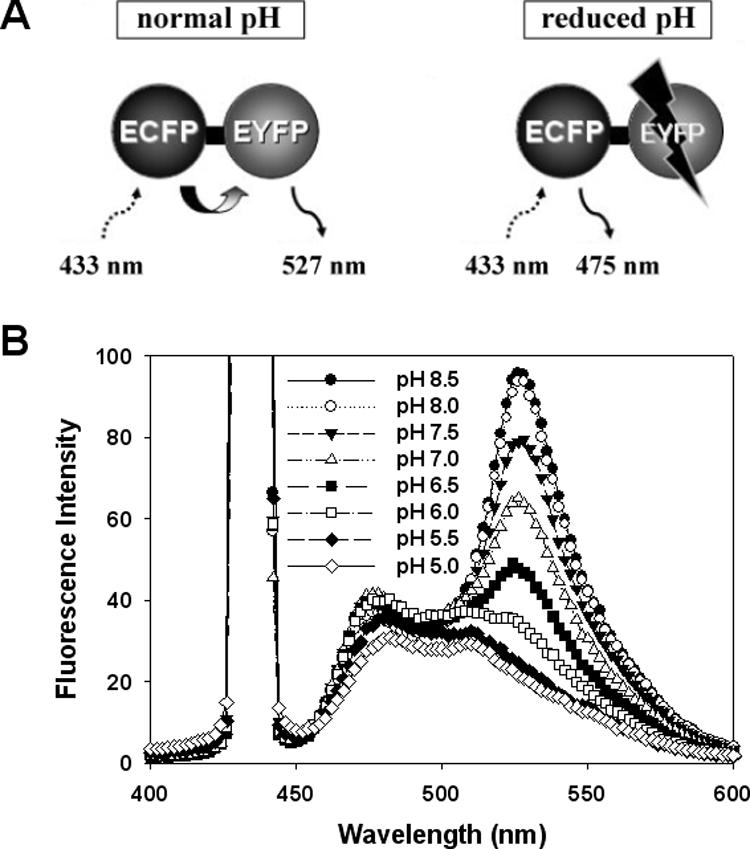
Basic concept of the pH-controlled FRET assay system (A) and emission spectra of ECFP-EYFP fusion proteins at different pHs (B). The emission spectra for ECFP-EYFP excited at 433 nm were monitored in buffers ranging from pH 5.0 to 8.5.
When we tested the effectiveness of various pH values in our system, we found that the FRET efficiency of ECFP-EYFP fusion molecules varied pH dependently over a pH range of 8.5 to 5.0 (Fig. 2B). Since a previous study showed that increasing the pHex from 7.0 to 8.5 triggered a corresponding increase in the pHi of E. coli, but this effect was not seen when the pHex was decreased below 7.0 (11), we chose to use a pHex below 7.0 (Fig. 2B). When we tested for pH 5.5 and 5.0, we found that although yellow FI at these pH values were sharply decreased versus that at pH 7.0, ECFP as well as EYFP were also severely unstable at these reduced pH values (Fig. 2B). In contrast, the fluorescence spectrum at pH 6.0 showed a significant difference from that at pH 7.0 without largely destabilizing the ECFP. Thus, a pHex of 6.0 was applied in subsequent experiments. In addition, our FRET system controlled energy transfer more effectively at pH 6.0 than the distance-based system using the calcium ion concentration (10).
Quantification of antimicrobial activities with yellow/cyan fluorescence intensity ratio.
We demonstrated the application of pH-based FRET for quantitative assay of AMPs by investigating the FRET spectrum and Y/C ratio, which is the FI ratio between EYFP at 527 nm and ECFP at 475 nm when emission is triggered from ECFP-EYFP fusion proteins by excitation at 433 nm, for various concentrations of three different proteins: BSA (negative control) and the AMPs halocidin 18-mer (Hal 18) (8, 14) and magainin II (16). Although antimicrobial activity is generally evaluated based on μg/ml amounts, we utilized the molar concentration in these experiments in an effort to equalize the number of effective molecules, because BSA has a much larger molecular mass (67 kDa) than Hal 18 (1.93 kDa) and magainin II (2.43 kDa). In the case of cells treated with BSA, no changes were observed in the spectrum at 527 nm (Fig. 3A) or the Y/C ratio plot (Fig. 3D), demonstrating that the negative control had no antimicrobial activity against E. coli, i.e., BSA did not alter the cellular entrapment of ECFP-EYFP. In contrast, treatment of cells with Hal 18 and magainin II dose dependently decreased emission at 527 nm (Fig. 3B and C) and dose dependently increased antimicrobial activity, as shown in the Y/C ratio plots (Fig. 3E and F). The linear slopes of these plots reflected the antimicrobial activity, indicating that this method could be used to quantitatively assess the antimicrobial activities of specific or unknown concentrations of target peptides. These findings strongly suggest that our novel pH-based FRET technique should be applicable for quantitative and/or comparative assay of AMPs. We also tested our pH-controlled FRET assay system using nonpeptide compounds; the enzyme lysozyme and the detergent SDS, which disrupt the bacterial membrane, and the chemical antibiotic kanamycin, which inhibits protein synthesis through binding to ribosome (Fig. 4). In both the case of lysozyme and of SDS, Y/C ratios were dose dependently decreased, that is, a tendency identical to that in the case of the antimicrobial peptide. In contrast, kanamycin did not show any changes of Y/C ratio. Therefore, these results demonstrate that our FRET-based assay system is specifically applicable for all cases of bacterial membrane disruption.
FIG. 3.

Antimicrobial activity assays using pH-controlled FRET with a fluorescence spectrophotometer. The emission spectra for samples treated with various concentrations of (A) BSA, (B) Hal 18, or (C) magainin II were scanned following excitation at 433 nm. The Y/C ratios for (D) BSA, (E) Hal 18, or (F) magainin II were plotted according to the applied amount of each agent. Each measured value and error bar represent the mean for three independent experiments and the standard deviation.
FIG. 4.

pH-controlled FRET assays with a fluorescence spectrophotometer for other nonpeptide compounds. The emission spectra for samples treated with various concentrations of (A) lysozyme, (B) SDS, or (C) kanamycin were scanned following excitation at 433 nm. The Y/C ratios for (D) lysozyme, (E) SDS, or (F) kanamycin were plotted according to the applied amount of each agent. Each measured value and error bar represents the mean for three independent experiments and the standard deviation.
Comparison between conventional and FRET-based antimicrobial assay methods.
We then compared our pH-based FRET method with the conventional colony counting method to assess both effectiveness and ease of use. Our method allowed observation of fluorescence changes within 2 h, far faster than methods based on CFU counts, which require a minimum half-day for bacterial colony formation. While pH-controlled FRET yielded a linear slope, plots derived from the colony-counting method showed a curved line (Fig. 5A), which could complicate quantitative comparison of antimicrobial activities. For a more direct comparison, we plotted the magainin II results obtained using the novel and conventional methods (Fig. 3F and 5A). The bidirectional error ranges of the resulting graph (Fig. 5B) clearly show that the conventional colony-counting method had a higher potential for introducing huge deviations, likely due to the required serial dilutions. Although the FRET-based method seemed to be less accurate than the colony-counting method in the case of a large AMP dose, errors were not overlapped with adjacent ranges, while the colony-counting method showed overlapped error ranges. Thus, our method appears simpler and more effective than the colony-counting method, especially for experiments involving small quantities of target peptides.
FIG. 5.

Conventional colony counting assay for antimicrobial activity (A) and direct comparison between pH-controlled FRET and colony counting (B). The two methods and their bidirectional error bars were compared by combining Fig. 3A and 2F. Each measured value and error bar represent the mean for three independent experiments and the standard deviation.
It is tempting to speculate about the possibility of developing a pH-dependent antimicrobial assay system that uses only a single fluorescent protein (e.g., EYFP), similar to the single-luciferase-based assays (6, 13). However, high deviations in initial cell numbers (Fig. 5) would be likely to cause huge variations in the FI values of a single-fluorescent- protein-based system. When two fluorescent proteins are employed, we can set the constant initial Y/C value as ∼1.6 and use this to compensate for variations in the absolute FI values. If we theoretically assume that a specific concentration of AMP can destroy 50 cells out of an initial cell number of 100, we can estimate that a 10% increase in the initial cell number could cause a 10% variation in the FI of a single-fluorescent-protein system but only a 4.5% variation in a two-fluorescent-protein system.
Employment of microplate reader system for high-throughput antimicrobial assay.
Recently, numerous groups have sought not only to identify new AMPs from various species but also to find artificially substituted peptide analogs capable of conferring improved activity (1, 7). One major limitation of substitution studies has been the enormous breadth of combinatorial peptide libraries and requirement of a high-throughput screening technique for antimicrobial activity. Thus, we tested whether our new pH-controlled FRET assay could be applied using a fluorescence microplate reader system as a first step towards scaling up to high-throughput screening applications. Indeed, our method could be easily applied using a microplate reader (Fig. 6A), which offered the important advantage of simultaneous excitation and detection. This resulted in an identical equilibration time for every sample, possibly reducing errors introduced by between-sample differences in the measuring interval. Furthermore, the microplate reader system allowed us to reduce the initial cell number by about 1/10 of the original requirement, providing higher sensitivity by increasing the relative peptide concentration per cell. We found that application of pH-controlled FRET with the microplate reader system yielded larger Y/C ratio reductions for both of the test AMPs. This allowed us to compare the antimicrobial activities of magainin II and Hal 18 (Fig. 6B), which we had been unable to manage using the fluorescence spectrophotometer (see Fig. 3). Our results revealed that magainin II had a higher antimicrobial activity than Hal 18; this finding was subsequently confirmed by the colony-counting method (data not shown). In the case of the MIC assay, magainin II had a slightly lower MIC than Hal 18 even though their values are similar (∼5 μM). Finally, we found that the Y/C ratio for magainin II reached the saturation value (∼0.8) at an amount of ∼0.75 nmol in the microplate reader system, suggesting that it might be possible to reduce the dose of treated peptides while still maintaining the Y/C ratio over the saturation value at pH 6.0 (which is required for accurate assay). However, although the initial cell number was reduced by 1/10 (from 1 × 109 to 1 × 108), we did not see a similar decrease in the amount of AMPs required for saturation of the Y/C ratio. This may indicate that a basal peptide concentration may be required for antimicrobial reactions against a specific number of cells.
FIG. 6.

Antimicrobial activity assays using pH-controlled FRET with a fluorescence microplate reader. (A) Overview of the high-throughput assay system. (B) Y/C ratios for BSA, Hal 18, and magainin II were plotted according to the applied amounts. Each measured value and error bar represent the mean for three independent experiments and the standard deviation.
In conclusion, we herein introduce a novel pH-based FRET system that is suitable for assaying the antimicrobial activities of antibiotic peptides. This system was based on our ability to control energy transfer efficiency by altering the outer environmental pH and the belief that AMP-induced increases in bacterial membrane permeability trigger cell disruption (i.e., the Shai-Matsuzaki-Huang model) (17). Indeed, the validity of our new system could be seen as indirectly supporting this proposed mechanism for AMP activity. Our assay was also precise enough to distinguish activity proportional to the concentration of AMPs. This improved quantitative sensitivity, and the subsequent ability to use smaller quantities of agents, might help counteract the high cost of chemical peptide synthesis and commercial AMPs, complications that have previously limited the use of larger-scale plate culture-based assays. Collectively, our findings indicate that our FRET-based assay system is easier and faster than conventional methods and may be scaled up with a fluorescence microplate reader system for high-throughput screening of numerous AMP candidates. Although the present system is designed to initially screen for antimicrobial activity against the gram-negative bacterium E. coli only, future work should allow the development of a pH-controlled FRET system for other bacterial cells (e.g., gram-positive strains) or in several recombinant bacteria at once, allowing for complete high-throughput screening. Thus, the present work introduces an important new system for quick and effective screening of de novo or improved AMP analogs from combinatorial peptide libraries.
Acknowledgments
We acknowledge support of this work by the Korea Science and Engineering Foundation through the Advanced Environmental Biotechnology Research Center at Pohang University of Science and Technology and by the Brain Korea 21 program issued from the Ministry of Education, Korea.
We thank In Hee Lee (Hoseo University, Korea) for providing chemically synthesized Hal 18.
REFERENCES
- 1.Blondelle, S. E., and K. Lohner. 2000. Combinatorial libraries: a tool to design antimicrobial and antifungal peptide analogues having lytic specificities for structure-activity relationship studies. Biopolymers 55:74-87. [DOI] [PubMed] [Google Scholar]
- 2.Cheung, T. C., and J. P. Hearn. 2003. Development of a baculovirus-based fluorescence resonance energy transfer assay for measuring protein-protein interaction. Eur. J. Biochem. 270:4973-4981. [DOI] [PubMed] [Google Scholar]
- 3.Chopra, I., C. Storey, T. J. Falla, and J. H. Pearce. 1998. Antibiotics, peptidoglycan synthesis and genomics: the chlamydial anomaly revisited. Microbiology 144:2673-2678. [DOI] [PubMed] [Google Scholar]
- 4.Fourmy, D., M. I. Recht, S. C. Blanchard, and J. D. Puglisi. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science 274:1367-1371. [DOI] [PubMed] [Google Scholar]
- 5.Hancock, R. E. W. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. i:156-164. [DOI] [PubMed] [Google Scholar]
- 6.Hilpert, K., R. Volkmer-Engert, T. Walter, and R. E. Hancock. 2005. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 23:1008-1012. [DOI] [PubMed] [Google Scholar]
- 7.Houghten, R. A., C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley, and J. H. Cuervo. 1991. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 354:84-86. [DOI] [PubMed] [Google Scholar]
- 8.Jang, W. S., K. N. Kim, Y. S. Lee, M. H. Nam, and I. H. Lee. 2002. Halocidin: a new antimicrobial peptide from hemocytes of the solitary tunicate, Halocynthia aurantium. FEBS Lett. 521:81-86. [DOI] [PubMed] [Google Scholar]
- 9.Llopis, J., J. M. McCaffery, A. Miyawaki, M. G. Farquhar, and R. Y. Tsien. 1998. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 95:6803-6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyawaki, A., J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura, and R. Y. Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388:882-887. [DOI] [PubMed] [Google Scholar]
- 11.Olsen, K. N., B. B. Budde, H. Siegumfeldt, K. B. Rechinger, M. Jakobsen, and H. Ingmer. 2002. Noninvasive measurement of bacterial intracellular pH on a single-cell level with green fluorescent protein and fluorescence ratio imaging microscopy. Appl. Environ. Microbiol. 68:4145-4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509-544. [DOI] [PubMed] [Google Scholar]
- 13.Vesterlund, S., J. Paltta, A. Laukova, M. Karp, and A. C. Ouwehand. 2004. Rapid screening method for the detection of antimicrobial substances. J. Microbiol. Methods 57:23-31. [DOI] [PubMed] [Google Scholar]
- 14.Wei, Q., Y. S. Kim, J. H. Seo, W. S. Jang, I. H. Lee, and H. J. Cha. 2005. Facilitation of expression and purification of an antimicrobial peptide by fusion with baculoviral polyhedrin in Escherichia coli. Appl. Environ. Microbiol. 71:5038-5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaccolo, M. 2004. Use of chimeric fluorescent proteins and fluorescence resonance energy transfer to monitor cellular responses. Circ. Res. 94:866-873. [DOI] [PubMed] [Google Scholar]
- 16.Zasloff, M. 1987. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 84:5449-5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395. [DOI] [PubMed] [Google Scholar]