

Abstract
The chemokine receptor CCR5 provides a portal of entry for human immunodeficiency virus type 1 (HIV-1) into susceptible CD4+ cells. Both monoclonal antibody (MAb) and small-molecule CCR5 inhibitors have entered human clinical testing, but little is known regarding their potential interactions. We evaluated the interactions between CCR5 MAbs, small-molecule CCR5 antagonists, and inhibitors of HIV-1 gp120, gp41, and reverse transcriptase in vitro. Inhibition data were analyzed for cooperative effects using the combination index (CI) method and stringent statistical criteria. Potent, statistically significant antiviral synergy was observed between the CCR5 MAb PRO 140 and the small-molecule CCR5 antagonists maraviroc (UK-427,857), vicriviroc (SCH-D), and TAK-779. High-level synergy was observed consistently across various assay systems, HIV-1 envelopes, CCR5 target cells, and inhibition levels. CI values ranged from 0.18 to 0.64 and translated into in vitro dose reductions of up to 14-fold. Competition binding studies revealed nonreciprocal patterns of CCR5 binding by MAb and small-molecule CCR5 inhibitors, suggesting that synergy occurs at the level of receptor binding. In addition, both PRO 140 and maraviroc synergized with the chemokine RANTES, a natural ligand for CCR5; however, additive effects were observed for both small-molecule CCR5 antagonists and PRO 140 in combination with other classes of HIV-1 inhibitors. The findings provide a rationale for clinical exploration of MAb and small-molecule CCR5 inhibitors in novel dual-CCR5 regimens for HIV-1 therapy.
The armamentarium for human immunodeficiency virus type 1 (HIV-1) infection currently includes 22 antiretroviral agents drawn from four mechanistic treatment classes: nucleoside reverse transcriptase inhibitors (NRTI), nonnucleoside reverse transcriptase inhibitors (NNRTI), protease inhibitors, and fusion inhibitors. The standard of care for HIV-1 infection involves combination use of three or more antiretroviral agents. Where available, such therapies have markedly reduced HIV-1 morbidity and mortality (34). However, current therapies are limited by the emergence of multidrug-resistant virus, by treatment-related toxicities, by unfavorable drug-drug interactions, and by often-complex dosing regimens that can reduce adherence to therapy. Consequently, many patients eventually exhaust their treatment options, and there is an urgent need for new agents that can be deployed in novel combination regimens.
In 1996, we and others demonstrated that the chemokine receptor CCR5 serves as an entry coreceptor for HIV-1 (1, 10, 12). HIV-1 entry proceeds through a cascade of events mediated by the HIV-1 envelope glycoproteins gp120 and gp41: gp120 sequentially binds CD4 and then CCR5 or another coreceptor molecule, thereby triggering gp41-mediated fusion of the viral and cellular membranes. CCR5 has emerged as an important target for novel HIV-1 therapies (reviewed in reference 35). Both small-molecule and monoclonal antibody (MAb) inhibitors of CCR5 have entered human testing, and the first of these has demonstrated potent antiviral effects in HIV-infected individuals (14, 21).
PRO 140 is a humanized CCR5 MAb that has entered phase 1b testing for HIV-1 therapy. PRO 140 and the parent mouse MAb (PA14) broadly and potently block CCR5-mediated HIV-1 entry in vitro (32, 33, 45). Although PRO 140 and small-molecule CCR5 antagonists target the same protein, their properties are complementary in a number of important respects. Whereas the available small-molecule CCR5 inhibitors potently block the natural activity of CCR5 (11, 39, 40, 48), antiviral concentrations of PRO 140 do not block CCR5 function in vitro (33). In addition, preliminary studies indicate that PRO 140 is highly active against viruses that are resistant to small-molecule CCR5 antagonists (20, 27). These functional differences are likely related to the distinct differences in CCR5 binding. Small-molecule CCR5 antagonists bind a hydrophobic pocket formed by the transmembrane helices of CCR5 and inhibit HIV-1 via allosteric mechanisms (13, 30, 47, 48), while PRO 140 binds an extracellular epitope on CCR5 and appears to act as a competitive inhibitor (33).
Given the mechanistic differences between PRO 140 and small-molecule CCR5 antagonists in clinical development and the need for novel combination regimens, we examined the interactions between these agents in vitro. PRO 140, structurally diverse small-molecule CCR5 antagonists, and other classes of HIV-1 inhibitors were tested alone and in combination for the ability to inhibit HIV-1 membrane fusion and viral entry. Surprisingly, we observed potent antiviral synergy for PRO 140 in combination with each of several small-molecule CCR5 antagonists but not for PRO 140 in combination with agents that target different stages of HIV-1 entry. Both PRO 140 and small-molecule CCR5 antagonists synergized with RANTES (CCL5), a natural ligand for CCR5, but purely additive effects were observed when different small-molecule CCR5 antagonists were combined. Competition binding experiments were conducted and offer a mechanism for the cooperative effects observed. Coupled with the available viral resistance data, these findings indicate that PRO 140 and small-molecule CCR5 drugs may represent distinct subclasses of CCR5 inhibitors.
MATERIALS AND METHODS
Inhibitors.
PRO 140 was expressed in mammalian cells and purified by protein A, ion exchange, and hydroxyapatite chromatographies. Maraviroc (UK-427,857; Pfizer) (11), vicriviroc (SCH-D; Schering-Plough Corporation) (39), TAK-779 (Takeda Pharmaceuticals) (3), enfuvirtide (T-20; Trimeris/Roche) (49), BMS-378806 (Bristol-Myers Squibb) (23), and PRO 542 (CD4-IgG2; Progenics) (2) were prepared according to published methods. Zidovudine (azidothymidine), RANTES, the CCR5 MAb 2D7, and the CD4 MAb Leu-3A were purchased from Sigma Chemicals (St. Louis, MO), R&D Systems (Minneapolis, MN), Pharmingen (San Diego, CA), and Becton Dickinson (Franklin Lakes, NJ), respectively. Maraviroc and vicriviroc were radiolabeled with tritium by GE Healthcare (Piscataway, NJ), and PRO 140 was conjugated to phycoerythrin (PE) by Southern Biotech, Inc. (Birmingham, AL).
HIV-1 membrane fusion assay.
HIV-1 envelope-mediated membrane fusion was examined using a fluorescence resonance energy transfer (RET) assay (24) with modifications. Briefly, HeLa cells that stably express HIV-1JR-FL gp120/gp41 (24) and CEM.NKR-CCR5 cells (NIH AIDS Research and Reference Reagent Program) (38, 46) were labeled separately overnight with fluorescein octadecyl ester (F18; Molecular Probes, Eugene, OR) and rhodamine octadecyl ester (R18; Molecular Probes), respectively. Cells were washed in phosphate-buffered saline containing 15% fetal bovine serum and coseeded at 15,000 cells/well into a 384-well plate. Inhibitors were added, and the plates were incubated in phosphate-buffered saline containing 15% fetal bovine serum plus 0.5% dimethyl sulfoxide for 4 h at 37°C prior to measurement of RET using a Victor2 plate reader (PerkinElmer, Boston, MA) as previously described (24). The CD4 MAb Leu3a was used as a control inhibitor, and percent inhibition was calculated as follows: (RET in the absence of inhibitor − RET in the presence of inhibitor)/(RET in the absence of inhibitor − RET in the presence of Leu3a) × 100.
HIV-1 pseudovirus assay.
A self-inactivating vector was derived from the pNL4-3ΔEnv-luciferase vector (12) by deleting 507 base pairs in the U3 region of the 3′ long terminal repeat so as to remove the TATA box and transcription factor binding sites. The human cytomegalovirus promoter was inserted upstream of the luciferase gene to enable expression of luciferase following integration.
Reporter viruses pseudotyped with HIV-1JR-FL or HIV-1SF162 envelopes were generated by cotransfection of 293T cells with the self-inactivating vector and the appropriate pcDNA Env-expressing vector as previously described (12). U87-CD4-CCR5 cells (8,000/well; NIH AIDS Research and Reference Reagent Program) were infected with 125 to 375 pg of HIV-1 pseudoviruses in 384-well plates in the presence or absence of inhibitor(s). Cultures were incubated for 72 h at 37°C in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum, 1 mg/ml puromycin, 0.3 mg/ml Geneticin, antibiotics, and 0.5% dimethyl sulfoxide. Luciferase activity (relative light units [RLU]) was measured using BrightGlo reagent (Promega, Madison, WI) according to the manufacturer's instructions. Percent inhibition was calculated as follows: (1 − RLU in the presence of inhibitor/RLU in the absence of inhibitor) × 100. Fifty percent inhibitory concentration (IC50) and IC90 were used to denote the respective concentrations required for 50% and 90% inhibition of HIV-1.
Synergy determinations.
Experimental design and data analysis were based on the combination index (CI) method (6, 7). Compounds were tested individually and in combination at a fixed molar ratio over a range of serial dilutions. Entry inhibitors were combined in equimolar amounts, whereas a 1:10 molar ratio was used for PRO 140 in combination with azidothymidine and nevirapine. Dose-response curves were fit using a four-parameter sigmoidal equation with upper and lower inhibition values constrained to 100% and 0%, respectively, in order to calculate concentrations required for 50% (IC50) and 90% (IC90) inhibition (GraphPad Prism; GraphPad Software, San Diego, CA). CI values for 50% (CI50) and 90% (CI90) inhibition were calculated as previously described (6, 7). For example, CI50 was calculated as
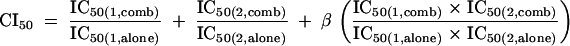 |
where IC50(1,alone) and IC50(1,comb) denote the concentrations of compound 1 required for 50% inhibition when the compound is used alone and in combination, respectively, and IC50(2,alone) and IC50(2,comb) have the corresponding meanings for compound 2. The mutually exclusive CI formula (β = 0) was used for combinations of CCR5 inhibitors, while the mutually nonexclusive formula (β = 1) was utilized for combinations of inhibitors to distinct targets (6). Again, using 50% inhibition and compound 1 as an example, the dose reduction was calculated as IC50(1,comb)/IC50(1,alone). Each test was conducted 4 to 12 times. Synergy, additivity, and antagonism are indicated by CI values of <1, 1, and >1, respectively.
Competition binding assays.
To examine inhibition of PRO 140 binding, CEM.NKR-CCR5 cells were suspended in phosphate-buffered saline with 0.1% sodium azide (PBSA) and incubated with various concentrations of unlabeled CCR5 antagonists at ambient temperature for 30 min. Azide was added to block CCR5 internalization during the assay. Cells were washed in PBSA and incubated with 5 nM PRO 140-PE for an additional 30 min prior to washing and analysis by flow cytometry using a FACSCalibur instrument (Becton Dickinson). The extent of PRO 140-PE binding was measured in terms of both the mean fluorescence intensity (MFI) and the percentage of cells gated for positive staining.
To examine inhibition of maraviroc binding, CEM.NKR-CCR5 cells were preincubated with unlabeled CCR5 inhibitors as described above prior to the addition of 2 nM 3H-maraviroc for an additional 30 min. The cells were washed in PBSA and lysed with 0.5 N HCl prior to scintillation counting using a Wallac1410 instrument. An additional study reversed the order of addition in order to examine the stability of maraviroc binding over the course of the assay. Cells were preincubated with 2 nM 3H-maraviroc for 30 min prior to washing, the addition of unlabeled inhibitors, and processing as described above. Fifty percent effective concentration (EC50) and EC90 were used to denote the concentrations of unlabeled compound required to inhibit binding of labeled compound by 50% and 90%, respectively.
Statistical analyses.
Two-tailed t tests were used to test mean CI50 and CI90 values for the null hypothesis H0 (CI = 1 [additivity]) using GraphPad Prism software. P values were corrected for multiple comparisons from an initial significance level (α) of 0.05 according to the Bonferroni method (9), excluding the PRO 140/PRO 140 mock combination that was included as an assay control. In the Bonferroni correction, P = α/n, where n is the number of comparisons. Twenty-two synergy comparisons (11 compounds × 2 CI values) were made based on data generated in the membrane fusion assay, resulting in a corrected P value of 0.0023. In the pseudovirus assay, 32 synergy comparisons (8 compounds × 2 viruses × 2 CI values) resulted in a corrected P value of 0.0016.
RESULTS
Inhibition of HIV-1 membrane fusion.
PRO 140 and maraviroc were used individually and together to inhibit HIV-1JR-FL envelope-mediated membrane fusion in the RET cell-cell fusion assay, and representative dose-response curves for the individual agents and combination are illustrated in Fig. 1A. Although both PRO 140 and maraviroc individually blocked HIV-1 fusion at low nanomolar potency, the combination was markedly more potent. In this assay, 50% inhibition was obtained using 2.9 nM PRO 140 alone, 5.0 nM maraviroc alone, or 2.1 nM of the combination (1.05 nM PRO 140 plus 1.05 nM maraviroc). This supra-additive effect is indicative of antiviral synergy between the two agents.
FIG. 1.

Dose-response curves for inhibition of HIV-1JR-FL envelope-mediated membrane fusion by combinations of CCR5 inhibitors. Dilutions were analyzed in triplicate wells, and the data points depict the means and standard deviations of replicates. (A) PRO 140 and maraviroc (UK-427,857) were tested individually and in a 1:1 fixed molar ratio over the indicated range of concentrations. In the experiment whose results are depicted, IC50 and IC90 values were 2.9 nM and 11 nM for PRO 140, 5.0 nM and 21 nM for maraviroc, and 2.1 nM and 4.6 nM for the combination, respectively. CI50 and CI90 values were 0.58 and 0.32, respectively. (B) Vicriviroc (SCH-D) and maraviroc were tested individually and in a 1:1 fixed molar ratio over the indicated range of concentrations. In the experiment whose results are depicted, IC50 and IC90 values were 5.5 nM and 34 nM for vicriviroc, 9.7 nM and 59 nM for maraviroc, and 6.1 nM and 31 nM for the combination, respectively. CI50 and CI90 values were 0.87 and 0.73, respectively.
In contrast, the combination of vicriviroc and maraviroc was no more potent than individual agents (Fig. 1B). In this example, the dose-response curves for the individual inhibitors and the combination were overlapping, with 50% inhibition requiring 9.7 nM maraviroc, 5.5 nM vicriviroc, and 6.1 nM of the combination. The data suggest purely additive effects for these inhibitors.
These studies were extended to additional CCR5 (TAK-779, RANTES, and 2D7), gp120 (BMS-378806 and PRO 542), and gp41 (enfuvirtide) inhibitors and were repeated four or more times for each condition. CI50 and CI90 values were calculated for each condition and averaged across the independent assays. Cooperativity was assessed using t tests to determine whether the CI50 and CI90 values were significantly different from 1. As a test of our methods, a PRO 140/PRO 140 mock combination was examined by adding PRO 140 to the assay wells in two separate additions. CI50 and CI90 values for the PRO 140/PRO 140 combination were 0.97 and 0.96, respectively (Table 1); therefore, purely additive effects were observed for this mock combination as expected.
TABLE 1.
CI values for inhibition of HIV-1JR-FL envelope-mediated membrane fusiona
| First inhibitor | Target | IC50 (nM) | IC90 (nM) | Second inhibitor | CI50 | P value | CI90 | P value |
|---|---|---|---|---|---|---|---|---|
| PRO 140 | CCR5 | 2.5 | 8.6 | PRO 140 | 0.97 ± 0.07 | 0.13 | 0.96 ± 0.14 | 0.37 |
| Maraviroc | CCR5 | 5.3 | 27 | PRO 140 | 0.61 ± 0.05 | <0.0001 | 0.40 ± 0.06 | <0.0001 |
| Vicriviroc | CCR5 | 3.2 | 16 | PRO 140 | 0.51 ± 0.05 | <0.0001 | 0.36 ± 0.06 | <0.0001 |
| TAK-779 | CCR5 | 11 | >200 | PRO 140 | 0.38 ± 0.08 | <0.0001 | NA | NA |
| RANTES | CCR5 | 2.4 | 38 | PRO 140 | 0.59 ± 0.08 | 0.0022 | 0.43 ± 0.05 | 0.0002 |
| RANTES | CCR5 | 2.4 | 38 | Maraviroc | 0.48 ± 0.03 | 0.0017 | 0.18 ± 0.01 | <0.0001 |
| Vicriviroc | CCR5 | 3.2 | 16 | Maraviroc | 0.86 ± 0.03 | 0.016 | 0.75 ± 0.02 | 0.0033 |
| Vicriviroc | CCR5 | 3.2 | 16 | TAK-779 | 1.3 ± 0.18 | 0.12 | NA | NA |
| 2D7 | CCR5 | 3.7 | 58 | PRO 140 | 1.0 ± 0.14 | 0.61 | 1.9 ± 0.61 | 0.024 |
| Enfuvirtide | gp41 | 8.6 | 66 | PRO 140 | 0.84 ± 0.16 | 0.040 | 0.89 ± 0.20 | 0.19 |
| PRO 542 | gp120 | 8.9 | 91 | PRO 140 | 0.96 ± 0.17 | 0.56 | 0.94 ± 0.19 | 0.45 |
| BMS-378806 | gp120 | 5.2 | 20 | PRO 140 | 1.3 ± 0.19 | 0.0015 | 1.1 ± 0.22 | 0.19 |
Statistically significant results (P < 0.0023 after application of the Bonferroni correction for multiple comparisons) are indicated in italicized bold text. IC50 and IC90 denote values for the first inhibitor. NA, not applicable. TAK-779 did not consistently achieve 90% inhibition in the assay. CI values represent the means and standard deviations of 4 to 12 independent assays.
Potent synergy was observed for PRO 140 in combination with each of three small-molecule CCR5 antagonists (maraviroc, vicriviroc, and TAK-779), and the findings were statistically significant even when the data were corrected for multiple comparisons via the Bonferroni method (Table 1). CI values ranged from 0.36 to 0.61, and these synergies translated into dose reductions ranging from three- to eightfold across the different conditions. Synergies were greater at 90% inhibition than at 50% inhibition. Synergy between PRO 140 and small-molecule CCR5 antagonists was robust in that it was observed at both the 50% and 90% inhibition levels in every instance. The exception was TAK-779, which did not mediate 90% inhibition when used individually, and therefore, a CI90 was not determined. Similarly, potent synergy was observed when RANTES was used in combination with either PRO 140 or maraviroc (Table 1).
Additional tests examined combinations of two small-molecule CCR5 antagonists (vicriviroc/maraviroc and vicriviroc/TAK-779) or two CCR5 MAbs (PRO 140/2D7). No significant synergy was observed for these combinations, although the vicriviroc/maraviroc CI90 values trended towards significance. The findings are consistent with prior observations of overlapping binding sites for PRO 140 and 2D7 (33) and for vicriviroc and TAK-779 (37).
PRO 140 was also tested in combination with the gp41 fusion inhibitor enfuvirtide and with the gp120 attachment inhibitors PRO 542 and BMS-378806 (Table 1). CI values ranged from 0.84 to 1.28, and none of these combinations demonstrated synergy that met our criteria for statistical significance. For the PRO 140/BMS-378806 combination, modest antagonism was observed at 50% but not 90% inhibition. The biological significance of this result is unclear.
Inhibition of HIV-1 pseudoviruses.
Next, we used single-cycle HIV-1 reporter viruses to examine whether the synergistic effects were limited to cell-cell fusion or whether they extended to other modes of HIV-1 entry. Signals in this assay require both viral entry and reverse transcription, enabling us to include NRTI and NNRTI in the analyses. Each combination was tested against reporter viruses pseudotyped with envelopes from HIV-1JR-FL and HIV-1SF162 in at least four independent assays per virus. A PRO 140/PRO 140 mock combination was again included as an assay control and demonstrated additive effects against both HIV-1JR-FL and HIV-1SF162 pseudoviruses as expected (Table 2).
TABLE 2.
CI values for inhibition of HIV-1 reporter viruses pseudotyped with envelopes from HIV-1JR-FL and HIV-1SF162a
| First inhibitor | Target | HIV-1 envelope | IC50 (nM) | IC90 (nM) | Second inhibitor | CI50 | P value | CI90 | P value |
|---|---|---|---|---|---|---|---|---|---|
| PRO 140 | CCR5 | JR-FL | 2.2 | 28 | PRO 140 | 1.2 ± 0.32 | 0.16 | 0.90 ± 0.15 | 0.047 |
| SF162 | 1.3 | 20 | PRO 140 | 1.0 ± 0.27 | 1.0 | 0.86 ± 0.33 | 0.21 | ||
| Vicriviroc | CCR5 | JR-FL | 2.4 | 44 | PRO 140 | 0.47 ± 0.15 | <0.001 | 0.18 ± 0.04 | <0.001 |
| SF162 | 0.34 | 14 | PRO 140 | 0.60 ± 0.17 | <0.001 | 0.28 ± 0.11 | <0.001 | ||
| Maraviroc | CCR5 | JR-FL | 7.4 | 46 | PRO 140 | 0.44 ± 0.06 | <0.001 | 0.24 ± 0.11 | <0.001 |
| SF162 | 0.87 | 13 | PRO 140 | 0.64 ± 0.07 | <0.001 | 0.31 ± 0.11 | <0.001 | ||
| Maraviroc | CCR5 | JR-FL | 7.4 | 46 | Vicriviroc | 0.71 ± 0.11 | 0.16 | 1.2 ± 0.15 | 0.32 |
| SF162 | 0.87 | 13 | Vicriviroc | 0.87 ± 0.06 | 0.19 | 0.86 ± 0.28 | 0.61 | ||
| 2D7 | CCR5 | JR-FL | 8.8 | >200 | PRO 140 | 1.5 ± 0.25 | 0.024 | NA | NA |
| SF162 | 2.2 | 74 | PRO 140 | 1.1 ± 0.47 | 0.61 | 1.0 ± 0.16 | 0.65 | ||
| PRO 542 | gp120 | JR-FL | 0.19 | 2.9 | PRO 140 | 1.2 ± 0.32 | 0.22 | 1.0 ± 0.18 | 0.92 |
| SF162 | 0.36 | 7.1 | PRO 140 | 0.98 ± 0.28 | 0.84 | 0.64 ± 0.26 | 0.010 | ||
| BMS-378806 | gp120 | JR-FL | 1.2 | 11 | PRO 140 | 1.2 ± 0.38 | 0.43 | 0.74 ± 0.23 | 0.059 |
| SF162 | 0.03 | 0.42 | PRO 140 | 1.1 ± 0.28 | 0.36 | 0.82 ± 0.21 | 0.068 | ||
| Nevirapine | RT | JR-FL | 30 | 310 | PRO 140 | 1.2 ± 0.38 | 0.36 | 0.73 ± 0.28 | 0.068 |
| SF162 | 42 | 280 | PRO 140 | 1.2 ± 0.34 | 0.30 | 0.63 ± 0.19 | 0.033 | ||
| Zidovudine | RT | JR-FL | 140 | 1900 | PRO 140 | 1.1 ± 0.38 | 0.37 | 0.85 ± 0.26 | 0.21 |
| SF162 | 86 | 2100 | PRO 140 | 0.99 ± 0.27 | 0.91 | 1.0 ± 0.38 | 1.0 |
Statistically significant results (P < 0.0016 after application of the Bonferroni correction for multiple comparisons) are indicated in italicized bold text. IC50 and IC90 refer to values for the first inhibitor. RT, reverse transcriptase; NA, not applicable. 2D7 did not consistently achieve 90% inhibition in the assay. CI values represent the means and standard deviations of four or more independent assays.
PRO 140 potently synergized with both maraviroc and vicriviroc in blocking virus-cell fusion, and the results met our criteria for statistical significance. Comparable levels of synergy were observed against both HIV-1JR-FL and HIV-1SF162 pseudoviruses at 50% and 90% inhibition (Table 2), with CI values ranging from 0.18 to 0.64. These synergies translated into dose reductions of up to 14-fold. These results are in good agreement with those obtained with the cell-cell fusion assay (Table 1). Neither TAK-779 nor RANTES mediated consistent, high-level inhibition of HIV-1 pseudovirus entry, and therefore, these compounds were not included in this analysis (data not shown).
Additive effects were observed for both the maraviroc/vicriviroc and PRO 140/2D7 combinations (Table 2). Similarly, additivity was observed for PRO 140 used in combination with the gp120 inhibitors PRO 542 and BMS-378806. No antagonism was observed for the PRO 140/BMS-378806 combination against either virus. Overall, these findings are consistent with those seen for cell-cell fusion. Lastly, additive effects were observed for PRO 140 in combination with either zidovudine (NRTI) or nevirapine (NNRTI).
Competition binding studies.
As described above, additive antiviral effects were observed for inhibitors known (PRO 140 and 2D7) or inferred (maraviroc and vicriviroc) to compete for CCR5 binding; however, little is known regarding the competitive binding of synergistic compounds (e.g., PRO 140/maraviroc and PRO 140/vicriviroc). Since noncompetitive binding provides a possible mechanism for synergy between CCR5 inhibitors, we explored this issue by using labeled forms of maraviroc and PRO 140.
Flow cytometry was used to examine inhibition of PRO 140-PE binding to CEM.NRK-CCR5 cells by unlabeled PRO 140, maraviroc, and vicriviroc. PRO 140-PE binding was efficiently inhibited by unlabeled PRO 140, as expected. Complete inhibition was observed in terms of both MFI values (Fig. 2A) and the percentages of cells gated for positive binding (Fig. 2B). The EC50 based on MFI data was 2.5 nM (Fig. 2A), and this value compares favorably with the antiviral IC50 of PRO 140 (Tables 1 and 2). Since the percentage of cells gated is a readout for essentially complete inhibition of binding, the EC90 value was calculated to be 17 nM, and this value is similar to the antiviral IC90 values observed for PRO 140 (Tables 1 and 2). 2D7 also completely inhibited binding of PRO 140-PE to CEM.NKR-CCR5 cells (data not shown). The CCR5 specificity of PRO 140-PE was also demonstrated by its inability to bind parental CEM.NKR-CCR5 cells (data not shown).
FIG. 2.

Inhibition of PRO 140-PE binding to CEM.NKR-CCR5 cells by unlabeled PRO 140, maraviroc, and vicriviroc. CEM.NKR-CCR5 cells were incubated with various concentrations of unlabeled PRO 140, maraviroc, or vicriviroc for 30 min at room temperature in PBSA buffer prior to the addition of 5 nM PRO 140-PE for an additional 30 min. Cells were washed and then analyzed by flow cytometry for both the MFI of binding and the percentage of cells gated for positive binding of PRO 140-PE. Inhibition was assessed on the basis of both MFI (A) and the percentage of cells gated (B).
In sharp contrast, modest levels of inhibition were observed for maraviroc and vicriviroc (Fig. 2). Micromolar concentrations of maraviroc and vicriviroc reduced PRO 140-PE MFI values by 50% or less (Fig. 2A). More dramatically, maraviroc and vicriviroc had little impact on the percentage of cells gated for positive binding of PRO 140-PE (Fig. 2B). The findings suggest that maraviroc and vicriviroc partially reduce the number of PRO 140-PE molecules bound per cell; however, these compounds do not reduce the number of cells that bind measurable amounts of PRO 140-PE. Therefore, maraviroc and vicriviroc represent partial antagonists of PRO 140 binding, and this finding provides a mechanism for the antiviral synergy observed between PRO 140 and these small-molecule CCR5 antagonists.
Next, we examined inhibition of 3H-maraviroc binding by unlabeled maraviroc, vicriviroc, and PRO 140. Binding of 3H-maraviroc to CEM.NKR-CCR5 cells was efficiently inhibited by unlabeled maraviroc (Fig. 3A). The EC50 for binding was 4.3 nM and is similar to the antiviral IC50 values observed for maraviroc (Tables 1 and 2).
FIG. 3.

Inhibition of 3H-maraviroc binding by unlabeled maraviroc, vicriviroc, and PRO 140. (A) CEM.NKR-CCR5 cells were preincubated with various concentrations of unlabeled maraviroc, vicriviroc, or PRO 140 for 30 min in PBSA buffer at ambient temperature prior to the addition of 2 nM 3H-maraviroc for an additional 30 min. Cells were washed and then analyzed for radioactivity by scintillation counting. (B) The stability of maraviroc binding under the assay conditions was examined by preincubating CEM.NKR-CCR5 cells with 2 nM 3H-maraviroc prior to washing, the addition of unlabeled compounds for 30 min, and processing as described above.
Vicriviroc also blocked 3H-maraviroc binding to background levels (Fig. 3A). However, there was no correlation between the compounds' antiviral potency and their potency in blocking 3H-maraviroc binding. For example, whereas vicriviroc demonstrated equal or slightly greater antiviral potency than maraviroc (Tables 1 and 2), vicriviroc was less potent in blocking 3H-maraviroc binding (EC50 = 17 nM) (Fig. 3A). This result is consistent with minor differences in the CCR5 binding sites of these compounds.
Surprisingly, PRO 140 also blocked 3H-maraviroc binding to background levels (Fig. 3A), and this result contrasts with the modest inhibition of PRO 140-PE binding by maraviroc (Fig. 2). PRO 140 inhibited 3H-maraviroc binding with an EC50 of 14 nM, which is 5- to 10-fold higher than the antiviral IC50 of PRO 140 (Tables 1 and 2).
A final experiment examined the stability of maraviroc binding to CEM.NKR-CCR5 cells under the conditions of the competition assay. For this, we preincubated cells with 3H-maraviroc and then examined its dissociation in the presence of unlabeled maraviroc, vicriviroc, and PRO 140. As indicated in Fig. 3B, there was minimal dissociation of 3H-maraviroc over 30 min at ambient temperature, and maraviroc wasn't displaced by either PRO 140 or vicriviroc. Therefore, the inability of maraviroc to efficiently compete for PRO 140 binding to CCR5 (Fig. 2) is not due to rapid dissociation of maraviroc from CCR5 during the course of the assay. Collectively, the data indicate that PRO 140 can bind CCR5 in the presence of prebound maraviroc.
DISCUSSION
This study is the first to examine combinations of CCR5 drugs that are currently in development for HIV-1 therapy. Surprisingly, we observed potent antiviral synergy between the CCR5 MAb PRO 140 and each of three structurally distinct small-molecule CCR5 antagonists. Consistent, high-level synergy was observed across various assay systems, viral isolates, target cells, and inhibition levels. PRO 140 and small-molecule CCR5 antagonists were more potently synergistic when used together rather than in combination with inhibitors that block other stages of HIV-1 entry. In contrast, additive effects were observed for combinations of two small-molecule CCR5 antagonists. Competition binding studies revealed complex and nonreciprocal patterns of CCR5 binding by MAb and small-molecule CCR5 inhibitors and suggest that the synergistic interactions occur at the level of receptor binding. Our findings have implications for the potential use of novel dual-CCR5 regimens for HIV-1 therapy.
Robust synergy between MAb and small-molecule CCR5 inhibitors was observed in this study, and this finding is consistent with that of a recent report (36). Potent synergy was observed for both cell-cell and virus-cell fusion, and there was a good concordance of findings in these two well-established assay systems. Comparable levels of synergy were observed for PRO 140 in combination with each of three small-molecule CCR5 antagonists from unrelated chemical series. In addition, consistent synergy was observed for each of two CCR5 target cells and two well-characterized HIV-1 envelopes. HIV-1JR-FL was isolated from the brain of an AIDS patient at autopsy (31). Like other late-stage neurovirulent viruses (17), HIV-1JR-FL encodes an envelope with high binding affinity for CCR5 (36). Lastly, similar levels of synergy were observed when PRO 140 and maraviroc were tested against HIV-1BaL pseudoviruses (15) in preliminary studies (data not shown).
Synergy increased with increasing levels of viral inhibition and translated into in vitro dose reductions of up to 14-fold. Viewed alternatively, this degree of synergy provides a corresponding increase in antiviral pressure at a given concentration of drugs, thereby improving viral suppression and potentially delaying the emergence of drug-resistant virus.
We also observed potent synergy for RANTES used in combination with either maraviroc or PRO 140. Endogenous levels of RANTES may afford some protection against HIV-1 disease progression during natural infection (16, 25), and therefore, our finding of synergy has important and positive implications for CCR5-targeted therapies of HIV-1. Antiviral synergy between RANTES and PRO 140 is not surprising based on our prior observation that RANTES signaling is not blocked by antiviral concentrations of murine PRO 140 (PA14) (33). Synergy between RANTES and maraviroc is less easily explained given that maraviroc is a potent CCR5 antagonist. However, our findings are consistent with prior observations of synergy between the small-molecule CCR5 antagonist SCH-C and aminooxypentane-RANTES (44), a RANTES derivative that has been evaluated as a potential topical microbicide (19).
In contrast to the robust synergy observed between MAb and small-molecule CCR5 antagonists, additive effects were observed for combinations of small-molecule CCR5 antagonists. A lack of cooperativity is consistent with the view that these molecules compete for binding to a common pocket on CCR5 (13, 30, 47, 48). However, synergy is not required to derive clinical benefit from combination therapy. No interference was observed between small-molecule CCR5 antagonists, and this finding has relevance when considering combining such agents in the clinic.
Similarly, we did not observe potent synergy between PRO 140 and inhibitors of HIV-1 attachment (PRO 542 and BMS-378806), fusion (enfuvirtide), or reverse transcriptase (zidovudine and nevirapine), and these findings underscore the significance of the synergy observed for PRO 140 and small-molecule CCR5 antagonists. A number of prior studies have examined interactions between various small-molecule CCR5 antagonists (maraviroc, SCH-C, TAK-220, TAK-652, and E913) and drugs from each of the existing HIV-1 treatment classes. Most (42-44) but not all (11, 26) studies have reported broad synergy between CCR5 inhibitors and the other HIV-1 treatment classes, and the divergent results may reflect differences in the compounds and methods used for antiviral testing as well as differences in the methods used for data analysis. When maraviroc was tested against 20 licensed antiretroviral agents, additive effects were observed in all but three cases, in which modest synergy was reported (11). This result is consistent with our findings for combinations of PRO 140 and HIV-1 inhibitors that do not target CCR5.
Synergy between anti-HIV-1 drugs may stem from a variety of mechanisms. In mixed virus cultures, one compound may inhibit virus resistant to a second compound (18), and NRTI/NNRTI combinations may overcome specific reverse transcriptase-mediated resistance mechanisms (4, 5). Metabolic interactions between inhibitors may increase their effective intracellular drug concentrations (28), and synergistic entry inhibitors may disrupt interdependent steps in the entry cascade (29, 41). The present study examined clonal viral envelopes rather than mixed populations, and the extracellular nature of the target argues against metabolic interactions. Multiple domains of gp120 contribute to CCR5 binding (8), but it is unclear at present whether these interactions represent separate or discrete events during infection.
Our findings indicate that antiviral synergy between MAb and small-molecule CCR5 inhibitors may occur at the level of the receptor, although the mechanism remains poorly defined. As discussed above, MAbs and small molecules bind distinct loci on CCR5 (13, 30, 33, 47, 48). When preincubated with CCR5 cells in the present study, PRO 140 completely blocked subsequent binding of maraviroc to the receptor, although the PRO 140 concentrations were higher than those needed to block HIV-1 entry into the same cells. In contrast, preincubation of CCR5 cells with supersaturating concentrations of maraviroc or vicriviroc reduced PRO 140 binding by 50% or less. As one possible explanation, PRO 140 could recognize CCR5 conformers that are not bound by maraviroc or vicriviroc. Although cell surface CCR5 exists in multiple conformations (22), it seems unlikely that the small-molecule antagonists could demonstrate potent antiviral activity while failing to bind a significant fraction of cell surface CCR5. In this regard, it is important to note that a common cellular background (CEM.NKR-CCR5 cells) was used for competition binding and antiviral studies, and therefore, the findings are not related to cell-specific differences in CCR5 expression.
In our view, a more plausible explanation for our findings is that PRO 140 is capable of forming a ternary complex with maraviroc-bound CCR5, and this ternary complex provides an increased barrier to HIV-1 entry. Within the context of this model, PRO 140 may bind maraviroc-bound CCR5 somewhat less efficiently than free CCR5, as evidenced by the modest reduction in PRO 140 binding in the presence of maraviroc. However, we note that our studies do not demonstrate the existence of a ternary complex, and additional studies will be required to test this model and to further define the mechanisms of synergy between MAb and small-molecule CCR5 inhibitors. To this end, time-of-addition studies have indicated that murine PRO 140 (PA14) acts at a later stage of the fusion cascade than does the small-molecule CCR5 antagonist SCH-C (36). This report proposed that SCH-C blocks gp120 binding to CCR5, while PA14 prevents ill-defined postbinding events.
The combination index method is widely used to assess drug-drug interactions. In this method, cooperativity is often defined on the basis of empirical CI values (e.g., <0.9 for synergy and >1.1 for antagonism) irrespective of interassay variability. Statistical analyses are performed infrequently, and even more rarely are adjustments made for multiple comparisons. In the absence of such analyses, there is increased potential to overestimate the number of synergistic combinations.
We adopted a rigorous approach for identifying synergistic effects. CI values were tested for statistical significance against the null hypothesis of additivity (CI = 1). In addition, our studies determined 20 to 30 different CI values per experiment (Tables 1 and 2), as is common in synergy studies. In order to reduce the potential for spurious positive results, we reduced the significance level using the Bonferroni correction. We also evaluated a mock combination as a control. We conclude that numerous apparent synergies (CI < 0.9) could not be distinguished from interassay variation based on the available data. Nevertheless, PRO 140 and small-molecule inhibitors demonstrated significant synergy under every test condition, lending credence to this finding. Combinations with CI values that trended towards significance in the present survey, such as the PRO 140/enfuvirtide combination, could be explored in future studies.
A growing body of data indicates that MAb and small-molecule CCR5 antagonists represent distinct subclasses of CCR5 inhibitors, and a number of important parallels can be drawn between NRTI and NNRTI on the one hand and between MAb and small-molecule CCR5 antagonists on the other. In each instance, there are distinct binding loci for the inhibitors on the target protein (reverse transcriptase or CCR5). One set of inhibitors (NNRTI or small-molecule CCR5 antagonists) acts via allosteric mechanisms, while the other set (NRTI or CCR5 MAbs) acts as a competitive inhibitor. Like NRTI and NNRTI, MAb and small-molecule CCR5 inhibitors are synergistic and possess complementary patterns of viral resistance in vitro in preliminary testing (20, 27). NRTI and NNRTI represent important and distinct treatment classes even though they target the same protein, and MAb and small-molecule CCR5 inhibitors similarly may offer distinct HIV-1 treatment modalities.
Acknowledgments
This work was supported by Public Health Service grants AI046871 and AI066329 from the National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, P. M. Murphy, and E. A. Berger. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955. [DOI] [PubMed] [Google Scholar]
- 2.Allaway, G. P., K. L. Davis-Bruno, G. A. Beaudry, E. B. Garcia, E. L. Wong, A. M. Ryder, K. W. Hasel, M.-C. Gauduin, R. A. Koup, J. S. McDougal, and P. J. Maddon. 1995. Expression and characterization of CD4-IgG2, a novel heterotetramer which neutralizes primary HIV-1 isolates. AIDS Res. Hum. Retrovir. 11:533-539. [DOI] [PubMed] [Google Scholar]
- 3.Baba, M., O. Nishimura, N. Kanzaki, M. Okamoto, H. Sawada, Y. Iizawa, M. Shiraishi, Y. Aramaki, K. Okonogi, Y. Ogawa, K. Meguro, and M. Fujino. 1999. A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc. Natl. Acad. Sci. USA 96:5698-5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basavapathruni, A., C. M. Bailey, and K. S. Anderson. 2004. Defining a molecular mechanism of synergy between nucleoside and nonnucleoside AIDS drugs. J. Biol. Chem. 279:6221-6224. [DOI] [PubMed] [Google Scholar]
- 5.Borkow, G., D. Arion, M. A. Wainberg, and M. A. Parniak. 1999. The thiocarboxanilide nonnucleoside inhibitor UC781 restores antiviral activity of 3′-azido-3′-deoxythymidine (AZT) against AZT-resistant human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 43:259-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chou, T. C., and D. C. Rideout. 1991. Synergism and antagonism in chemotherapy. Academic Press, New York, N.Y.
- 7.Chou, T. C., and P. Talalay. 1984. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 22:27-55. [DOI] [PubMed] [Google Scholar]
- 8.Cormier, E. G., and T. Dragic. 2002. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein interactions with the CCR5 coreceptor. J. Virol. 76:8953-8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cudeck, R., and L. L. O'Dell. 1994. Applications of standard error estimates in unrestricted factor analysis: significance tests for factor loadings and correlations. Psychol. Bull. 115:475-487. [DOI] [PubMed] [Google Scholar]
- 10.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. Di Marzio, S. Marmon, R. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661. [DOI] [PubMed] [Google Scholar]
- 11.Dorr, P., M. Westby, S. Dobbs, P. Griffin, B. Irvine, M. Macartney, J. Mori, G. Rickett, C. Smith-Burchnell, C. Napier, R. Webster, D. Armour, D. Price, B. Stammen, A. Wood, and M. Perros. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49:4721-4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Nagashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667-673. [DOI] [PubMed] [Google Scholar]
- 13.Dragic, T., A. Trkola, D. A. Thompson, E. G. Cormier, F. A. Kajumo, E. Maxwell, S. W. Lin, W. Ying, S. O. Smith, T. P. Sakmar, and J. P. Moore. 2000. A binding pocket for a small molecule inhibitor of HIV-1 entry within the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. USA 97:5639-5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fatkenheuer, G., A. L. Pozniak, M. A. Johnson, A. Plettenberg, S. Staszewski, A. I. M. Hoepelman, M. S. Saag, F. D. Goebel, J. K. Rockstroh, B. J. Dezube, T. M. Jenkins, C. Medhurst, J. F. Sullivan, C. Ridgway, S. Abel, I. T. James, M. Youle, and E. van der Ryst. 2005. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat. Med. 11:1170-1172. [DOI] [PubMed] [Google Scholar]
- 15.Gartner, S., P. Markovits, D. M. Markovitz, M. H. Kaplan, R. C. Gallo, and M. Popovic. 1986. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science 233:215-219. [DOI] [PubMed] [Google Scholar]
- 16.Garzino-Demo, A., R. B. Moss, J. B. Margolick, F. Cleghorn, A. Sill, W. A. Blattner, F. Cocchi, D. J. Carlo, A. L. DeVico, and R. C. Gallo. 1999. Spontaneous and antigen-induced production of HIV-inhibitory beta-chemokines are associated with AIDS-free status. Proc. Natl. Acad. Sci. USA 96:11986-11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorry, P. R., J. Taylor, G. H. Holm, A. Mehle, T. Morgan, M. Cayabyab, M. Farzan, H. Wang, J. E. Bell, K. Kunstman, J. P. Moore, S. M. Wolinsky, and D. Gabuzda. 2002. Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. J. Virol. 76:6277-6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson, V. A., D. P. Merrill, J. A. Videler, T. C. Chou, R. E. Byington, J. J. Eron, R. T. D'Aquila, and M. S. Hirsch. 1991. Two-drug combinations of zidovudine, didanosine, and recombinant interferon-alpha A inhibit replication of zidovudine-resistant human immunodeficiency virus type 1 synergistically in vitro. J. Infect. Dis. 164:646-655. [DOI] [PubMed] [Google Scholar]
- 19.Kawamura, T., S. S. Cohen, D. L. Borris, E. A. Aquilino, S. Glushakova, L. B. Margolis, J. M. Orenstein, R. E. Offord, A. R. Neurath, and A. Blauvelt. 2000. Candidate microbicides block HIV-1 infection of human immature Langerhans cells within epithelial tissue explants. J. Exp. Med. 192:1491-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuhmann, S. E., P. Pugach, K. J. Kunstman, J. Taylor, R. L. Stanfield, A. Snyder, J. M. Strizki, J. Riley, B. M. Baroudy, I. A. Wilson, B. T. Korber, S. M. Wolinsky, and J. P. Moore. 2004. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J. Virol. 78:2790-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalezari, J., M. Thompson, P. Kumar, P. Piliero, R. Davey, K. Patterson, A. Shachoy-Clark, K. Adkison, J. Demarest, Y. Lou, M. Berrey, and S. Piscitelli. 2005. Antiviral activity and safety of 873140, a novel CCR5 antagonist, during short-term monotherapy in HIV-infected adults. AIDS 19:1443-1448. [DOI] [PubMed] [Google Scholar]
- 22.Lee, B., M. Sharron, C. Blanpain, B. J. Doranz, J. Vakili, P. Setoh, E. Berg, G. Liu, R. H. Guy, S. R. Durell, M. Parmentier, C. N. Chang, K. Price, M. Tsang, and R. W. Doms. 1999. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J. Biol. Chem. 274:9617-9626. [DOI] [PubMed] [Google Scholar]
- 23.Lin, P. F., W. Blair, T. Wang, T. Spicer, Q. Guo, N. Zhou, Y. F. Gong, H.-G. H. Wang, R. Rose, G. Yamanaka, B. Robinson, C. B. Li, R. Fridell, C. Deminie, G. Demers, Z. Yang, L. Zadjura, N. Meanwell, and R. Colonno. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 100:11013-11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litwin, V., K. A. Nagashima, A. M. Ryder, C. H. Chang, J. M. Carver, W. C. Olson, M. Alizon, K. W. Hasel, P. J. Maddon, and G. P. Allaway. 1996. Human immunodeficiency virus type 1 membrane fusion mediated by a laboratory-adapted strain and a primary isolate analyzed by resonance energy transfer. J. Virol. 70:6437-6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, H., D. Chao, E. E. Nakayama, H. Taguchi, M. Goto, X. Xin, J. K. Takamatsu, H. Saito, Y. Ishikawa, T. Akaza, T. Juji, Y. Takebe, T. Ohishi, K. Fukutake, Y. Maruyama, S. Yashiki, S. Sonoda, T. Nakamura, Y. Nagai, A. Iwamoto, and T. Shioda. 1999. Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proc. Natl. Acad. Sci. USA 96:4581-4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maeda, K., K. Yoshimura, S. Shibayama, H. Habashita, H. Tada, K. Sagawa, T. Miyakawa, M. Aoki, D. Fukushima, and H. Mitsuya. 2001. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J. Biol. Chem. 276:35194-35200. [DOI] [PubMed] [Google Scholar]
- 27.Marozsan, A. J., S. E. Kuhmann, T. Morgan, C. Herrera, E. Rivera-Troche, S. Xu, B. M. Baroudy, J. Strizki, and J. P. Moore. 2005. Generation and properties of a human immunodeficiency virus type 1 isolate resistant to the small molecule CCR5 inhibitor, SCH-417690 (SCH-D). Virology 338:182-199. [DOI] [PubMed] [Google Scholar]
- 28.Molla, A., H. Mo, S. Vasavanonda, L. Han, C. T. Lin, A. Hsu, and D. J. Kempf. 2002. In vitro antiviral interaction of lopinavir with other protease inhibitors. Antimicrob. Agents Chemother. 46:2249-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagashima, K. A., D. A. D. Thompson, S. I. Rosenfield, P. J. Maddon, T. Dragic, and W. C. Olson. 2001. Human immunodeficiency virus type 1 entry inhibitors PRO 542 and T-20 are potently synergistic in blocking virus-cell and cell-cell fusion. J. Infect. Dis. 183:1121-1125. [DOI] [PubMed] [Google Scholar]
- 30.Nishikawa, M., K. Takashima, T. Nishi, R. A. Furuta, N. Kanzaki, Y. Yamamoto, and J. Fujisawa. 2005. Analysis of binding sites for the new small-molecule CCR5 antagonist TAK-220 on human CCR5. Antimicrob. Agents Chemother. 49:4708-4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Brien, W. A., Y. Koyanagi, A. Namazie, J. Q. Zhao, A. Diagne, K. Idler, J. A. Zack, and I. S. Chen. 1990. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature 348:69-73. [DOI] [PubMed] [Google Scholar]
- 32.Olson, W. C., and P. J. Maddon. 2003. Resistance to HIV-1 entry inhibitors. Curr. Drug Targets Infect. Disord. 3:255-262. [DOI] [PubMed] [Google Scholar]
- 33.Olson, W. C., G. E. E. Rabut, K. A. Nagashima, D. N. H. Tran, D. J. Anselma, S. P. Monard, J. P. Segal, D. A. D. Thompson, F. Kajumo, Y. Guo, J. P. Moore, P. J. Maddon, and T. Dragic. 1999. Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J. Virol. 73:4145-4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palella, F. J., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, S. D. Holmberg, and the HIV Outpatient Study Investigators. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338:853. [DOI] [PubMed] [Google Scholar]
- 35.Ray, N., and R. W. Doms. 2006. HIV-1 coreceptors and their inhibitors. Curr. Top. Microbiol. Immunol. 303:97-120. [DOI] [PubMed] [Google Scholar]
- 36.Safarian, D., X. Carnec, F. Tsamis, F. Kajumo, and T. Dragic. An anti-CCR5 monoclonal antibody and small molecule CCR5 antagonists synergize by inhibiting different stages of human immunodeficiency virus type 1 entry. Virology, in press. [DOI] [PubMed]
- 37.Seibert, C., W. Ying, S. Gavrilov, F. Tsamis, S. E. Kuhmann, A. Palani, J. R. Tagat, J. W. Clader, S. W. McCombie, B. M. Baroudy, S. O. Smith, T. Dragic, J. P. Moore, and T. P. Sakmar. 2006. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology 349:41-54. [DOI] [PubMed] [Google Scholar]
- 38.Spenlehauer, C., C. A. Gordon, A. Trkola, and J. P. Moore. 2001. A luciferase-reporter gene-expressing T-cell line facilitates neutralization and drug-sensitivity assays that use either R5 or X4 strains of human immunodeficiency virus type 1. Virology 280:292-300. [DOI] [PubMed] [Google Scholar]
- 39.Tagat, J. R., S. W. McCombie, D. Nazareno, M. A. Labroli, Y. Xiao, R. W. Steensma, J. M. Strizki, B. M. Baroudy, K. Cox, J. Lachowicz, G. Varty, and R. Watkins. 2004. Piperazine-based CCR5 antagonists as HIV-1 inhibitors. IV. Discovery of 1-[(4,6-dimethyl-5-pyrimidinyl)carbonyl]-4-[4-[2-methoxy-1(R)-4-(trifluoromethyl)phenyl]ethyl-3(S)-methyl-1-piperaz inyl]-4-methylpiperidine (Sch-417690/Sch-D), a potent, highly selective, and orally bioavailable CCR5 antagonist. J. Med. Chem. 47:2405-2408. [DOI] [PubMed] [Google Scholar]
- 40.Takashima, K., H. Miyake, N. Kanzaki, Y. Tagawa, X. Wang, Y. Sugihara, Y. Iizawa, and M. Baba. 2005. Highly potent inhibition of human immunodeficiency virus type 1 replication by TAK-220, an orally bioavailable small-molecule CCR5 antagonist. Antimicrob. Agents Chemother. 49:3474-3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tremblay, C., C. Kollman, F. Giguel, T. C. Chou, and M. S. Hirsch. 2000. Strong in vitro synergy observed between the fusion inhibitor T-20 and a CXCR4 blocker AMD-3100. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 25:99-102. [DOI] [PubMed] [Google Scholar]
- 42.Tremblay, C. L., F. Giguel, T. C. Chou, H. Dong, K. Takashima, and M. S. Hirsch. 2005. TAK-652, a novel CCR5 inhibitor, has favourable drug interactions with other antiretrovirals in vitro. Antivir. Ther. 10:967-968. [PubMed] [Google Scholar]
- 43.Tremblay, C. L., F. Giguel, Y. Guan, T.-C. Chou, K. Takashima, and M. S. Hirsch. 2005. TAK-220, a novel small-molecule CCR5 antagonist, has favorable anti-human immunodeficiency virus interactions with other antiretrovirals in vitro. Antimicrob. Agents Chemother. 49:3483-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tremblay, C. L., F. Giguel, C. Kollmann, Y. Guan, T.-C. Chou, B. M. Baroudy, and M. S. Hirsch. 2002. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 46:1336-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trkola, A., T. J. Ketas, K. A. Nagashima, L. Zhao, T. Cilliers, L. Morris, J. P. Moore, P. J. Maddon, and W. C. Olson. 2001. Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Virol. 75:579-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trkola, A., J. Matthews, C. Gordon, T. Ketas, and J. P. Moore. 1999. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J. Virol. 73:8966-8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsamis, F., S. Gavrilov, F. Kajumo, C. Seibert, S. Kuhmann, T. Ketas, A. Trkola, A. Palani, J. W. Clader, J. R. Tagat, S. McCombie, B. Baroudy, J. P. Moore, T. P. Sakmar, and T. Dragic. 2003. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 77:5201-5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watson, C., S. Jenkinson, W. Kazmierski, and T. Kenakin. 2005. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol. Pharmacol. 67:1268-1282. [DOI] [PubMed] [Google Scholar]
- 49.Wild, C., T. Oas, C. McDanal, D. Bolognesi, and T. Matthews. 1992. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 89:10537-10541. [DOI] [PMC free article] [PubMed] [Google Scholar]