

Abstract
Bacteria residing in sediments have key functions in the marine food web. However, it has been difficult to correlate the identity and activity of bacteria in sediments due to lack of appropriate methods beyond cultivation-based techniques. Our aim was to use a combination of molecular approaches, bromodeoxyuridine incorporation and immunocapture, terminal restriction fragment length polymorphism, and cloning and sequencing of 16S rRNA genes to assess the composition of growing bacteria in Baltic Sea sediments. The study site was a highly polluted area off the Swedish coast. The sediments were sampled in two consecutive years, before and after remediation, by dredging of the top sediments. Levels of polyaromatic hydrocarbons (PAHs), mercury, and polychlorinated biphenyls were dramatically reduced as a result of the cleanup project. The compositions of growing members of the communities were significantly different at the two sampling periods. In particular, members from the class Deltaproteobacteria and genus Spirochaeta were more dominant before dredging, but members of the classes Gammaproteobacteria and the Flavobacteria represented the most dominant growing populations after dredging. We also cultivated isolates from the polluted sediments that could transform the model PAH compound, phenanthrene. Some of these isolates were confirmed as dominant growing populations by the molecular methods as well. This suite of methods enabled us to link the identity and activity of the members of the sediment communities.
Bacterial communities in sediments are instrumental in the marine food web, where they are responsible for recycling of nutrients and degradation of pollutants (32, 35). However, prior to the advent of molecular approaches, not much was known about the composition of bacteria in marine sediments other than the small fraction of the community that could be cultivated. We recently used a molecular approach, terminal restriction fragment length polymorphism (T-RFLP), to investigate microbial communities in sediments along a Swedish coastal region of the Baltic Sea and found that the bacterial and archaeal communities responded differently to pollutant levels (12). Although T-RFLP is useful for providing a fingerprint of the composition of the dominant members of a community, the presence of 16S rRNA genes corresponding to particular organisms does not necessary imply that those organisms are active. However, due to the lack of sufficient methodology, it has been difficult to correlate the identity and activity of microorganisms in environmental samples.
The classical approach for identification of viable microorganisms in environmental samples, including sediment, is plate counting on agar medium. In the Baltic water column, a large number of bacteria have been successfully isolated (20, 25, 37). However, the number of microbially diverse populations in sediments is predicted to be much higher than that in the water column (45), and the composition of viable microorganisms in Baltic Sea sediments is still largely unexplored.
Approaches for studying activity of microbial communities in sediments include measurements of net rates of biochemical processes (e.g., O2 uptake, CO2 production, ATP production, etc.). Another approach for assessing bacterial community productivity in marine environments is the measurement of microbial incorporation of radioactively labeled thymidine into newly synthesized DNA (14, 15). However, this method cannot discriminate the relative contributions of different microbial taxa to community productivity.
In recent years, there have been developments in molecular approaches for linking function or activity with identity. For example, Mar-FISH is based on combination of uptake of radioactive substrates with fluorescent in situ hybridization (FISH) (24), and stable isotope probing can be combined with molecular fingerprinting approaches to link microbial identity (biomarker) and activity (isotope assimilation) (38). Another promising molecular approach that has recently been used for identification of growing cells in environmental samples is based on incorporation of the thymidine analogue bromodeoxyuridine (BrdU) into the DNA of cells during DNA replication (3, 46). The DNA with incorporated BrdU can be selectively extracted by immunocapture and analyzed by molecular fingerprinting techniques, such as T-RFLP, to determine the composition of the growing members of the community (3, 4, 21).
In this study, we used a combination of BrdU immunocapture and T-RFLP fingerprinting to assess the composition of growing bacteria in Baltic Sea sediments. We sampled sediments outside a paper mill located south of Västervik, on the southeast coast of Sweden. This site was the focus of a large Swedish remediation initiative from 2001 to 2003. Sediment samples were collected from the inner parts of the bay in the former industrial area where the Västervik paper mill operated between 1915 and 1980. The same site was sampled twice. The first sampling was in 2002, from an area of the bay where the sediments were highly polluted with polycyclic aromatic hydrocarbons (PAHs), polychlorinated biphenyls (PCBs), and mercury (Hg), which also were reported to leak to surrounding water, air, and soil environments (34). The second sampling was performed exactly 1 year later after removal of the top highly polluted sediment layers by dredging. When dredging was completed, only low levels of pollutants could be detected in the sediments.
This study was performed to monitor the community structures and to identify actively growing bacteria in surface sediments before and after the Västervik remediation project. The same region of the Baltic Sea was studied on two consecutive years, but with two drastically different environmental conditions due to removal of the surface polluted sediment layers. A combination of molecular approaches was used to examine the actively growing bacteria in these sediments.
MATERIALS AND METHODS
Sediment sampling.
Approximately 170,000 m3 of polluted benthic sediment was dredged from the bay of Örserumsviken, on the southeast coast of Sweden (57°43.5′N, 16°40.5′E), from June 2001 to September 2003 by the construction company Vägverket Produktion Syd (Hisingsbacka, Sweden). Triplicate sediment cores were collected using a Limnos sediment sampler (Limnos AB, Åbo, Finland) from four sites in Örserumsviken on 5 October 2002 and on 5 October 2003, before and after dredging of this region of the bay, respectively. The surface layers (0 to 2 cm) of the sediment cores were collected in sterile 50-ml Falcon tubes for analysis. Immediately after sampling, 1-ml portions of samples were supplemented with 2 mM BrdU (Roche, Mannheim, Germany) in 2-ml microcentrifuge tubes and incubated at 5°C (the in situ temperature in the sediment at the time of sampling) in the dark for 10 h. The BrdU-incubated samples and the rest of the sediment samples without added BrdU were frozen and stored at −80°C until DNA extraction was performed as described below.
Chemical analysis of pollutants.
The Swedish Environmental Institute (IVL; Gothenburg, Sweden) collected samples and performed all chemical analyses prior to dredging from 1999 to 2000 and after dredging in July 2003. To quantify PCBs, sediments were pretreated with concentrated sulfuric acid and purified using an alumina column (150 mesh, chromatographic grade, combusted at 400°C for 4 h and stored at 120°C before use) and eluted with pentane for removal of matrix interference. The purified extracts were concentrated to a final volume of 1.0 ml using a Kuderna Danish evaporator. Analysis was performed by gas chromatography on a Varian model 3800 (Palo Alto, CA), with an electron capture detector, which was equipped with a 50-m-long capillary column (CP-Sil 8 CB; Chrompack, Holland). PCB components were identified and quantified using internal and external standards (certified standard solution from Larodan-Sweden, NO813). For PAH quantification, sediment samples were purified in a silicate column and eluted with pentane as described above and then reextracted to an organic state, hexane, by additions of water. The purified extracts were then concentrated to 1 ml. PAH concentrations were analyzed using a mass spectrometer, Varian Saturn 2000 Ion-Trap (Palo Alto), equipped with a 25-m-long capillary column (CP-Sil 8 CB; Chrompack, Holland). PAH components were identified and quantified using internal and external standards (certified standard solution from Larodan-Sweden, SMR 1491). The total mercury concentration was determined by gas chromatography (6, 8). Before gas chromatography, samples were incubated with nitric acid during autoclaving. After reduction of the sediment samples with tin chloride, the mercury concentration was analyzed using a Perkin-Elmer 2100 flameless atomic absorption spectrophotometer (Perkin-Elmer, Norwalk, CT). The level of detection was 0.01 mg mercury/kg dry weight (DW) sediment.
DNA extraction and BrdU immunocapture.
DNA extractions were performed from each of the three separate, replicate sediment cores taken from the four sampling stations on the two sampling occasions. DNA was extracted from 1-ml portions of the upper sediment layers in each core that were either preincubated with BrdU as described above, or without BrdU, using a FastDNASPIN kit (for soil) following the manufacturer's instructions (Q-BIOgene, Carlsbad, CA). Immunochemical purification of BrdU-labeled DNA was performed by a modification of the methods described by Urbach et al. (46) and Jansson and coworkers (3, 4, 21). Monoclonal anti-BrdU antibodies (3 μl; Roche, Basel, Switzerland) were mixed at a 1:9 ratio with herring sperm DNA (1.25 mg ml−1 in phosphate-buffered saline [PBS]; Promega, Madison, WI) which had been denatured at 100°C for 5 min, quickly transferred to ice, and kept on ice for 5 min. The mixture was incubated for 1 h at room temperature. Magnetic beads (Dynabeads) coated with goat anti-mouse immunoglobulin G (DYNAL, Oslo, Norway) were washed three times with 1 mg ml−1 acetylated bovine serum albumin (BSA) in PBS buffer (PBS-BSA) using a magnetic particle concentrator (DYNAL) and resuspended in PBS-BSA to the initial concentration. Sediment DNA samples (1 μg total in 25 μl of PBS) were denatured (heated for 5 min at 100°C, quickly transferred to ice, and kept on ice for 5 min), mixed with 30 μl of the herring sperm-DNA antibody mixture, and incubated for 1 h in the dark at room temperature with constant agitation. The samples were mixed with 10-μl portions of Dynabeads, and the incubation was continued for an additional 1 h. To elute the BrdU-containing DNA fraction, 100 μl of 1.7 mM BrdU (in PBS-BSA) was added, and the samples were incubated for 1 h in the dark at room temperature with constant agitation. The DNA was separated from the beads with the magnetic particle concentrator, isolated by two rounds of ethanol precipitation, and dissolved in 20 μl of sterile distilled water.
The efficacy of BrdU immunocapture was confirmed by comparing visible bands on agarose gels after DNA extraction, immunocapture, and PCR amplification with control sediment to which no BrdU was added and no PCR-amplified product was visible (data not shown).
T-RFLP.
16S rRNA genes (BrdU-labeled or nonlabeled DNA) were PCR amplified from all DNA extracts from sediment samples (see above) using the following bacterial primers, as previously described (12): fD1-Hex (AGAGTTTGATCMTGGCTCAG) (48), labeled at the 5′ end with hexachlorofluorescein; and 926r (CCGTCAATTCCTTTRAGTTT) (31). The PCR products (12 μl, approximately 20 ng DNA) were digested for 2 h (50 μl total volume) using HaeIII restriction enzyme (Promega), and the fragments were separated by electrophoresis on an ABI PRISM 377 DNA sequencer (Applied Biosystems, Foster City, CA). The lengths of the fluorescent terminal restriction fragments (TRFs) were determined using GeneScan, version 3.1, analysis software (Applied Biosystems), and their relative peak areas were determined by dividing the individual TRF area by the total area of peaks within the thresholds of 60 to 490 bp. Only peaks with relative fluorescence intensities over 50 U and that contributed to the total community abundance at 1.5% or more were included in the later analyses.
Cloning.
Cloning and sequencing of 16S rRNA genes PCR amplified from BrdU-extracted DNA were performed to enable possible species identification of TRFs of interest in the T-RFLP profiles. The same PCR products from DNA extracts used for T-RFLP were used for cloning so that the results could be directly compared. The PCR products from the three replicates and four sampling sites at each sampling time were pooled since the T-RFLP results showed that there was minimal variation between these samples, resulting in two samples corresponding to DNA from actively growing community members before and after dredging, respectively. The amplified, approximately 900-bp BrdU-incubated 16S rRNA gene fragments (total of 5 ng) were cloned in separate reactions using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Cloned inserts were screened by RFLP using the r926 and fD1 primers and amplification conditions described. The PCR products were digested with HaeIII and RsaI (Promega) in individual reactions and then pooled together. The resulting restriction fragments were separated by gel electrophoresis in 2.2% agarose gels. Gels were stained with ethidium bromide (1 mg liter−1). In total, 220 clones before dredging and 210 clones after dredging were screened. The relative abundance of each individual phylotype, which was represented by a unique restriction pattern in RFLP analysis, was calculated by dividing the numbers of individual clone types by the total number of clones that were analyzed.
Isolation and identification of PAH-degrading bacteria.
One hundred sixty milliliters of frozen (−80°C) polluted surface sediments that were collected in four 50-ml Falcon tubes was thawed at 4°C overnight. These sediment samples were pooled and homogenized by gentle stirring and spiked with 10 ppm phenanthrene as described by Brinch and colleagues (7). Three replicates, each containing 50 ml of the spiked sediment slurry samples, were poured into 200-ml Pyrex flasks with a tightened screw-cap top and incubated in darkness at 5°C for 7 days. After 7 days of incubation, samples (1 g) were homogenized in 10 ml PBS buffer and serially diluted in PBS before plating onto 10% tryptic soy agar media (Difco Laboratories, Detroit, MI). After inoculation, the plates were covered with a thin layer of phenanthrene by the sublimation method as previously described (1). After 12 h of incubation at 5°C in the dark, colonies and surrounding agar were assessed for color changes and clearing zones under UV light illumination indicative of phenanthrene degradation (26). Potentially positive colonies were also tested for catechol dioxygenase activity by spraying 0.5 M catechol solution over the plates and assessing eventual color change of colonies or/and agar medium, as previously described (50).
Sequencing and phylogenetic analysis.
16S rRNA genes of isolates cultivated from the sediment samples and from one clone of each RFLP group in each sample were partially sequenced on an Applied Biosystems International PRISM 377 DNA sequencer using the fD1 and r926 primers described above. Sequences were compared to sequences in the GenBank database by using the blastn software (2) and also the BLAST software from NCBI's Map Viewer to find the closest relatives. Multiple sequence alignments were performed with the MUSCLE software, version 3.5, available at http://www.drive5.com/muscle (11). Maximum likelihood trees (13) were calculated using the software PHYML version 2.4 (19). One thousand bootstrap resamplings were analyzed. Trees were edited in BBEdit, version 6.1 (Bare Bones Software, Inc., Bedford, MA), and drawn in NJplot (36). Reference 16S rRNA gene sequences derived from NCBI or from bacteria with completely sequenced genomes were downloaded from NCBI's Map Viewer. All 16S rRNA gene sequences were also tested for chimeras in Chimera Check, version 2.7, Ribosomal Database Project II (9). Chimeric 16S rRNA gene sequences that were detected within the data set were discarded. Sequences with 97% sequence similarity or more were considered to belong to the same species as the currently deposited sequences in the database. This takes into account microvariations, which may be induced by PCR and cloning (42), as well as variations between 16S rRNA gene copies, which usually differ by 1% or less (28).
Statistical analyses.
Correspondence analysis (CA) (30) was performed on the T-RFLP data resulting from three DNA extractions from each of three sediment cores taken at each sampling site. T-RFLP data from BrdU-incubated and -nonincubated samples were prepared in the same matrix and analyzed using ADE-4 software (44). TRF data were entered according to TRF size (bp) and relative abundance. TRF peaks were clustered together in the range of ±1.0 bp. The statistical significance (P < 0.001) of clustering patterns in CA plots was determined by one-way analysis of variance (ANOVA) using Prism 4 software (GraphPad Software, Inc., San Diego, CA).
Completeness of clone libraries was calculated by using coverage (C) values from the equation C = 1 − (n1/N) × 100 (18), where n is the number of unique clones and N is the total number of clones examined.
Nucleotide sequence accession number.
The nucleotide sequences have been deposited in the EMBL nucleotide sequence database under accession no. AM167975 to AM167991, AM167995, and AM167997 to AM168001 for clones and AM260538 to AM260540 and AM286417 to AM286420 for isolates.
RESULTS
Environmental variables before and after dredging.
PAH, PCB, and mercury concentrations in benthic sediments decreased after dredging of the bay of Örserumsviken (Fig. 1). The average PAH concentration for the sampling stations before dredging was 1.7 mg/kg (DW) sediment, whereas after dredging it was below the detection limit. PCB and mercury concentrations also decreased dramatically from 2.3 to 0.1 and 5.to 0.1 mg/kg (DW) sediment, respectively.
FIG. 1.

Pollutant concentration in DW sediment from Örserumsviken, Västervik, Sweden, before (gray bars) and after (black bars) dredging was commenced.
CA of 16S rRNA gene profiles.
According to CA of 16S rRNA gene profiles derived from T-RFLP data, the microbial community structures at the different sampling sites in the bay were similar and clustered together for a given sampling date (Fig. 2). We expected to see differences in the community patterns before and after dredging of the sediments considering the drastic changes in the environmental parameters, and this was reflected in the CA plots by distinctly separate clustering of the communities before and after dredging. In addition, there were distinct differences in the actively growing communities (BrdU-incorporated 16S rRNA genes) and directly extracted community DNA, which we here define as the “total” community, keeping in mind that only dominant members of the community are represented and that there are some biases in the molecular methods used. The total and actively growing communities clustered separately (P < 0.001), even on the same sampling date. The community profiles were highly reproducible, as confirmed by basically the same distribution patterns for the independent replicates taken at the different sites. Our results confirmed previous studies that have demonstrated that T-RFLP is a highly reproducible and robust technique that yields high-quality fingerprints consisting of fragments of precise size (33). We also calculated eigenvalues from the CA plots. The eigenvalues describe how much variation in the data set can be explained by environmental factors. The eigenvalues in this study were 37% and 32%, for the first and second ordination axis, respectively, indicating that 69% of the variation in the T-RFLP data sets could be explained by unknown environmental factors and not by random events. The first ordination axis in the CA plot statistically describes which environmental factor or factors have the primary influence on the data set, whereas the second ordination axis describes those variables that are still significantly impacting the data set, but to a lesser extent.
FIG. 2.

Ordination diagram from CA of T-RFLP data derived from surface sediment (0 to 2 cm) of triplicate sediment cores collected from Örserumsviken before and after dredging. Numbers correspond to sampling sites. Color labeling: red, actively growing bacterial community before dredging; blue, actively growing bacterial community after dredging; green, total bacterial community before dredging; black, total bacterial community after dredging. Distances between the profiles that clustered together were significant (P < 0.001). Eigenvalues for first and second ordination axes were 37% and 32%, respectively.
Identification of growing bacteria.
The PCR products from BrdU-immunocaptured DNA were cloned, resulting in approximately 1,500 clones from each sampling occasion. A subset of clones was screened by RFLP using HaeIII and RsaI in restriction digestions, and 13 and 10 unique restriction patterns were obtained before and after dredging, respectively. In total, 430 clones were analyzed from both sampling occasions and 23 unique sequences were defined (see Fig. 4), indicating a low number of actively growing community members. Coverage was 100% for both clone libraries before and after dredging, respectively. This implies that all phylotypes present in clone libraries were detected. All sequences resulting from clones in this study were named with the prefix VAS (abbreviation for Västervik).
FIG. 4.

Phylogenetic classification of sequenced BrdU-incorporated16S rRNA genes. Clones sequenced in this study have the abbreviation VAS printed in plain and bold text, representing samples taken before and after dredging, respectively. Alignments of approximately 900 characters were used to generate the tree. The phenogram was drawn by the maximum likelihood method, and bootstrap values are presented as percentage of 1,000 resamplings. “U” indicates the bootstrap value is lower than 50%. The number of clones of individual sequences in each clone library is indicated inside parentheses. The predicted length of HaeIII-generated TRFs, based on in silico restriction of clone sequences, is indicated to the right of the brackets. Uncl., uncultured.
Several differences were seen in the identities of the actively growing bacteria in the sediment before and after dredging. Before dredging, Deltaproteobacteria were the most prevalent growing bacteria represented in the clone library, but no representatives of this group were found after dredging. Also, a member from the Spirochaeta group was growing and detected before dredging (Fig. 3). After dredging, Gammaproteobacteria were the most prevalent growing members of the community, although these were much less prevalent prior to dredging (Fig. 3). Members from the Flavobacteria were also detected as growing members of the community after dredging. Cyanobacteria were growing in the sediments both before and after dredging with relative abundances of 12% and 18% at the two sampling occasions, respectively. Actinobacteria were also represented in both clone libraries, with relative abundances of 12% and 15% before and after dredging, respectively, indicating that members of this group were growing under both conditions.
FIG. 3.
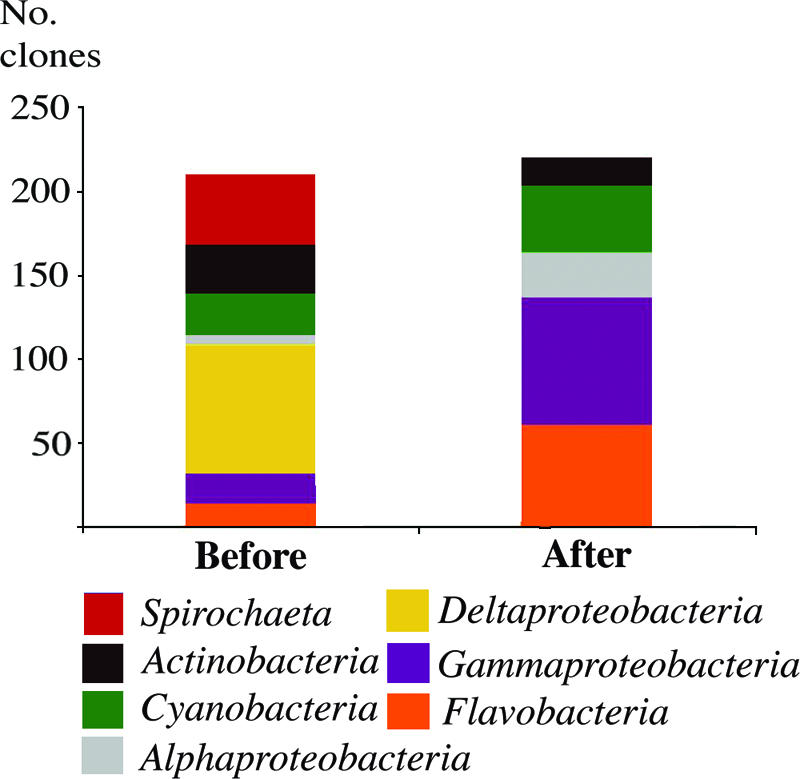
Clone frequency of actively growing bacterial community members from sediment collected from Öreserumsviken before and after dredging. Color labeling of bacterial genus or class: red, Spirochaeta; black, Actinobacteria; green, Cyanobacteria; gray, Alphaproteobacteria; yellow, Deltaproteobacteria; blue, Gammaproteobacteria; orange, Flavobacteria.
Matching of active clones to T-RFLP data.
Approximately 50% of the cloned sequences from the BrdU-extracted DNA could be matched to specific TRFs in the T-RFLP profiles (Fig. 4, and Table 1). Since these data were obtained from the same DNA extracts, the results could be directly compared and the sequence information could be used to provide possible identities of growing bacterial populations. In turn, the T-RFLP data could be used to determine the relative abundance of specific species identified by cloning and sequencing. Some of the most abundant sequences had similar relative abundance values using both the T-RFLP and cloning approaches: for example, before dredging, VAS6, a member of the Corynebacterium genus, represented by fragment length 73, a Spirochaeta sp. sequence, VAS4, represented by fragment length 222, and an unclassified member of Gammaproteobacteria, VAS7, represented by fragment length 224; and after dredging, the unclassified actinobacterium, VAS21, represented by fragment length 173 (Fig. 4. and Table 1). Some sequences could not be correlated to the T-RFLP profiles, and in most cases this was probably due to them having terminal restriction sites outside the investigated 16S rRNA gene sequence delimited by T-RFLP, i.e., 60 to 500 bp. For example, VAS1, a sequence which had its closest match to Pseudomonas sp. (accession no. AF521651), had its terminal HaeIII restriction site at a location 39 bp from the 5′ end of the gene.
TABLE 1.
Relative abundances of 16S rRNA gene sequences of actively growing bacteria represented in T-RFLP profiles and clone libraries
| No. of bp before/after dredging | Closest match (NCBI accession no.) | % Identity | Relative abundance (%)
|
|
|---|---|---|---|---|
| T-RFLP profilea | Clone libraryb | |||
| Before | ||||
| 73 | VAS6, Corynebacterium sp. (AY592796) | 100 | 8.5 | 10.0 |
| 122 | VAS10, Prochlorococcus sp. (NC005072) | 98 | 3.8 | 4.8 |
| 194 | VAS9, Nitrobacter sp. (CP000115) | 97 | 1.8 | 2.9 |
| 215 | VAS14, Arthrobacter sp. (DQ341426) | 99 | 5.5 | 0.5 |
| 222 | VAS4, Spirochaeta sp. (AB121100) | 99 | 14.0 | 10.5 |
| 224 | VAS7, γ-proteobacterium (AY539822)c | 99 | 10.4 | 14.8 |
| 319 | VAS3, Flavobacterium sp. (AM167564) | 98 | 3.2 | 3.3 |
| 373 | VAS11, uncultured Cyanobacterium (AP008231) | 95 | 1.0 | 4.8 |
| After | ||||
| 71 | VAS25, Flavobacterium sp. (DQ007435) | 98 | 5.8 | 4.5 |
| 161 | VAS15, α-proteobacterium (DQ336968) | 99 | 1.6 | 5.9 |
| 173 | VAS21, Actinobacterium (UAC575535) | 97 | 5.1 | 6.8 |
| 203 | VAS23, uncultured bacterium (AB234247) | 99 | 2.4 | 4.5 |
| 293 | VAS16, Sphingomonas sp. (AF385529) | 100 | 14.6 | 17.8 |
Data represent the average relative abundance values of TRFs from three replicate T-RFLP profiles. The standard deviation for TRF replicates was ±0.4 to 1.2%.
rRNA gene sequences were digested in silico using the recognition site for restriction enzyme HaeIII.
This bacterium also was cultivated on PAH agar medium.
Isolation of microorganisms.
Bacterial isolates (2,400 colonies/ml sediment slurry) were cultivated from the polluted sediments on agar medium containing phenanthrene as a model PAH compound. The isolates were obtained from frozen and thawed samples, which probably reduced the number of isolates obtained. Nevertheless, after incubation, clear zones were observed around approximately 200 of the colonies under UV light illumination, suggesting that those isolates were capable of phenanthrene transformation. The 16S rRNA genes of 70 representative colonies were analyzed by RFLP and screened for catechol dioxygenase activity. Four isolates accumulated an orange/brown color in the colonies and in the surrounding agar after catechol application, indicative of catechol transformation. Their 16S rRNA genes were sequenced and had the following matches: Pseudomonas sp., accession no. AF521651 (99% sequence similarity [12/70 colonies]); a γ-proteobacterium, accession no. AY539822 (99% sequence similarity [5/70 colonies]); Exiguobacterium oxidotolerans, accession no. AB105164 (100% sequence similarity [14/70 colonies]), and Arthrobacter sp., accession no. AJ967024 (99% sequence similarity [12/70 colonies]). The pseudomonad and the gammaproteobacterium were also identified in the clone library of the active community in the polluted sediments (clones VAS1 and VAS7, each contributing with 5/70 and 7/70 colonies, respectively), indicating in these cases good matches between the cultivation-based and molecular approaches for assessing growing cells. However, no representatives of Exiguobacterium oxidotolerans or Arthrobacter sp. were present in the clone library. The remaining five isolates that were isolated from phenanthrene-incubated agar plates showed no catechol dioxygenase activity. Their 16S rRNA gene sequences had the following matches: Serratia sp., accession no. AY689059 (98% sequence similarity [4% of relative colony abundance]); a γ-proteobacterium, accession no. DQ226202 (96% sequence similarity [10% of relative colony abundance]); Pseudomonas putida, accession no. DQ178233 (98% sequence similarity [7% of relative colony abundance]); Stenotrophomonas maltophilia, accession no. AY581129 (96% sequence similarity [7% of relative colony abundance]), and Arctic Sea Ice bacterium, accession no. AF468434 (97% sequence similarity [7% of relative colony abundance]).
Community analyses.
The numbers of TRFs detected in T-RFLP electropherograms were used to gauge the relative richness of the bacterial communities, although one should keep in mind that only dominant members of the community are detected using this approach and that there is some bias in the methodology used. Before dredging, the numbers of TRFs were 15 and 60 for actively growing and for total community members, respectively. After dredging, the numbers of TRFs were 14 and 64 for actively growing and for total community members, respectively. There were therefore considerably fewer TRFs represented in the growing community compared to the total community at both sampling periods. This low number of growing bacteria was also reflected by comparison to the low numbers of unique sequences in clone libraries created from BrdU-labeled 16S rRNA genes, with only 13 and 10 unique sequences obtained at the first and second sampling years, respectively. Hence, approximately the same numbers of actively growing bacteria were detected using the two molecular approaches, T-RFLP and cloning and sequencing.
DISCUSSION
Here we studied the community structures of actively growing bacteria in a specific region of the Baltic Sea in two consecutive years under two highly different conditions, before and after dredging of a polluted bay. This is the first study where the molecular approaches BrdU immunocapture coupled to T-RFLP and cloning and sequencing were applied to determine the composition of actively growing bacteria in a marine environment. The T-RFLP data were used for statistical comparison of the communities, while the clone libraries provided information on the possible identities of species represented in the T-RFLP profiles. These data were further supported by 16S rRNA gene sequences obtained from isolates.
Based on results from clone libraries and T-RFLP analysis of 16S rRNA gene fragments, we found that the number of actively growing bacterial communities was approximately 75% lower than for the dominant members of the total communities. Also, none of the TRFs in the BrdU-extracted DNA were present in electropherograms of directly extracted DNA. The low number of 16S rRNA genes in the clone libraries was striking, since screening of approximately 200 clones per library (Fig. 4) was sufficient to reach complete coverage for each library (i.e., no new 16S rRNA gene sequences could be detected). One possible explanation for the low numbers could be insufficient BrdU incorporation by all growing cells in the community. In previous studies, it was found that some anaerobic bacteria did not incorporate the similar molecule, thymidine, when it was added exogenously during DNA replication (17, 22, 49). However, in our study, representatives of the Deltaproteobacteria with close phylogenetic matches to anaerobes were capable of BrdU uptake (Fig. 4). Also, in this study, we probably selected for the fast-growing bacterial community members since sediments were incubated with BrdU for only 10 h. However, this was done to limit the ability of the bacteria to use BrdU as a carbon source, as previously observed to be problematic for radioactively labeled thymidine during production and growth rate measurements (40). In a separate study, we confirmed that BrdU was not being used as a carbon source in these sediments (Edlund and Jansson, unpublished). Interestingly, we recently found that there were very few growing bacteria detected in soil using the BrdU immunocapture approach (21), reinforcing the hypothesis that most bacteria in the environment persist in a dormant, nongrowing physiological state.
We observed significant differences in the compositions of both the actively growing communities and the total/dominant members of the communities at the two different sampling periods, and there are several possible reasons for these observations. For example, the difference could be due to removal of pollutants that repressed some bacterial populations (5, 16) or that conversely served as carbon sources for specific populations (10, 29), or the difference could be due to the selection of resistant (e.g., mercury resistant) populations before dredging. Another explanation for the significant differences in microbial community structures before and after dredging could be due to succession since the upper sediment layers were completely removed. Therefore, the community profiles representing sediment samples after dredging may reflect a new niche that was recolonized by immigration and recruitment (39) of Baltic bacteria from the water column and the surrounding environment.
Sequences corresponding to Deltaproteobacteria were only obtained in the clone library from the polluted sediments, supporting previous findings that this group is ubiquitous in organic-rich marine sediments (23). Two sequences, VAS2 and VAS5, belonging to the purple sulfur bacteria (Chromatiaceae), known to require anoxic conditions, hydrogen sulfide, and illumination, were obtained from sediments before dredging, suggesting that that these photosynthetic sulfide oxidizers were also actively growing in the polluted sediments. However, these species were probably metabolizing sulfur and carbohydrate resources that were stored inside their cells, as previously described by Stal and Moezelaar (43), during the BrdU incubation period since the samples were incubated in darkness.
Members of the Gammaproteobacteria were growing in the sediments on both sampling occasions (Fig. 3). However, only one operational taxonomic unit (OTU), with closest identity to a 16S rRNA gene sequence with NCBI accession no. AY539822, dominated the community after dredging while four OTUs were present before dredging (Fig. 4). In previous studies concerning PAH-polluted environments, members of the Gammaproteobacteria were also shown to be metabolically active and capable of degrading low-molecular-weight PAHs (27, 41, 47). In line with these findings, we identified a Pseudomonas sp. (accession no. AF521651) and an unnamed species belonging to Gammaproteobacteria (accession no. AY539822) by cloning DNA from cells growing in the polluted sediments. We also cultivated a matching Pseudomonas sp. and an isolate of Gammaproteobacteria that were capable of phenanthrene transformation. Another isolate in this study, Exiguobacterium oxidotolerans (accession no. AB105164), was able to transform phenanthrene but was not represented in the clone library. However, we previously isolated a similar strain that had 99% 16S rRNA sequence identity to this bacterium in sediments from a different region of the Baltic Sea along a eutrophication gradient (12), indicating that this species may be common in coastal Baltic Sea surface sediments.
Several of the 16S rRNA gene sequences from the clone libraries that could be matched to specific TRFs had similar relative abundance values in T-RFLP profiles and in clone libraries (Table 1), demonstrating the utility of this combination of culture-independent approaches. The T-RFLP data enabled us to determine which of the phylogroups were most dominant, whereas the sequence information enabled us to ascertain their identities. Thus, a combination of approaches is useful to obtain more information about the identities of key microbial species in complex environmental samples, rather than relying on one technique alone. This study may serve as a good base for future studies of key bacterial species involved in recycling nutrients and in degradation of pollutants in marine sediments.
Acknowledgments
We thank Christer Ramström and Christer Hermansson at Västervik's County Environment and Building Office, Västervik, for help with background information about the sampling area and for providing us with samples. We also thank Sven-Erik Gustavsson, AB Shakt & Transport, Växjö, for supplying us with sampling equipment, a boat, and assistance with sampling. We also thank Magnus Rosenquist at the Swedish Agricultural University, Uppsala, and Krishanu Mukherjee, Karolinska Institute, for assistance with bioinformatics and Sara Sjöling at Södertörn University College, Huddinge, for critical reading of the manuscript.
This project was financed by The Baltic Sea Foundation.
Footnotes
Published ahead of print on 1 September 2006.
REFERENCES
- 1.Alley, J. F., and L. R. Brown. 2000. Use of sublimation to prepare solid microbial media with water-insoluble substrates. Appl. Environ. Microbiol. 66:439-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schaeffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artursson, V., and J. K. Jansson. 2003. Use of bromodeoxyuridine immunocapture to identify active bacteria associated with arbuscular mycorrhizal hyphae. Appl. Environ. Microbiol. 69:6208-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artursson, V., R. D. Finlay, and J. K. Jansson. 2005. Combined bromodeoxyuridine immunocapture and terminal-restriction fragment length polymorphism analysis highlights differences in the active soil bacterial metagenome due to Glomus mosseae inoculation or plant species. Environ. Microbiol. 7:1952-1966. [DOI] [PubMed] [Google Scholar]
- 5.Bååth, E. 1989. Effects of heavy metals in soil on microbial processes and populations. Water Air Soil Pollut. 47:335-379. [Google Scholar]
- 6.Bloom, N. S., and W. F. Fitzgerald. 1988. Determination of volatile mercury species at the picogram level by low temperature gas chromatography with cold-vapor atomic fluorescence detection. Anal. Chim. Acta 208:151-161. [Google Scholar]
- 7.Brinch, U. C., F. Ekelund, and C. S. Jacobsen. 2002. Method for spiking soil samples with organic compounds. Appl. Environ. Microbiol. 68:1808-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brosset, C. 1997. The behavior of mercury in physical environments. Water Air Soil Pollut. 34:145-166. [Google Scholar]
- 9.Cole, J. R., B. Chai, R. J. Farris, Q. Wang, S. A. Kulam, and D. M. McGarrell. 2003. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 31:442-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denaro, R., G. D. Auria, G. Di Marco, M. Genovese, M. Troussellier, M. M. Yakimov, and L. Giuliano. 2005. Assessing terminal restriction fragment length polymorphism suitability for the description of bacterial community structure and dynamics in hydro-carbon marine environments. Environ. Microbiol. 7:78-87. [DOI] [PubMed] [Google Scholar]
- 11.Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edlund, A., T. Soule, S. Sjöling, and J. K. Jansson. 2006. Microbial community structure in polluted Baltic Sea sediments. Environ. Microbiol. 8:223-232. [DOI] [PubMed] [Google Scholar]
- 13.Felsenstein, J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17:368-376. [DOI] [PubMed] [Google Scholar]
- 14.Fuhrman, J. A., and F. Azam. 1980. Bacterioplankton secondary production estimates for coastal waters of Brittish Columbia, Antarctica, and California. Appl. Environ. Microbiol. 39:1085-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuhrman, J. A., and F. Azam. 1982. Thymidine incorporation as a measure of heterotrophic bacterioplankton production in marine surface waters: evaluation and field results. Mar. Biol. 66:109-120. [Google Scholar]
- 16.Giller, K. E., E. Witter, and S. P. McGrath. 1998. Toxicity of heavy metals to microorganisms and microbial processes in agricultural soils: a review. Soil Biol. Biochem. 30:1389-1414. [Google Scholar]
- 17.Gilmour, C. C., M. E. Leavitt, and M. P. Shiaris. 1990. Evidence against incorporation of exogenous thymidine by sulfate-reducing bacteria. Limnol. Oceanogr. 35:1401-1409. [Google Scholar]
- 18.Good, I. J. 1953. The population frequencies of species and the estimation of population parameters. Biometrika 40:237-264. [Google Scholar]
- 19.Guindon, S., F. Lethiec, P. Duroux, and O. Gascuel. 2005. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33:557-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hagström, Å., J. Pinhassi, and U. L. Zweifel. 2000. Biogeographical diversity among marine bacterioplankton. Aquat. Microbiol. Ecol. 21:231-244. [Google Scholar]
- 21.Hjort, K., A. Lembke, A. Speksnijder, K. Smalla, and J. K. Jansson. Community structure of actively growing bacterial populations in plant pathogen suppressive soil. Microb. Ecol., in press. [DOI] [PubMed]
- 22.Johnstone, B. H., and R. D. Jones. 1989. A study on the lack of (methyl-3H) thymidine uptake and incorporation by chemolithotrophic bacteria. Microb. Ecol. 18:73-77. [DOI] [PubMed] [Google Scholar]
- 23.Jørgensen, B. B. 1977. The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark). Limnol. Oceanogr. 22:814-832. [Google Scholar]
- 24.Kindaichi, T., T. Ito, and S. Okabe. 2004. Ecophysiological interaction between nitrifying bacteria and heterotrophic bacteria in autotrophic nitrifying biofilms as determined by microautoradiography-fluorescence in situ hybridization. Appl. Environ. Microbiol. 70:1641-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kisand, V., C. Rocio, and J. Wikner. 2002. Phylogeny of culturable estuarine bacteria catabolizing riverine organic matter in the northern Baltic Sea. Appl. Environ. Microbiol. 61:379-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiyohara, H., K. Nagao, and K. Yana. 1982. Rapid screen for bacteria degrading water-insoluble, solid hydrocarbons on agar plates. Appl. Environ. Microbiol. 43:454-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu, T., T. Omori, and T. Kodama. 1993. Microbial degradation of the polycyclic aromatic hydrocarbons acenaphthene and acenaphthylene by a pure bacterial culture. Biosci. Biotechnol. Biochem. 57:864-865. [DOI] [PubMed] [Google Scholar]
- 28.Kroes, I. P., W. Lepp, and D. A. Relman. 1999. Bacterial diversity within the human subgingival crevice. Proc. Natl. Acad. Sci. USA 96:14547-14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindstrom, J. E., R. P. Barry, and J. F. Braddock. 1999. Long-term effects on microbial communities after a subarctic oil spill. Soil Biol. Biochem. 31:1677-1689. [Google Scholar]
- 30.Müller-Schneider, T. 1994. The visualization of structural change by means of correspondence analysis, p. 267-279. In M. Greenacre and J. Blasius (ed.), Correspondence analysis in the social sciences. Academic Press, London, United Kingdom.
- 31.Muyzer, G., A. Teske, C. O. Wirsen, and H. W. Jannasch. 1995. Phylogenetic relationship of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 164:162-172. [DOI] [PubMed] [Google Scholar]
- 32.Osborn, A. M., K. D. Bruce, P. Strike, and D. A. Ritchie. 1997. Distribution, diversity and evolution of the bacterial mercury resistance (mer) operon. FEMS Microbiol. Rev. 19:239-262. [DOI] [PubMed] [Google Scholar]
- 33.Osborn, A. M., E. R. B. Moore, and K. M. Timmis. 2000. An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ. Microbiol. 2:39-50. [DOI] [PubMed] [Google Scholar]
- 34.Palm, A., I. Wängberg, and E. Broström-Lundén. 2001. Kvicksilver och organiska miljögifter i Örserumsviken, IVL. Report no. B 1433. Swedish Environmental Research Institute Ltd., Stockholm, Sweden.
- 35.Pazlarová, J., K. Demnerová, M. Macková, and J. Burkhard. 1997. Analysis of PCB-degrading bacteria: physiological aspects. Appl. Microbiol. 24:334-336. [DOI] [PubMed] [Google Scholar]
- 36.Perrière, G., and M. Gouy. 1996. WWW-Query: an on-line retrieval system for biological sequence banks. Biochimie 78:364-369. [DOI] [PubMed] [Google Scholar]
- 37.Pinhassi, J., U. L. Zweifel, and Å. Hagström. 1997. Dominant marine bacterioplankton species found among colony-forming bacteria. Appl. Environ. Microbiol. 63:3359-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radajewski, S., I. R. McDonald, and J. C. Murrell. 2003. Stable-isotope probing of nucleic acids: a window to the function of uncultured microorganisms. Curr. Opin. Biotechnol. 14:296-302. [DOI] [PubMed] [Google Scholar]
- 39.Reice, S. R. 1994. Nonequilibrium determinants of biological community structure. Am. Sci. 82:424-435. [Google Scholar]
- 40.Robarts, R. D., and T. Zohary. 1993. Fact or fiction—bacterial growth rates and production as determined by [methyl-3H]thymidine? Adv. Microb. Ecol. 13:371-425. [Google Scholar]
- 41.Selifnov, S. A., M. Grifoll, J. E. Gurst, and P. J. Chapman. 1993. Isolation and characterization of (+)-1,1 a-dihydroxy-1-hydrofluoren-9-one formed by angular dioxygenation in the bacterial catabolism of fluorine. Biochem. Biophys. Res. Commun. 193:67-76. [DOI] [PubMed] [Google Scholar]
- 42.Speksnijder, A. G. C. L., G. A. Kowalchuk, S. De Jong, E. Kline, J. R. Stephen, and H. J. Laanbroek. 2001. Microvariation artifacts introduced by PCR and cloning of closely related 16S rRNA gene sequences. Appl. Environ. Microbiol. 67:469-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stal, L. J., and R. Moezelaar. 1997. Fermentation in cyanobacteria. FEMS Microbiol. Rev. 21:179-211. [Google Scholar]
- 44.Thioulouse, J., D. Chessel, S. Dolédec, and J. M. Olivier. 1997. ADE-4: a multivariate analysis and graphical display software. Stat. Comput. 7:75-83. [Google Scholar]
- 45.Torsvik, V., L. Øvreås, and T. F. Thingstad. 2002. Prokaryotic diversity—magnitude, dynamics, and controlling factors. Science 296:1064-1065. [DOI] [PubMed] [Google Scholar]
- 46.Urbach, E., K. L. Vergin, and S. J. Giovannoni. 1999. Immunochemical detection and isolation of DNA from metabolically active bacteria. Appl. Environ. Microbiol. 65:1207-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utkin, I. B., M. M. Yakimov, L. N. Matveeva, E. I. Kozlyak, I. S. Rogozhin, Z. G. Solomon, and A. M. Bezborodov. 1990. Catabolism of naphthalene and salicylate by Pseudomonas fluorescens. Folia Microbiol. 35:557-560. [Google Scholar]
- 48.Weisburg, W. G., S. M. Barns, D. A. Pelletier, and D. J. Lane. 1991. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173:697-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winding, A. 1992. [3H]thymidine incorporation to estimate growth rates of anaerobic bacterial strains. Appl. Environ. Microbiol. 58:2660-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zukowski, M. M., D. F. Gaffney, D. Speck, M. Kanuffmann, A. Findeli, A. Wisecup, and J. P. Lecocq. 1983. Chromogenic identification of genetic regulatory signals in Bacillus subtilis based on expression of a cloned Pseudomonas gene. Proc. Natl. Acad. Sci. USA 80:1101-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]