

Abstract
Phospholemman (PLM), a member of the FXYD family of small ion transport regulators, inhibits cardiac Na+/Ca2+ exchanger (NCX1). NCX1 is made up of N-terminal domain consisting of the 1st five transmembrane segments (residues 1-217), a large intracellular loop (residues 218-764), and a C-terminal domain comprising the last four transmembrane segments (residues 765-938). Using GST pulldown assay, we demonstrated that the intracellular loop, but not the N- or C-terminal transmembrane domains of NCX1, was associated with PLM. Further analysis using protein constructs of GST fused to various segments of the intracellular loop of NCX1 suggest that PLM bound to residues 218-371 and 508-764 but not 371-508. Split Na+/Ca2+ exchangers consisting of N- or C-terminal domains with different lengths of the intracellular loop were co-expressed with PLM in HEK293 cells that are devoid of endogenous PLM and NCX1. While expression of N-terminal but not C-terminal domain alone resulted in correct membrane targeting, co-expression of both N- and C-terminal domains was required for correct membrane targeting and functional exchange activity. NCX1 current measurements indicate that PLM decreased NCX1 current only when the split exchangers contained residues 218-358 of the intracellular loop. Co-immunoprecipitation experiments with PLM and split exchangers suggest that PLM associated with the N-terminal domain of NCX1 when it contained intracellular loop residues 218-358. TM43, a PLM mutant with its cytoplasmic tail truncated, did not co-immunoprecipitate with wild-type NCX1 when co-expressed in HEK293 cells, confirming little to no interaction between the transmembrane domains of PLM and NCX1. We conclude that PLM interacted with the intracellular loop of NCX1, most likely at residues 218-358.
Introduction
Phospholemman (PLM), a 72 amino acid sarcolemmal protein with a single transmembrane (TM) domain (1), is the 1st member of the FXYD gene family of regulators of ion transport (2). In addition to its well-known effects on modulation of Na+-K+-ATPase activity (3–8), PLM has recently been shown to co-localize and associate with the cardiac Na+/Ca2+ exchanger (NCX1), as well as regulate NCX1 activity (9–14). Studies using deletion mutants demonstrate that the cytoplasmic domain of PLM is required for its modulatory effects on NCX1 (11). In addition, serine63 and serine68 in the cytoplasmic domain of PLM are important residues, not only because they are the physiological substrates for protein kinases (PK) A (serine68) and C (serine63 and serine68)(15), but also because they are involved in the regulation of cardiac contractility and NCX1 activity (11,13). Specifically, PLM when phosphorylated at serine68, inhibits Na+/Ca2+ exchange (13).
The cardiac Na+/Ca2+ exchanger is a sarcolemmal protein that exchanges 3 Na+ ions for 1 Ca2+ ion (16). It is intimately involved in cardiac excitation-contraction coupling in that it regulates both Ca2+ influx and efflux during a contraction cycle (17). NCX1 consists of N-terminal domain comprising the 1st five TM segments, a large cytoplasmic loop, and a C-terminal domain consisting of the last four TM segments (18,19). The transport activity of NCX1 can be regulated by many factors including but not limited to phosphorylation, Ca2+, Na+, exchange inhibitory peptide (XIP) and phosphatidylinositol 4,5-bisphosphate (PIP2)(19). The large intracellular loop, while not required for ion transport activity (20), contains the high affinity Ca2+ binding domain (residues 371-508)(21), the endogenous XIP region (residues 219-238)(22) associated with regulation of NCX1 activity by Na+, Ca2+, and PIP2 (23,24), and the interaction site for endogenous XIP (residues 562-679)(25). It is not known which part of the NCX1 molecule is involved in regulation of its activity by PLM.
Here we demonstrated by GST pulldown assay that neither the N- nor the C-terminal TM domains of NCX1 associated with PLM. Rather, PLM associated with residues 218-371 and 508-764 but not residues 371-508 of the intracellular loop of NCX1. In HEK293 cells, co-expression of split Na+/Ca2+ exchangers consisting of N- and C-terminal TM domains with varying lengths of intracellular loop resulted in correct plasma membrane targeting and functional NCX1 current (INaCa). PLM inhibited INaCa only in split exchangers containing residues 218-358 of the intracellular loop. In addition, PLM co-immunoprecipitated with split exchangers containing intracellular loop residues 218-358. Deleting cytoplasmic tail of PLM abolished its association with wild-type NCX1. We conclude that PLM interacts with NCX1 at residues 218-358 of the intracellular loop.
Experimental Procedures
GST fusion expression constructs
The full-length cDNA of rat NCX1 inserted in pcDNA3.1 was obtained from Dr. Jonathon Lytton (University of Calgary) and used by us previously (26). Forward and reverse PCR primers were designed and constructed for amplifying discrete regions of the NCX1 open reading frame (ORF) in PCR reactions using Taq polymerase. Number designations for each NCX1 fragment is indicated as amino acid positions along the mature NCX1 protein sequence (1-939aa; Fig. 1) in the schematic presented below. In the schematic, linker sequences containing restriction enzyme recognition sites are underlined and their identities indicated following each sequence.
Fig. 1. Model of the Na+/Ca2+ exchanger and schematic for GST-NCX1Δ fusion protein constructs.
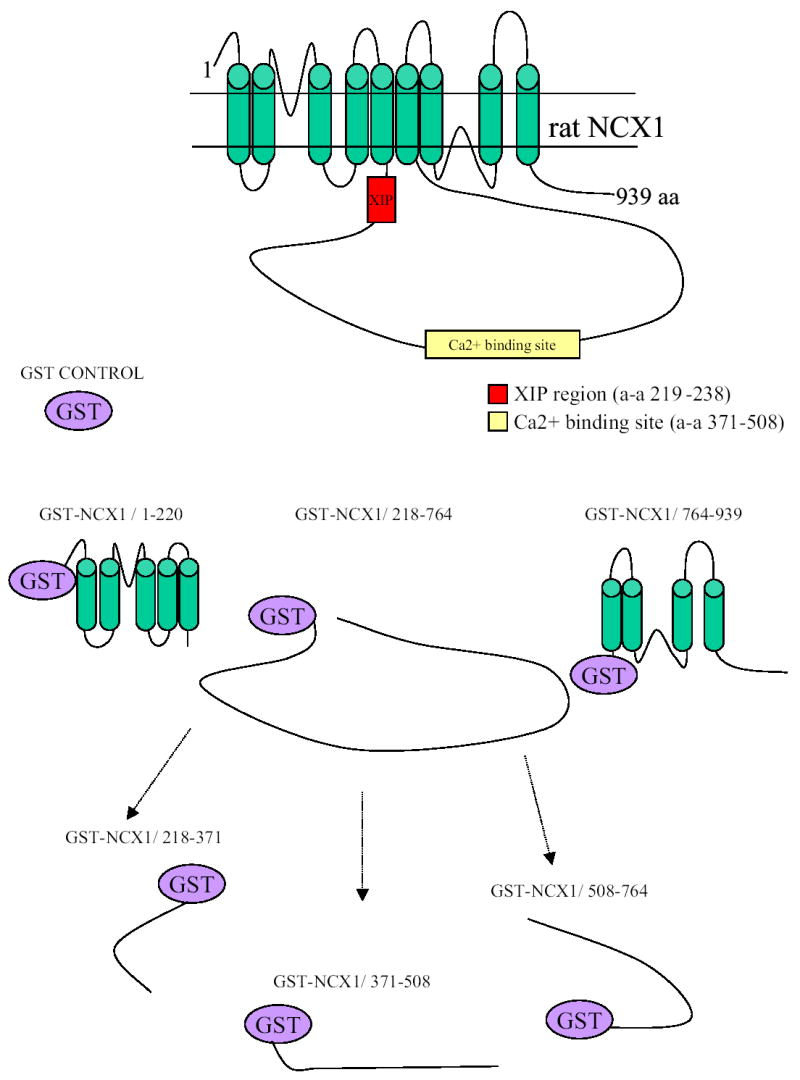
The exchanger is modeled to have 9 transmembrane (TM) segments and a large intracellular loop between TM5 and TM6 according to Philipson and Nicoll (19). The rat (939 amino acids) rather than dog (938 amino acids) NCX1 is represented, with the XIP region (residues 219-238) and high affinity Ca2+ binding domain (residues 371-508) highlighted. Schematics for GST-NCX1 fusion protein constructs are also shown. GST-NCX1/1-220, GST-NCX1/218-764, and GST-NCX1/764-939 are fusion constructs containing the N-terminal 1st 5 TM segments (predicted mw 48 kDa), intracellular loop (81 kDa) and C-terminal last 4 TM segments (43 kDa), respectively. GST-NCX1/218-371 (41 kDa), GST-NCX1/371-508 (40 kDa) and GST-NCX1/508-764 (52 kDa) are fusion constructs containing different regions of the intracellular loop.
NCX1/1-220
Forward 5′-d[AAGCTTGGATCCGATACAGAGGCAGAAACAGG]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTGAATTCCCGCCTTGCTGCGACCCA AGC]-3′ HindIII and EcoRI
NCX1/218-371
Forward 5′-d[AAGCTTGGATCCGCAAGGCGGCTTCTCTTT TAC]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTCTCGAGGACTGCGTCATTTTCAAC C]-3′ HindIII and XhoI
NCX1/371-508
Forward 5′-d[AAGCTTGGATCCGTCAGTAAGGTCTTC]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTCTCGAGCTCAAAAGTAAAGATGC]-3′ HindIII and XhoI
NCX1/508-764
Forward 5′-d[AAGCTTGGATCCGAGGAACCCGTGACTCAC G]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTCTCGAGCCAATATTCTGTAGGTGG] -3′ HindIII and XhoI
NCX1/218-764
Forward 5′-d[AAGCTTGGATCCGCAAGGCGGCTTCTCTTTTAC]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTCTCGAGCCAATATTCTGTAGGTGG] -3′ HindIII and XhoI
NCX1/764-939
Forward 5′-d[AAGCTTGGATCCTGGCTGGGCCTGCTTCAT TGTC]-3′ HindIII and BamHI
Reverse 5′-d[AAGCTTGTCGACTTAGAAGCCTTTTATGTG GC]-3′ HindIII and SalI
The newly amplified NCX1 PCR fragments were cloned in pCR2.1-TOPO (Invitrogen) according to the manufacturer’s protocol. DNA sequencing confirmed the sequence fidelity of fragments with the published cDNA sequence. The cDNA sequence was subsequently released from pCR2.1-TOPO and subcloned into pGEX-6P-1 (Pharmacia LKB Biotechnology) to generate pGEX-6P-1-NCX1 truncated mutants. After transformation of DH5α E. coli, clones with cDNA inserts in the appropriate orientation were selected by restriction endonuclease mapping.
The reading frame of the GST-NCX1 junction sequence was confirmed by DNA sequencing using Amersham Biosciences sequencing primers: 5′ pGEX sequencing primer (5′-d[GGGCTGGCAAGCCACGTTTGGTG]-3′) and 3′ pGEX sequencing primer (5′-d[CCGGGAGCTGCATGTGTCAGAGG]-3′). After transformation into BL21 E. coli, constructs were screened under the control of an IPTG inducible promoter for the expression of GST-NCX1 fusion proteins at the predicted MW.
GST fusion proteins
The expression and purification of the GST fusion proteins was performed as described with modification for purification of insoluble fusion proteins (27). BL21 E. coli carrying pGEX-6P-1 or the fusion protein expression constructs was grown in sterile 2X YTA medium (16g tryptone, 10g yeast extract and 5g NaCl per liter, pH 7.0) containing 0.2% glucose and 100 μg/ml ampicillin at 37°C until the bacterial cultures reached mid-logarithmic growth phase. IPTG (600 μM) was added, and the cultures incubated at 30°C for 4 h.
GST fusion proteins were purified as follows (27). After centrifugation, bacterial pellets were resuspended in 25 ml STE buffer (in mM: 10 Tris, pH 8.0, 150 NaCl, 2 EDTA, and 1 PMSF) containing pepstatin, leupeptin and aprotinin (10 μg/ml each). The bacteria were lysed by incubation with 100 μg/ml lysozyme at 4°C for 15 min, followed by the addition of 5 mM DTT and 1.5% N-laurylsarcosine. After brief sonication on ice, 2% Triton X-100 was added and the lysate clarified at 4°C by centrifugation at 10,000 rpm for 10 min. Clarified supernatants were incubated at 4°C overnight with 400 μl GSH-Sepharose (50% slurry, Pharmacia LKB Biotechnology). The matrix was recovered by centrifugation and washed five times with wash buffer (in mM: 50 Tris, pH 8; 150 NaCl, 0.05% Tween and complete protease and phosphatase inhibitor cocktails) followed by 2 washes with 500 mM KCl.
GST fusion protein integrity was analyzed on Coomassie blue-stained SDS-polyacrylamide gels and also by Western blot using anti-GST antibody (Amersham Biosciences). Protein concentration was determined by Lowry assay (BioRad).
GST Fusion protein binding assays
For pulldown experiments with GST alone, GST-NCX1/1-220, GST-NCX1/218-764 and GST-NCX1/764-939, crude membranes (100–600 μg) were prepared from HEK293 cells overexpressing PLM (12) and resuspended in 400 μl of Buffer III (in mM: 140 NaCl, 25 imidazole, 1 EDTA, pH 7.4; and a combination of complete protease and phosphatase inhibitor cocktails) with 5 mg/ml of C12E8. Membranes were precleared with 40 μl of GSH-sepharose (50% slurry) for 2 h at 4°C, after which they were combined with prepared GST-fusion protein-GSH sepharose matrices and incubated overnight at 4°C. The bead matrices were collected by centrifugation and washed 4 times with Buffer III containing C12E8. Bead matrices were boiled for 5 min with 40 μl of 2X sample buffer (0.12 M Tris-HCl (pH 6.8), 3.4% SDS, 12% glycerol, 0.2M DTT, 0.004% bromophenol blue). Proteins were resolved on either 10% (GST-NCX1 fusion proteins) or 15% SDS-polyacrylamide gels (PLM). After transfer to PVDF membranes, GST and PLM were detected by Western blotting with anti-GST antibodies (1:1,000) and C2 antibody (1:5,000)(28), respectively.
For pulldown experiments with GST-NCX1/218-371, GST-NCX1/371-508, GST-NCX1/508-764, GST-NCX1/218-764, and GST alone, His-tagged PLM (mw ~22 kDa) obtained from transformed BL21(DE3)pLysS bacteria (28) was used. His-tagged PLM (1 μg) in 50 mM Tris (pH 8) and 150 mM NaCl containing complete protease and phosphatase inhibitor cocktails was incubated with the prepared GST-fusion proteins at 4°C for 30 min. After centrifugation, bead matrices were washed 5 times with wash buffer containing 0.1% NP-40, after which they were subjected to SDS-polyacrylamide electrophoresis. Anti-His antibodies (1:1,000) rather than C2 antibody were used to detect PLM in these experiments.
Split Na+/Ca2+ exchangers
Three split exchanger chimeras (N265-YFP, N358-GFP and GFP-532C) were generated from dog NCX1 (938 amino acids)(19) and two (CFP-732C and GST-764C) from rat NCX1 (939 amino acids)(29). Two fusion proteins were made with either YFP or GFP inserted at the C-terminal end of split exchanger proteins. They are indicated with the letter N followed by a number, which designates the last residue of the peptide, and for N265-YFP and N358-GFP, the position to which YFP or GFP was linked. Three fusion proteins were made with GFP, CFP, or GST inserted at the N-terminal end of split exchanger proteins. The number preceding the letter C indicates the first amino acid of the peptide, and for GFP-532C, CFP-732C and GST-764C, the position to which GFP, CFP or GST was linked (see Figs. 5 & 6). N358-GFP (in pEGFPN-1 vector) was constructed as described previously (20). N265-YFP was constructed using pYFPN-1 vector. To obtain GFP-532C, the NCX1 fragment (amino acids 532-938) was amplified from pGEM-GFP-532C (20) by PCR using primers containing Bgl II and Sal I restriction enzyme linkers on either end. After digestion with restriction enzymes, the PCR product was subcloned by ligation into Bgl II/Sal I linearized pEGFPC-1. To construct CFP-732C, the rat NCX1 fragment from amino acids 732-939 was amplified from pAdTrack-CMV-NCX1 (26) by PCR using primers with Xho I and Hind III restriction enzyme linkers on either end. After digestion, the PCR product was subcloned by ligation into Xho I and Hind III linearized pECFPC-1. For GST-764C, the GST-NCX1 fragment in GST-NCX1/764-939 (pGEX-6P-1) was amplified by PCR using primers containing Kpn I and Xba I restriction enzyme linkers on either end. After digestion, the PCR product was subcloned by ligation into Kpn I and Xba I linearized pAdTrack vector. All inserts were confirmed by restriction digest and sequencing.
Fig. 5. Inhibition of INaCa by PLM requires interaction with residues 218-358 of the intracellular loop of NCX1.
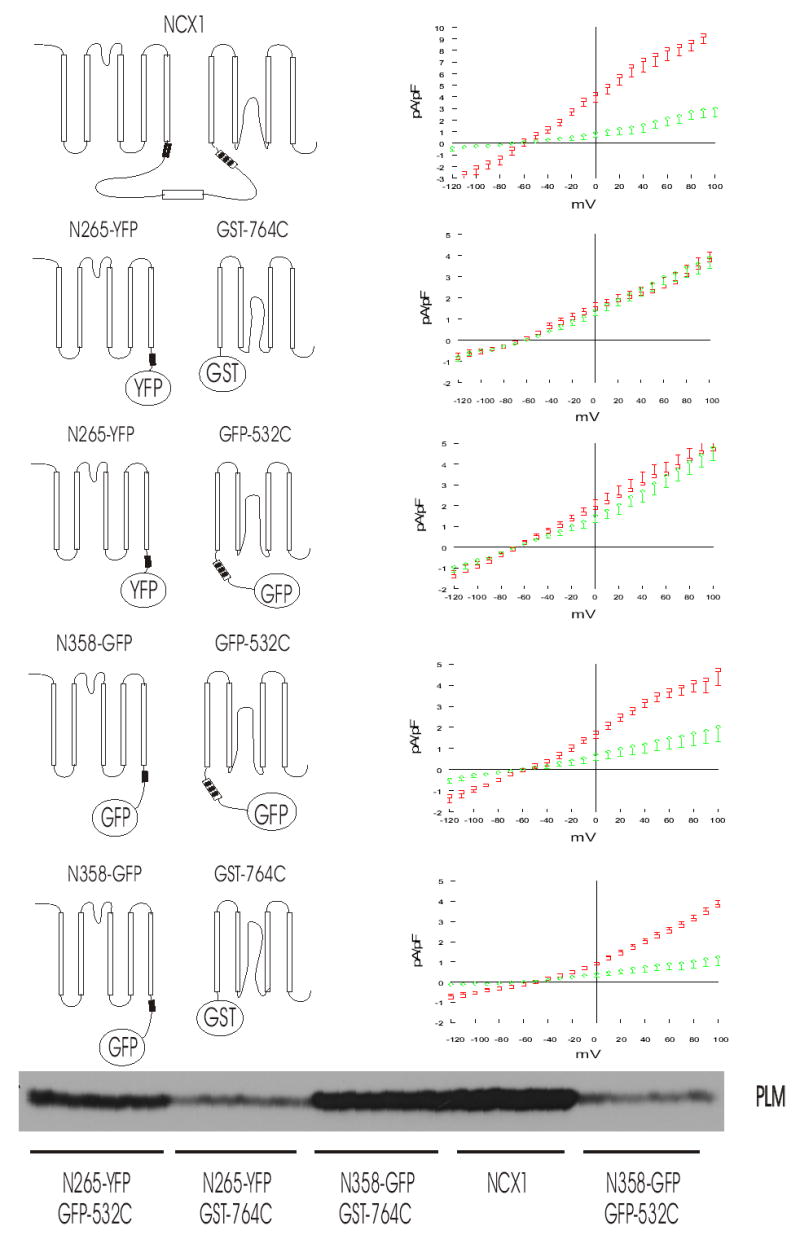
Left, schematic illustration of wild-type NCX1 and combinations of split exchanger chimeras. XIP region (filled box), Ca2+ binding domain (open box) and interaction site for endogenous XIP (striped box) are shown. Right, INaCa measured in HEK293 cells transfected with wild-type NCX1, or combination of N-terminal and C-terminal split exchanger chimeras with varying lengths of intracellular loop, with (diamonds) or without (squares) PLM. Symbols represent mean ± SE of 6 to 9 cells in each I–V curve with the exception of NCX1 + PLM (n=3). Bottom, Western blot demonstrating that PLM is expressed in cells whether they are co-transfected with wild-type NCX1 or combination of N-terminal and C-terminal split exchanger chimeras.
Fig. 6. Inhibition of INaCa by PLM in HEK293 cells transfected with CFP-732C and N358-GFP but not with CFP-732C and N265-YFP.
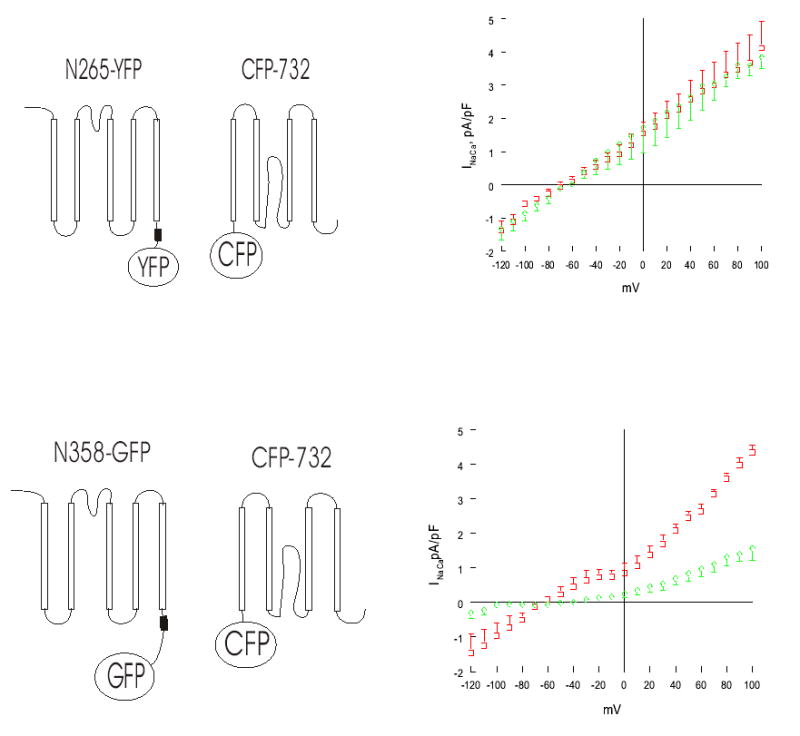
Left, schematic illustration of combinations of split exchanger chimeras. XIP region (filled box) is shown. Right, INaCa measured in HEK293 cells transfected with CFP-732C and N358-GFP or N265-YFP, with (diamonds) or without (squares) PLM. Symbols represent mean ± SE of 3 to 5 cells in each I–V curve.
Transfection of HEK293 cells
HEK293 cells (American Type Culture Collection; Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 (Cellgro; Herndon, VA), containing 10% heat-inactivated fetal bovine serum (FBS), at a density of 1.2 × 106 cells per 100 mm dish. After 24 h, medium was changed and cells were transfected with 25 μl Lipofectamine (Invitrogen, Carlsbad, CA) and total of 3μg plasmid DNA per dish: either control vector alone (3μg), combinations of N-terminal + C-terminal split exchangers + control vector (1μg each), combinations of N-terminal + C-terminal split exchangers + pAdTrack-CMV-PLM (1μg each), and N265-YFP or N358-GFP or GFP-532C (1μg each) alone with control vector (2μg), according to the manufacturer’s instructions. Levels of DNA and lipid were optimized in transfection assays to ensure minimal toxicity to cells. The lipid-DNA complex was left on cells for 5 h at 37ºC/5% CO2. Medium was then replaced with DMEM/Ham’s F12 + 10% FBS and cells were cultured for additional 48h before experiments. For patch-clamp applications, cells were trypsinized at 24 h post-transfection using Trypsin-EDTA (Invitrogen), transferred to 35 mm dishes containing sterile glass coverslips and incubated a further 24 h prior to experimentation. Cells for Western blot and co-immunoprecipitation applications were left in 100 mm dishes until 48 h post-transfection.
Confocal microscopy
HEK293 cells were transiently transfected with N265-YFP + GFP-532C, N265-YFP + CFP-732C, N358-GFP + GFP-532C, N358-GFP + CFP732C, N265-YFP alone, N358-GFP alone and GFP-532C alone on glass slide chambers (Nunc, Lab-Tek Division; Naperville, IL). Adherent cells were washed 3 times with PBS containing 2 mM EGTA and then fixed for 30 min in 3% paraformaldehyde in PBS with 2 mM EGTA. After two rinses with PBS, cells were permeabilized for 2 min in 0.05% Triton X-100, followed by 2 additional rinses with PBS. A coverslip was applied over mounting solution (90% glycerol in PBS + p-phenylaminediamine). Images of HEK293 cells (GFP, ex. 488, em. 505–525 nm; YFP, ex. 514, em. 525–600 nm; CFP, ex. 458, em. 465–510 nm) were acquired with a Leica TCS SP2 confocal microscope and processed with LCS software.
Na+/Ca2+ exchange current (INaCa) measurements
Whole cell patch-clamp recordings were performed at 30ºC as described previously (12,13). Briefly, fire-polished pipettes (tip-diameter 2–3 μm) were filled with a buffered Ca2+ solution containing (in mM): 100 Cs+ glutamate, 7.25 Na+ HEPES, 1 MgCl2, 12.75 HEPES, 2.5 Na2ATP, 10 EGTA, and 6 CaCl2, pH 7.2. Free Ca2+ in the pipette solution was 205 nM, measured fluorimetrically with fura 2. Cells were bathed in an external solution containing (in mM): 130 NaCl, 5 CsCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 CaCl2, 10 HEPES, 10 Na+ HEPES, and 10 glucose, pH 7.4. Verapamil (1 μM), ouabain (1mM), and niflumic acid (30 μM) were used to block Ca2+, Na+-K+-ATPase, and Cl− currents, respectively. K+ currents were minimized by Cs+ substitution for K+ in both pipette and external solutions. Only cells that fluoresced green (ex. 380, em. 510 nm), indicating successful transfection with NCX1 or its split exchanger chimeras, were selected for current measurements. Membrane potential (Em) was held at the calculated reversal potential of INaCa (−73 mV) for 5 min before stimulation. A descending voltage ramp (from +100 to −120 mV; 500 mV/s) was immediately followed by an ascending voltage ramp (from –120 to +100 mV; 500 mV/s). The voltage ramp was repeated after addition of 1 mM CdCl2 to the external solution. Currents were derived from measurements during the descending voltage ramp. INaCa was defined as the difference current measured in the absence and presence of Cd2+ (12,13). To facilitate comparison of NCX1 currents, INaCa of each cell was divided by Cm to account for variations in cell sizes.
Co-Immunoprecipitation of PLM and NCX1, and PLM and split exchanger chimeras
HEK293 cells were transfected with pAdTrack-CMV-PLM (1μg) + pAdTrack-CMV-NCX1 (2μg), or N358-GFP and GFP-532C (1μg each) + pAdTrack-CMV-PLM (1μg), or N358-GFP (2μg) + pAdTrack-CMV-PLM (1μg). After 48h, crude membrane pellets from transfected HEK293 cells were prepared as described previously (12). They were resuspended in a minimal volume of Buffer III, and then adjusted to 2 mg in 300 μl of Buffer III and combined with C12E8 detergent in 100 μl Buffer III (at a detergent: protein ratio of 2:1) at room temperature for 20 min. After addition of another 400 μl of Buffer III, samples were subjected to ultracentrifugation at 37,000g and 4°C using a Beckman TLA 100.3 rotor. The supernatant was transferred to a fresh tube and the protein content was determined. 300μg of solubilized crude membrane preparation was used in both pre-immune control and antibody immunoprecipitation experiments. Samples were precleared before addition of relevant antibodies by preincubation of supernatants with 40 μl of protein-A-agarose for 3h at 4ºC. Precleared supernatants were incubated with either 5 μg of preimmune rabbit IgG (polyclonal Ab control) or 5 μg of polyclonal PLM antibody (C2 Ab) overnight at 4ºC. The next day, 40 μl (50% slurry) of washed suspended protein-A-agarose beads were added to each sample and incubated for a further 2 h at 4ºC. Beads were pelleted, washed 4–6 times with 1.5 ml Buffer III containing 0.05% C12E8 and resuspended in 40 μl of 2x Laemmli sample buffer (+DTT). Beads were boiled for 5 min at 95ºC and immunoblotting was performed.
Primary antibody incubation was performed overnight at 4ºC in 5% non-fat milk/TBS-T containing 1:250 R3F1, 1:200 anti-GFP antibody, or 1:12,000 C2 Ab. Secondary antibodies used were either 1:2,000 donkey anti-rabbit IgG conjugated horseradish peroxidase (HRP*, Amersham Biosciences; Uppsala, Sweden) or 1:2,000 sheep anti-mouse IgG-HRP* (Amersham). Immunoreactivity was detected using an enhanced chemiluminescence kit, HyBlotCL autorad film (Denville Scientific; Metuchen, NJ) and a developer.
Co-immunoprecipitation of TM43 and NCX1
HEK293 cells were transfected with pAdTrack-CMV + pAdTrack-CMV-NCX1 (1.5μg each); pAdTrack-CMV-TM43 + pAdTrack-CMV-NCX1 (1.5μg each); or pAdTrack-CMV-PLM + pAdTrack-CMV-NCX1 (1.5μg each). TM43 is a dog PLM mutant whose cytoplasmic tail has been deleted (11). Crude membranes were prepared and co-immunoprecipitation experiments performed using either B8 antibody (recognizes the N-terminus of dog but not rat PLM)(11) or R3F1 antibody (recognizes intracellular loop of NCX1)(30).
Statistical analysis
All results are expressed as means ± SE. For analysis of a parameter (e.g., INaCa) as a function of group (e.g., split exchanger chimeras with and without co-expressed PLM) and voltage, two-way ANOVA was used to determine statistical significance. A commercial software package (JMP version 4.0.5, SAS Institute; Cary, NC) was used. In all analyses, p<0.05 was taken to be statistically significant.
Results
GST pulldown assays suggest that intracellular loop but not transmembrane domains of NCX1 associates with PLM
GST fusion proteins were constructed to contain the N-terminal domain, intracellular loop or C-terminal domain of rat NCX1 (Fig. 1). Although there were some minor non-specific PLM binding to GSH-sepharose matrix (despite preclearing), PLM was associated primarily with the intracellular loop rather than with the transmembrane domains of NCX1 (Fig. 2). Additional GST fusion proteins were then constructed to contain various segments of the intracellular loop of NCX1 (Fig. 1). PLM associated with residues 218-371 and 508-764 but not 371-508 of the intracellular loop (Fig. 3). This suggests that PLM did not bind to the Ca2+ regulatory site (residues 371-508) of the cardiac Na+/Ca2+ exchanger.
Fig. 2. PLM associates with the intracellular loop but not TM domains of NCX1.
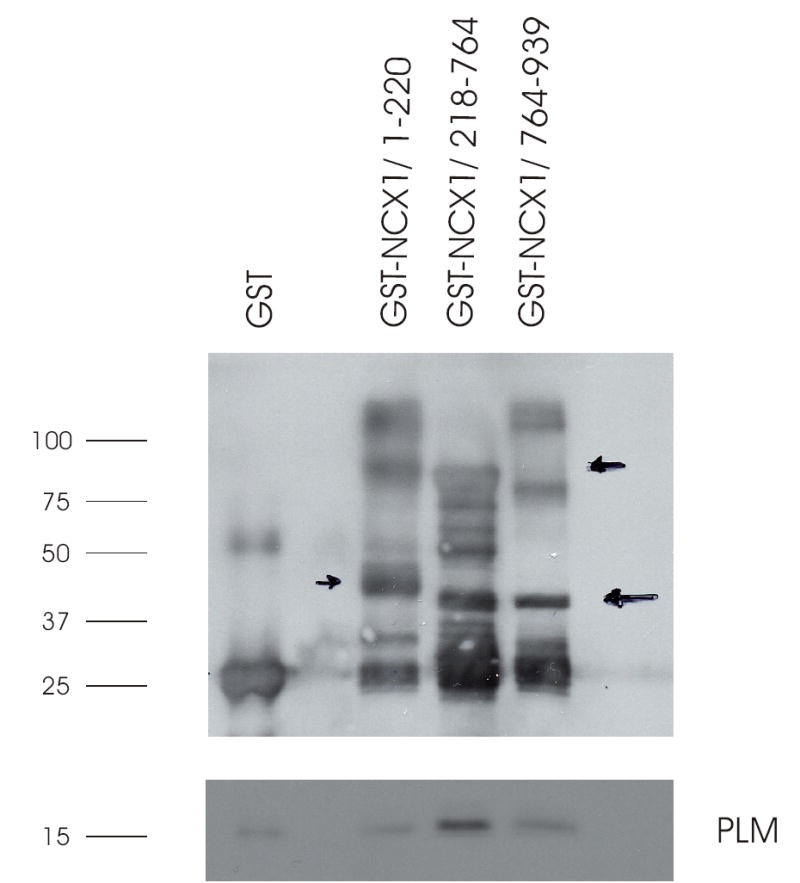
Purified GST or GST-NCX1 fusion proteins linked to GSH-sepharose beads were incubated with crude membranes prepared from HEK293 cells overexpressing PLM, and GST pulldown assay performed as described in Experimental Procedures. Upper panel: GST and GST-NCX1 fusion proteins detected by anti-GST antibody. Arrows (drawn on the original gel) point to the predicted mw of GST-NCX1/1-220 (48 kDa), GST-NCX1/218-764 (81 kDa), and GST-NCX1/764-939 (43 kDa). Lower panel: PLM detected by C2 antibody. This experiment was performed 4 times with similar results.
Fig. 3. PLM associates with proximal and distal regions of intracellular loop by GST pulldown.
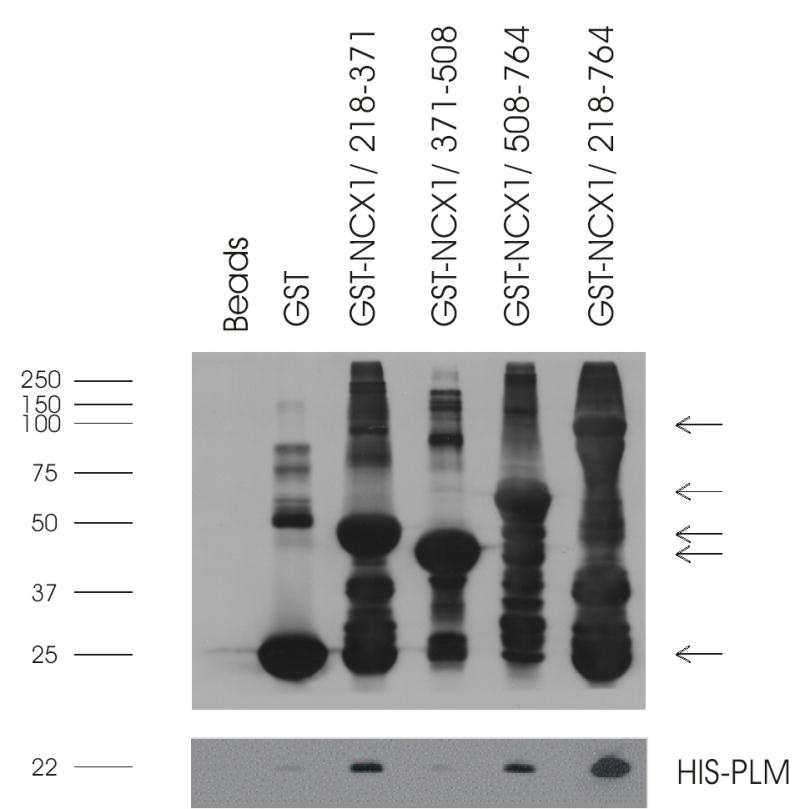
GST pulldown assays were performed as described in Fig. 2, except that the GST-NCX1 fusion proteins contained varying segments of the intracellular loop of NCX1 and that bacterial derived His-tagged PLM (28) was used. Upper panel: GST and GST-NCX1 fusion proteins detected by anti-GST antibody. Arrows (in descending order) point to the predicted mw of GST-NCX1/218-764 (81 kDa), GST-NCX1/508-764 (52 kDa), GST-NCX1/218-371 (41 kDa), GST-NCX1/371-508 (40 kDa) and GST (26 kDa). Lower panel: His-tagged PLM detected by anti-His antibody. This experiment was performed 4 times with similar results.
Co-expression of N- and C-terminal split exchanger chimeras results in correct membrane targeting
Previous studies demonstrated that co-expression of both N- and C-terminal domains is required for proper membrane targeting in Xenopus oocytes (20). Transfection of GFP-532C and N265-YFP in HEK293 cells resulted in both split exchanger chimeras being expressed at the membrane (Fig. 4A – C). Likewise, transfection of N358-GFP and GFP-532C resulted in correct membrane targeting in HEK293 cells (Fig. 4D). Transfection of N358-GFP (Fig. 4E) or N265-YFP (picture not shown) alone also resulted in correct membrane localization. By contrast, transfection of GFP-532C alone resulted in diffuse intracellular distribution of the split exchanger chimera (Fig. 4F). Transfection of N265-YFP and CFP-732C resulted in both split exchanger chimeras expressed at the plasma membrane (Fig. 4G – I). These results suggest that expression of N-terminal domain alone is sufficient for correct membrane targeting. C-terminal domain requires co-expression of N-terminal domain for proper targeting to the plasma membrane. We could not detect any GST-764C signals using anti-GST antibodies (data not shown).
Fig. 4. Localization of NCX1 split exchanger chimeras by confocal microscopy in transfected HEK293 cells.
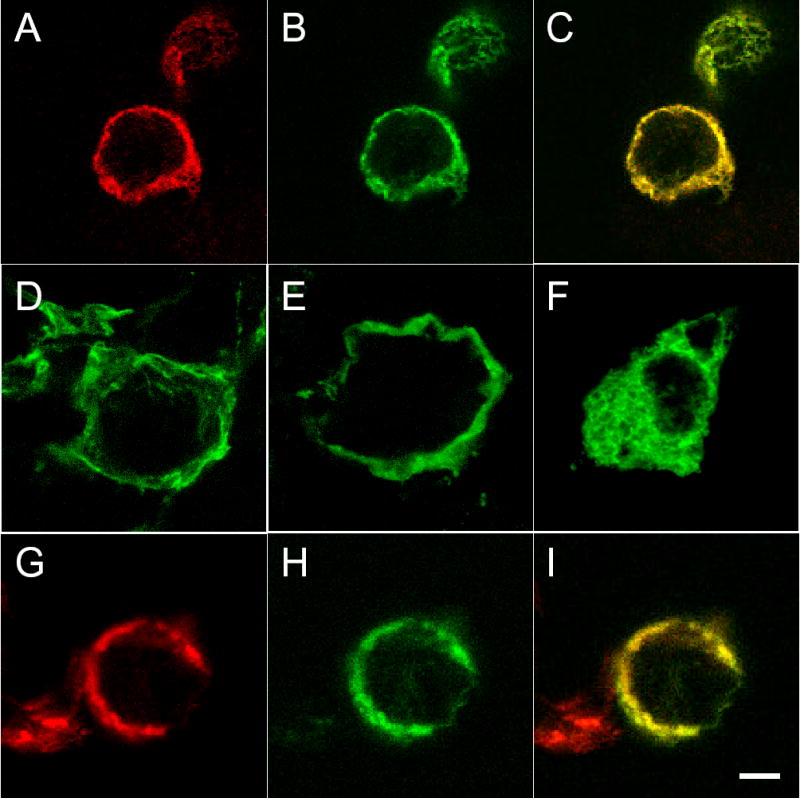
Cells were transiently transfected with either N-terminal or C-terminal split exchanger chimera alone, or combination of N-terminal and C-terminal split exchanger chimeras. After 48h, cells were fixed, permeabilized, and visualized with a Leica TCS SP2 confocal microscope. In cells transfected with N265-YFP + GFP-532C (A to C), both N265-YFP (A) and GFP-532C (B) split exchanger chimeras are primarily localized to the plasma membrane, and the two split exchanger chimeras co-localized with each other (C : merged image of A and B). Similarly, the split exchanger chimeras in cells transfected with N358-GFP + GFP-532C are correctly targeted to the plasma membrane (D). When transfected alone, only N358-GFP (E) and N265-YFP (not shown) but not GFP-532C (F) is correctly targeted to the membrane. In cells transfected with N265-YFP + CFP-732C (G to I), both N265-YFP (G) and CFP-732P (H) split exchanger chimeras are primarily localized to the plasma membrane and they co-localized with each other (I: merged image of G and H). Scale bar = 5 μm.
Co-expression of N- and C-terminal split exchanger chimeras results in functional INaCa
We have previously demonstrated that under the conditions used in our electrophysiological studies, the Cd2+-sensitive current is INaCa (12,13). Co-expression of any N-terminal with any C-terminal split exchanger chimeras in HEK293 cells resulted in INaCa, although the magnitude of the current was ~1/2 that measured in cells expressing the intact exchanger (Fig. 5). This finding is similar to that observed when split exchanger chimeras were expressed in Xenopus oocytes (20). By contrast, when N265-YFP alone was expressed in HEK293 cells, despite correct membrane targeting, no significant current was detected (data not shown). We did not measure currents in HEK293 cells expressing GFP-532C alone since the split exchanger chimera was not properly targeted to the plasma membrane.
PLM inhibits INaCa by interacting with residues 218 – 358 of the intracellular loop
Co-expression of PLM with wild-type NCX1 resulted in significant suppression of INaCa (Fig. 5), in agreement with our previous observations (12,13). Co-expression of PLM with the N- and C-terminal TM domains (N265-YFP + GST-764C) without the intracellular loop did not result in any inhibition of INaCa (Fig. 5), indicating that PLM acts on NCX1 by interacting with the intracellular loop. Co-expression of PLM with N265-YFP + GFP-532C, in which the C-terminal chimera contains residues 532–764 of the intracellular loop, also did not result in suppression of INaCa. Therefore, despite association with PLM (Fig. 3), this segment of intracellular loop spanning residues 532–764 was likely not the site at which interaction with PLM resulted in NCX1 inhibition. Note that the absence of inhibition by PLM on INaCa in cells co-expressing N265-YFP + GST-764C or N265-YFP + GFP-532C was not due to lack of expression of PLM (Fig. 5). Co-expression of PLM with N358-GFP + GFP-532C, in which the N-terminal chimera contains residues 218-358 of the intracellular loop, resulted in significant inhibition of INaCa. Finally, co-expression of PLM with N358-GFP + GST-764C also resulted in significant inhibition of INaCa. These results, when taken altogether, suggest that PLM inhibits NCX1 by interacting with residues 218-358 of the intracellular loop.
It should be noted that although there is variability in PLM expression among cells transfected with various combination of split exchanger chimeras or wild-type NCX1 (likely due to use of multiple plasmid vectors on the same cells), INaCa was clearly suppressed by PLM in cells transfected with N358-GFP + GFP-532C but not affected at all in cells transfected with N265-YFP + GST-764C, despite similar levels of PLM expression (Fig. 5). In addition, levels of PLM expression in cells transfected with N265-YFP + GFP-532C were much higher than those in cells transfected with N358-GFP + GFP-532C, but INaCa was not suppressed by PLM in the former while it was clearly inhibited in the latter. These considerations support our conclusion that residues 218-358 of the intracellular loop of NCX1 are important for PLM’s inhibitory effects.
Using anti-GST antibodies, we were unable to detect GST-764C in transfected cells either by confocal microscopy or by Western blotting (data not shown), despite clear presence of INaCa (Fig. 5). We therefore constructed another split exchanger chimera, CFP-732C, and showed unequivocally that it was expressed on the plasma membrane when co-transfected with N265-YFP (Fig. 4G–I). In HEK293 cells transfected with N265-YFP + CFP-732C, PLM did not inhibit INaCa (Fig. 6). By contrast, co-expression of PLM with N358-GFP + CFP-732C resulted in clear suppression of INaCa. Expression of PLM was similar in cells transfected with N265-YFP + CFP-732C and in those transfected with N358-GFP + CFP 732C (data not shown), despite clear suppression of INaCa in the latter but not in the former (Fig. 6). These additional experiments confirmed our hypothesis that PLM inhibited INaCa by interacting with residues 218-358 of the exchanger.
PLM associates with residues 218-358 of the intracellular loop of NCX1
To detect physical association between PLM and residues 218-358 of the intracellular loop of NCX1, we performed co-immunoprecipitation experiments. We used C2 antibody to immunoprecipitate PLM from crude membranes prepared from HEK293 cells co-transfected with PLM and NCX1 or its split exchanger chimeras. We then used anti-GFP antibody to detect presence of split exchanger chimeras in the immunoprecipitates. We also used R3F1, a monoclonal antibody that detects residues 560-629 and 649-705 of the intracellular loop of NCX1 (30), to screen for presence of wild-type NCX1 and GFP-532C in the immunoprecipitate. Using R3F1, we showed that PLM was able to co-immunoprecipitate wild-type NCX1 (mw 160 kDa) and N358-GFP + GFP-532C (Fig. 7, upper panel). In the immunoprecipitates, the lane labeled N358-GFP + GFP-532C shows a ~60 kDa band detected by R3F1 and corresponds to the same molecular weight band detected by anti-GFP antibody (Fig. 7, lower panel). This band represents the GFP-532C chimera. Using anti-GFP antibodies, we demonstrated that PLM associated with N358-GFP alone, and N358-GFP + GFP-532C chimeras (Fig. 7, lower panel). Note the slight difference in molecular weights between N358-GFP and GFP-532C in the membrane input lanes. We do not know the identity of the high molecular weight ( ~150–160 kDa) signal detected by R3F1 in the lane labeled N358-GFP + GFP-532C. Likewise, the origins of the high molecular weight signals detected by anti-GFP antibody in lanes labeled N358-GFP and N358-GFP + GFP-532C are unknown. We speculated that the higher molecular weight bands (~150–160 kDa) may represent multimers of N358-GFP or the complex formed by N358-GFP and GFP-532C chimeras.
Fig. 7. Association of PLM with wild-type NCX1 or N358-GFP + GFP-532C in transfected HEK293 cells.
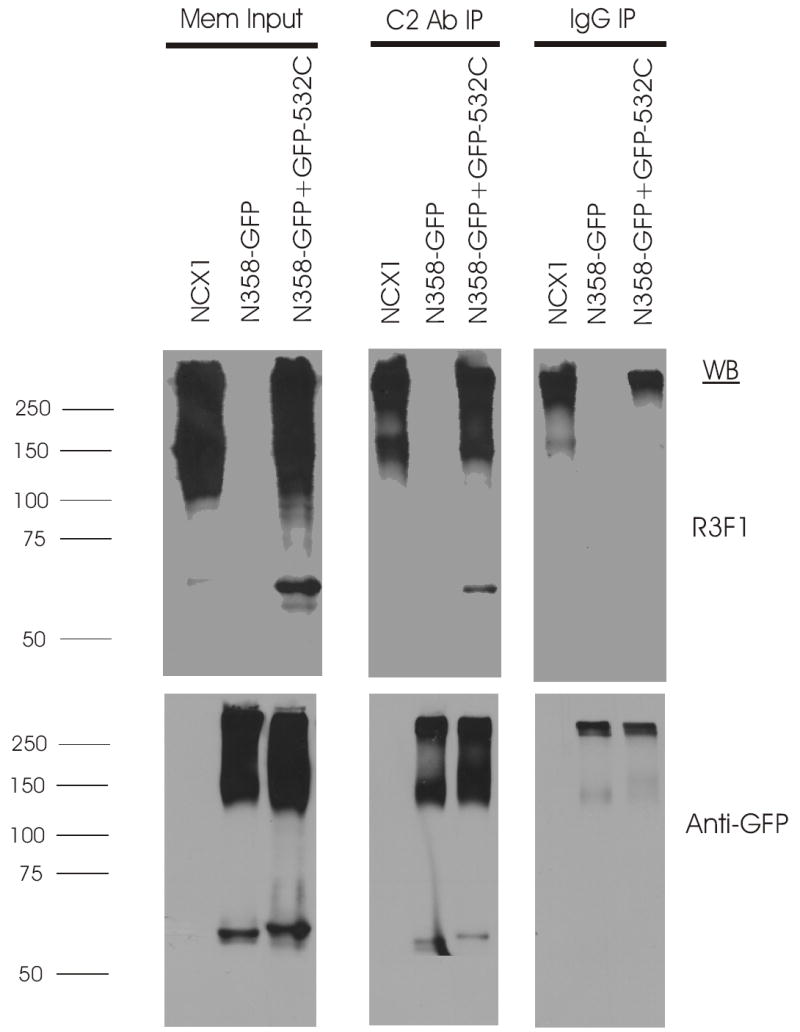
Top panel, immunoblot of NCX1 or split exchanger chimera immunoprecipitates from 300 μg of solubilized crude membrane preparations using 5 μg of anti-PLM antibody (C2 Ab) or control rabbit serum (IgG), and probed with antibody to NCX1 (R3F1) which recognizes the epitope formed by residues 560-629 and 649-705 of the intracellular loop (30). Using R3F1, we usually detected a higher molecular weight band (~250 kDa) in addition to the expected 160 kDa band in both transfected HEK293 cells and native rat heart membranes. Increased protein loading in the membrane input lanes (35μg/lane) often resulted in smearing of the 160 and 250 kDa signals so that they overlap (12). The important point is that C2 Ab co-immunoprecipitated NCX1 whose 160 kDa signal is clearly separable from the non-specific 250 kDa signal. Bottom panel, after stripping, the same blot was probed with anti-GFP antibody. Crude membrane preparations (Mem Input) were derived from HEK293 cells transiently transfected (48h) with PLM and NCX1 or PLM with combination of split exchanger chimeras as indicated. The antibodies used in the immunoblots are indicated on the right, and molecular mass markers (in kDa) are shown on the left. While individual N358-GFP and GFP-532C signals can readily be discerned, we could not simultaneously detect both N358-GFP and GFP-532C signals in the N358-GFP + GFP-532C lane. Rather, a higher molecular weight band (~160 kDa) is detected by the anti-GFP antibody. The identity of this higher molecular weight band is unknown but we speculate that it may be derived from N358-GFP homodimers or N358-GFP + GFP-532C heterodimers. This experiment was performed 3 times with similar results.
Association of NCX1 with PLM requires the cytoplasmic domain of PLM
Our observations so far suggest that PLM interacts with the proximal, N-terminal end of the intracellular loop of NCX1, specifically residues 218-358. It is known that the TM domain of PLM interacts with the TM domains of Na+-K+-ATPase (31). It is therefore of interest to determine if the TM domain of PLM is necessary for its interaction with NCX1. Using B8 antibody which is specific for the N-terminus of dog (but not rat) PLM (11), we confirmed our previous observations in transfected HEK293 cells (12) that wild-type PLM co-immunoprecipitated NCX1 (Fig. 8). TM43, a dog PLM mutant in which the cytoplasmic tail is deleted (11), was unable to associate with NCX1 (Fig. 8). Reciprocal co-immunoprecipitation experiments using R3F1 to immunoprecipitate NCX1 confirmed association of NCX1 with wild-type PLM but not the truncation mutant TM43 (Fig. 9). Our results indicate that there is little interaction between the TM domain of PLM and those of NCX1.
Fig. 8. Association of PLM with wild-type NCX1 requires cytoplasmic tail of PLM.
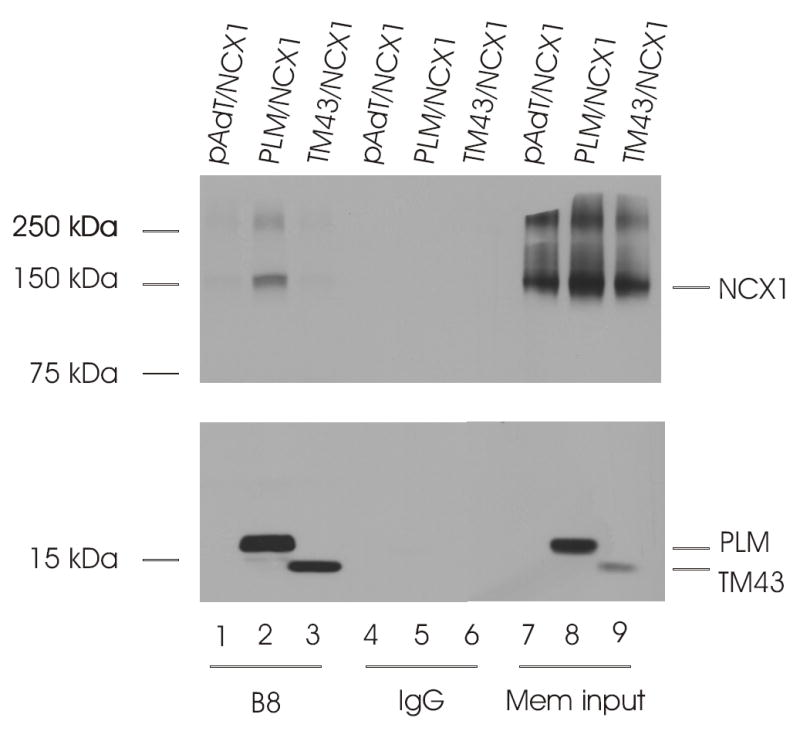
Top panel, immunoblot of NCX1 immunoprecipitates from 300 μg of solubilized membrane preparations using 5μl of anti-PLM antibody (B8) or control rabbit serum (IgG), and probed with antibody to NCX1 (R3F1). Bottom panel, immunoblot of PLM immunoprecipitates from 300 μg of solubilized crude membrane preparations using 5μl of anti-PLM antibody (B8) or control rabbit serum (IgG), and probed with antibody to PLM (B8). Crude membrane preparations (Mem input) were derived from HEK293 cells transiently transfected (48h) with pAdTrack-CMV and NCX1 (pAdT/NCX1), PLM/NCX1, or NCX1 with the PLM cytoplasmic tail truncation mutant TM43. The locations of NCX1, PLM and TM43 are indicated on the right, and molecular mass markers (in kDa) are shown on the left. This experiment was performed 2 times with similar results.
Fig. 9. Reciprocal co-purification of PLM with NCX1 in HEK293 crude membrane extracts.
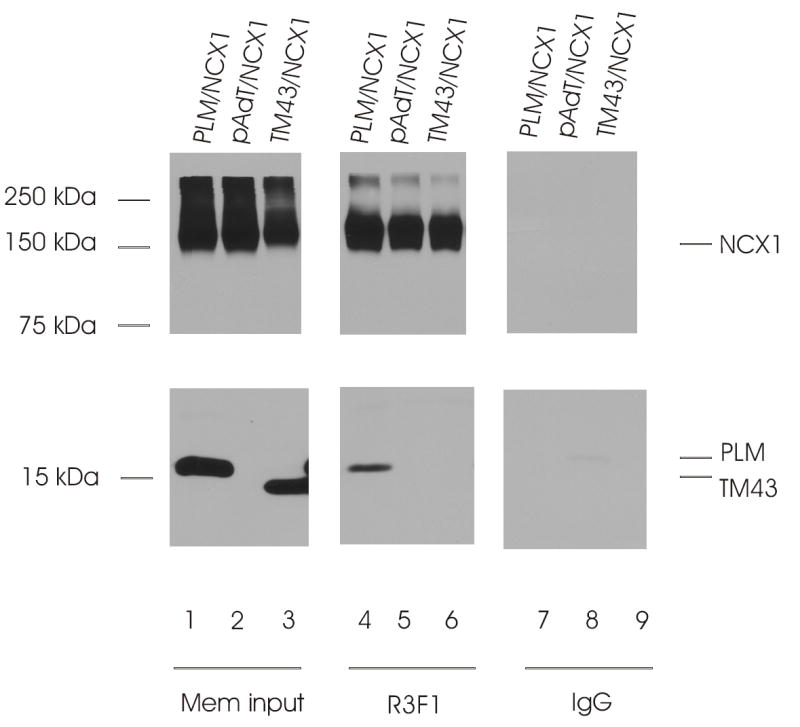
Top panel, immunoblot of NCX1 immunoprecipitates from 300 μg of solubilized membrane preparations using 4 μl of anti-NCX1 antibody (R3F1) or control rabbit serum (IgG) and probed with R3F1. Bottom panel, immunoblot of corresponding PLM/TM43 immunoprecipitates probed with anti-PLM antibody (B8). Crude membranes (Mem input) were prepared as in Fig. 8. The locations of NCX1, PLM and TM43 are indicated on the right, and molecular markers (in kDa) are shown on the left.
Discussion
Phospholemman or FXYD1 is mainly expressed in the heart, skeletal muscle, liver and juxtaglomerular apparatus of the kidney (32). It is a major sarcolemmal substrate for protein kinases A and C in heart and skeletal muscle (1,33,34). Early work based on overexpression of PLM in Xenopus oocytes suggest that PLM is a hyperpolarization-activated anion-selective channel (35). When reconstituted in lipid bilayers, PLM forms a channel that is highly permeable to taurine (36). In support of the channel concept, recent structural studies indicate that synthetic peptides representing the TM domain of PLM form tetramers in reconstituted proteoliposomes (37). The channel-like properties of PLM is thought to be a major mechanism by which PLM regulates cell volume in non-cardiac tissues (38-40).
PLM (FXYD1) and other members of the FXYD gene family including the γ-subunit of the Na+-K+-ATPase (FXYD2)(41), channel inducing factor (CHIF; FXYD4)(42), FXYD7(43) and PLMS (a 15 kDa homologue of PLM isolated from shark rectal glands)(44), associate with and regulate the activity of the α-subunits of Na+-K+-ATPase. When co-expressed with α- and β-subunits of Na+-K+-ATPase in Xenopus oocytes, PLM modulates Na+-K+-ATPase activity, primarily by decreasing apparent affinities for Na+ and K+ without affecting Vmax (3). In the heart, PLM co-immunoprecipitates with α-subunits of Na+-K+-ATPase (3,8) and regulates its activity either by affecting Km for Na+ (4) and/or Vmax (5,7,8). Mutational analysis suggests that FXYD proteins (FXYD2, 4 and 7) interact with TM9 segment of Na+-K+-ATPase (45). Co-immunoprecipitation and covalent cross-linking studies demonstrate the proximity of TM segment of PLM to TM2 segment of Na+-K+-ATPase (31). Molecular modeling based on the Ca2+-ATPase crystal structure in the E1ATP bound conformation, is consistent with location of the single TM segment of FXYD proteins between TM segments 2, 6 and 9 of the α-subunit of Na+-K+-ATPase (31).
In adult rat cardiac myocytes, overexpression of PLM results in contraction and [Ca2+]i transient abnormalities (28) similar to those in which NCX1 is downregulated (46), leading us to suggest that in addition to inhibition of Na+-K+-ATPase, another function of PLM is to regulate NCX1 activity. This is because theoretically, inhibition of Na+-K+-ATPase by PLM would be expected to raise [Na+]i, thereby decreasing the thermodynamic driving force for Ca2+ efflux via NCX1, and resulting in increase [Ca2+]i and cardiac contractility. This hypothesis is not supported by data which show at high extracellular Ca2+ concentrations ([Ca2+]o), PLM overexpression in rat cardiac myocytes leads to reduction, rather than enhancement, of both [Ca2+]i transient and contraction amplitudes (9,28). By contrast, when PLM is downregulated by adenovirus-mediated antisense transfer (10) or eliminated (14) by engineering a phospholemman-deficient mouse (6), both [Ca2+]i transient and contraction amplitudes are increased, rather than decreased, at high [Ca2+]o. Indeed, investigations using adenovirus-mediated gene transfer in adult rat myocytes (9–11), heterologous expression model system (12,13), and cardiac myocytes isolated from phospholemman-deficient mice (14) provide strong support that PLM inhibits NCX1. First, PLM co-localizes with and co-immunoprecipitates NCX1, both in cardiac myocytes (9,10,12) and in transfected HEK293 cells (12). Second, PLM inhibits INaCa (measured under conditions in which Na+-K+-ATPase activity is absent because of presence of ouabain and absence of K+) in rat (9–11) and mouse (13) cardiac myocytes, and in HEK293 cells expressing NCX1 and PLM (12,13). Third, PLM inhibits Na+-dependent Ca2+ uptake in HEK293 cells co-expressing NCX1 and PLM (12). Fourth, while phosphorylation relieves inhibition of Na+-K+-ATPase by PLM (4,5,7), PLM phosphorylated at serine68 is the PLM form that inhibits NCX1 (11–13). The different mechanisms by which PLM inhibits Na+-K+-ATPase (dephosphorylation) and NCX1 (phosphorylation) argue that PLM exerts direct effects on NCX1.
Molecular and biochemical studies indicate that PLM and other FXYD proteins interact with TM2 and TM9 of Na+-K+-ATPase (31,45). Molecular modeling suggests that PLM docks into the groove between TM2, TM6 and TM9 of Na+-K+-ATPase. It is also of interest that phospholamban (PLB), which shares a short region of sequence similarity with PLM (RSSIRRLST69 in PLM and RSAIRRAST17 in PLB), also docks between TM2, TM6 and TM9 of the sarcoplasmic reticulum Ca2+-ATPase which it regulates (47). From these studies, it appears that single span, small membrane proteins such as PLM and PLB regulate ATPase pump activity via interaction of TM domains. Thus a major finding of the present study is that by GST pulldown assay, co-immunoprecipitation and INaCa measurements, the TM domains of NCX1 did not interact with the TM domain of PLM. This may be because while both Na+-K+-ATPase and sarcoplasmic reticulum Ca2+-ATPase belong to the P-type ATPase family, NCX1 belongs to the exchanger superfamily (19). Whether other proteins such as sorcin (48) and calcineurin (49) that have recently been reported to regulate NCX1 activity require interactions with TM domains of NCX1 is unknown at present.
A second major finding is that PLM exerted its modulatory influences on NCX1 by interacting with its intracellular loop, specifically, the proximal segment containing residues 218-358. Both GST pulldown assays and split exchanger studies indicate that the high affinity Ca2+ binding domain (residues 371-508) of the intracellular loop was unlikely to mediate the inhibitory effects of PLM. In addition, based on the observation the PLM did not inhibit INaCa in HEK293 cells expressing N265-YFP + GFP-532C, it is unlikely that the endogenous XIP region (residues 219-238) and the interaction site for endogenous XIP (residues 562-679) were involved in regulation of NCX1 by PLM.
A third finding is that the cytoplasmic tail of PLM was required for its modulatory influences on NCX1 activity. Previous studies demonstrate that unlike wild-type PLM, the cytoplasmic tail truncation mutant of PLM (TM43) does not affect cardiac contractility when overexpressed in adult rat cardiac myocytes (11). Our current finding that TM43, unlike wild-type PLM, was unable to co-immunoprecipitate NCX1 provides additional support that the cytoplasmic tail of PLM interacts with the intracellular loop of NCX1.
In HEK293 cells co-expressing PLM and NCX1, activation of PKC results in large increases in INaCa (13). Mutating serine68 of PLM to alanine or glutamic acid results in complete loss of increases in INaCa when PKC is activated (13). Mutating serine63 of PLM to alanine (in which serine68 is intact) preserves the ability of activated PKC to increase INaCa (13). Our results on the serine68 and serine63 PLM mutants suggest that changes in conformation in PLM by mutating serine68 alters its interaction with NCX1, resulting in NCX1 not being accessible to PKC action. PKC activation is associated with increased NCX1 phosphorylation at serine249, serine250 and serine357 (50). Our current observation that the cytoplasmic tail of PLM interacts with the N-terminal end of intracellular loop (residues 218-358) of NCX1 is consistent with the hypothesis that serine68 of PLM intimately associates with or hinders access to serine249, serine250 and serine357 of NCX1.
In adult rat cardiac myocytes, both α1- and α2-subunitis of Na+-K+-ATPase are expressed in the sarcolemma and t-tubules, with the α1-subunit perhaps more abundant in the t-tubules (51,52). Na+/Ca2+ exchanger is also distributed largely within the t-tubules (53). In addition, in cardiac sarcolemma, both α1- and α2-subunitis of Na+-K+-ATPase form distinct protein complexes with the Na+/Ca2+ exchanger (54). In guinea pig myocytes, immunoprecipitation and indirect immunofluorescence localization techniques indicate that PLM associates with α1- but not α2-subunits of Na+-K+-ATPase (5,7). Since PLM also co-localizes (9) and associates with (10,12) the cardiac Na+/Ca2+ exchanger, it is likely that PLM, Na+/Ca2+ exchanger and Na+-K+-ATPase form a macromolecular complex, especially in the t-tubules. For example, PLM may dock in the groove formed by TM2, TM6 and TM9 of the α-subunit of Na+-K+-ATPase (31), while its cytoplasmic tail interacts with the intracellular loop of Na+/Ca2+ exchanger. Phosphorylation of PLM at serine68 alters its conformation and interactions such that Na+-K+-ATPase is disinhibited (4,5,7) while Na+/Ca2+ exchanger is inhibited (13). Upon β-adrenergic stimulation, the coordinated action of phosphorylated PLM prevents Na+ overload (by disinhibiting Na+-K+-ATPase) and cellular Ca2+ depletion (by inhibiting Na+/Ca2+ exchange), thereby preserving both the chronotropic and inotropic responses under the circumstances of fight or flight.
In summary, we have demonstrated by 3 fundamentally different techniques that the cytoplasmic domain of PLM interacted with the N-terminal end (residues 218-358) of the intracellular loop of NCX1. We could not detect associations or functional interactions between the TM domains of NCX1 and the single TM domain of PLM. We speculate that PLM, Na+/Ca2+ exchanger and Na+-K+-ATPase form a macromolecular complex, especially in the t-tubules of cardiac myocytes.
Acknowledgments
We would like to thank Drs. Kenneth D. Philipson, Scott John and Michela Ottolia of the David Geffen School of Medicine at UCLA for providing N265-YFP, N358-GFP and pGEM-GFP-532C constructs.
This study was supported in part by the National Institutes of Health Grants HL-58672 & HL-74854 (J.Y. Cheung), DK-46678 (J.Y. Cheung, co-investigator), GM-69841 & HL-77814 (L.I. Rothblum), NS-21925 & NS-41363 (D.J. Carey); American Heart Association Pennsylvania Affiliate Grants-in-Aid 0265426U (X. Zhang) and 0355744U (J.Y. Cheung); American Heart Association Pennsylvania Affiliate PostDoctoral Fellowship 0425319U (B.A. Ahlers); and by grants from the Geisinger Foundation (L.I. Rothblum and D.J. Carey).
References
- 1.Palmer CJ, Scott BT, Jones LR. J Biol Chem. 1991;266:11126–11130. [PubMed] [Google Scholar]
- 2.Sweadner KJ, Rael E. Genomics. 2000;68:41–56. doi: 10.1006/geno.2000.6274. [DOI] [PubMed] [Google Scholar]
- 3.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Proc Natl Acad Sci U S A. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, Tucker AL, Bers DM. Circ Res. 2005;97:252–259. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- 5.Fuller W, Eaton P, Bell JR, Shattock MJ. Faseb J. 2004;18:197–199. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 6.Jia LG, Donnet C, Bogaev RC, Blatt RJ, McKinney CE, Day KH, Berr SS, Jones LR, Moorman JR, Sweadner KJ, Tucker AL. Am J Physiol Heart Circ Physiol. 2005;288:H1982–1988. doi: 10.1152/ajpheart.00142.2004. [DOI] [PubMed] [Google Scholar]
- 7.Silverman BZ, Fuller W, Eaton P, Deng J, Moorman JR, Cheung JY, James AF, Shattock MJ. Cardiovasc Res. 2005;65:93–103. doi: 10.1016/j.cardiores.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XQ, Moorman JR, Ahlers BA, Carl LL, Lake DE, Song J, Mounsey JP, Tucker AL, Chan YM, Rothblum LI, Stahl RC, Carey DJ, Cheung JY. J Appl Physiol. 2006;100:212–220. doi: 10.1152/japplphysiol.00757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang XQ, Qureshi A, Song J, Carl LL, Tian Q, Stahl RC, Carey DJ, Rothblum LI, Cheung JY. Am J Physiol Heart Circ Physiol. 2003;284:H225–233. doi: 10.1152/ajpheart.00698.2002. [DOI] [PubMed] [Google Scholar]
- 10.Mirza MA, Zhang XQ, Ahlers BA, Qureshi A, Carl LL, Song J, Tucker AL, Mounsey JP, Moorman JR, Rothblum LI, Zhang TS, Cheung JY. Am J Physiol Heart Circ Physiol. 2004;286:H1322–1330. doi: 10.1152/ajpheart.00997.2003. [DOI] [PubMed] [Google Scholar]
- 11.Song J, Zhang XQ, Ahlers BA, Carl LL, Wang J, Rothblum LI, Stahl RC, Mounsey JP, Tucker AL, Moorman JR, Cheung JY. Am J Physiol Heart Circ Physiol. 2005;288:H2342–2354. doi: 10.1152/ajpheart.01133.2004. [DOI] [PubMed] [Google Scholar]
- 12.Ahlers BA, Zhang XQ, Moorman JR, Rothblum LI, Carl LL, Song J, Wang J, Geddis LM, Tucker AL, Mounsey JP, Cheung JY. J Biol Chem. 2005;280:19875–19882. doi: 10.1074/jbc.M414703200. [DOI] [PubMed] [Google Scholar]
- 13.Zhang XQ, Ahlers BA, Tucker AL, Song J, Wang J, Moorman JR, Mounsey JP, Carl LL, Rothblum LI, Cheung JY. J Biol Chem. 2006;281:7784–7792. doi: 10.1074/jbc.M512092200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker, A. L., Song, J., Zhang, X. Q., Wang, J., Ahlers, B. A., Carl, L. L., Mounsey, J. P., Moorman, J. R., Rothblum, L. I., and Cheung, J. Y. (2006) Am J Physiol Heart Circ Physiol [DOI] [PMC free article] [PubMed]
- 15.Waalas SI, Czernik AJ, Olstad OK, Sletten K, Walaas O. Biochem J. 1994;304 ( Pt 2):635–640. doi: 10.1042/bj3040635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaustein M, Lederer W. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 17.Bers DM. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 18.Nicoll DA, Ottolia M, Lu L, Lu Y, Philipson KD. J Biol Chem. 1999;274:910–917. doi: 10.1074/jbc.274.2.910. [DOI] [PubMed] [Google Scholar]
- 19.Philipson KD, Nicoll DA. Annu Rev Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- 20.Ottolia M, John S, Qiu Z, Philipson KD. J Biol Chem. 2001;276:19603–19609. doi: 10.1074/jbc.M101489200. [DOI] [PubMed] [Google Scholar]
- 21.Levitsky DO, Nicoll DA, Philipson KD. J Biol Chem. 1994;269:22847–22852. [PubMed] [Google Scholar]
- 22.Li Z, Nicoll DA, Collins A, Hilgemann D, Filoteo A, Penniston J, Weiss J, Tomich J, Philipson KD. J Biol Chem. 1991;266:1014–1020. [PubMed] [Google Scholar]
- 23.He Z, Feng S, Tong Q, Hilgemann DW, Philipson KD. Am J Physiol Cell Physiol. 2000;278:C661–666. doi: 10.1152/ajpcell.2000.278.4.C661. [DOI] [PubMed] [Google Scholar]
- 24.Matsuoka S, Nicoll DA, He Z, Philipson KD. J Gen Physiol. 1997;109:273–286. doi: 10.1085/jgp.109.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maack C, Ganesan A, Sidor A, O’Rourke B. Circ Res. 2005;96:91–99. doi: 10.1161/01.RES.0000151334.48676.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang XQ, Song J, Rothblum LI, Lun M, Wang X, Ding F, Dunn J, Lytton J, McDermott PJ, Cheung JY. Am J Physiol Heart Circ Physiol. 2001;281:H2079–2088. doi: 10.1152/ajpheart.2001.281.5.H2079. [DOI] [PubMed] [Google Scholar]
- 27.Frangioni JV, Neel BG. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- 28.Song J, Zhang X, Carl L, Qureshi A, Rothblum L, Cheung J. Am Journal of Physiol Heart Circ Physiol. 2002;283:H576–H583. doi: 10.1152/ajpheart.00197.2002. [DOI] [PubMed] [Google Scholar]
- 29.Low W, Kasir J, Rahamimoff H. FEBS Lett. 1993;316:63–67. doi: 10.1016/0014-5793(93)81737-k. [DOI] [PubMed] [Google Scholar]
- 30.Porzig H, Li Z, Nicoll DA, Philipson KD. Am J Physiol Cell Physiol. 1993;265:C748–C756. doi: 10.1152/ajpcell.1993.265.3.C748. [DOI] [PubMed] [Google Scholar]
- 31.Lindzen M, Gottschalk KE, Fuzesi M, Garty H, Karlish SJ. J Biol Chem. 2006;281:5947–5955. doi: 10.1074/jbc.M512063200. [DOI] [PubMed] [Google Scholar]
- 32.Geering K. Am J Physiol Renal Physiol. 2006;290:F241–250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 33.Lindemann JP. J Biol Chem. 1986;261:4860–4867. [PubMed] [Google Scholar]
- 34.Presti CF, Jones LR, Lindemann JP. J Biol Chem. 1985;260:3860–3867. [PubMed] [Google Scholar]
- 35.Moorman JR, Ackerman SJ, Kowdley GC, Griffin M, Mounsey JP, Chen Z, Cala SE, O’Brian JJ, Szabo G, Jones LR. Nature. 1995;377:737–740. doi: 10.1038/377737a0. [DOI] [PubMed] [Google Scholar]
- 36.Chen Z, Jones LR, O’Brian JJ, Moorman JR, Cala SE. Circ Res. 1998;82:367–374. doi: 10.1161/01.res.82.3.367. [DOI] [PubMed] [Google Scholar]
- 37.Beevers AJ, Kukol A. Protein Sci. 2006;15:1127–1132. doi: 10.1110/ps.051899406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis CE, Patel MK, Miller JR, John JE, 3rd, Jones LR, Tucker AL, Mounsey JP, Moorman JR. Neurochem Res. 2004;29:177–187. doi: 10.1023/b:nere.0000010447.24128.ac. [DOI] [PubMed] [Google Scholar]
- 39.Morales-Mulia M, Pasantes-Morales H, Moran J. Biochem Biophys Acta. 2000;1496:252–260. doi: 10.1016/s0167-4889(00)00023-9. [DOI] [PubMed] [Google Scholar]
- 40.Moran J, Morales-Mulia M, Pasantes-Morales H. Biochem Biophys Acta. 2001;1538:313–320. doi: 10.1016/s0167-4889(01)00082-9. [DOI] [PubMed] [Google Scholar]
- 41.Therien AG, Goldshleger R, Karlish SJ, Blostein R. J Biol Chem. 1997;272:32628–32634. doi: 10.1074/jbc.272.51.32628. [DOI] [PubMed] [Google Scholar]
- 42.Beguin P, Crambert G, Guennoun S, Garty H, Horisberger JD, Geering K. Embo J. 2001;20:3993–4002. doi: 10.1093/emboj/20.15.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beguin P, Crambert G, Monnet-Tschudi F, Uldry M, Horisberger JD, Garty H, Geering K. Embo J. 2002;21:3264–3273. doi: 10.1093/emboj/cdf330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahmmoud YA, Vorum H, Cornelius F. J Biol Chem. 2000;275:35969–35977. doi: 10.1074/jbc.M005168200. [DOI] [PubMed] [Google Scholar]
- 45.Li C, Grosdidier A, Crambert G, Horisberger JD, Michielin O, Geering K. J Biol Chem. 2004;279:38895–38902. doi: 10.1074/jbc.M406697200. [DOI] [PubMed] [Google Scholar]
- 46.Tadros GM, Zhang XQ, Song J, Carl LL, Rothblum LI, Tian Q, Dunn J, Lytton J, Cheung JY. Am J Physiol Heart Circ Physiol. 2002;283:H1616–1626. doi: 10.1152/ajpheart.00186.2002. [DOI] [PubMed] [Google Scholar]
- 47.MacLennan DH, Asahi M, Tupling AR. Ann N Y Acad Sci. 2003;986:472–480. doi: 10.1111/j.1749-6632.2003.tb07231.x. [DOI] [PubMed] [Google Scholar]
- 48.Seidler T, Miller SL, Loughrey CM, Kania A, Burow A, Kettlewell S, Teucher N, Wagner S, Kogler H, Meyers MB, Hasenfuss G, Smith GL. Circ Res. 2003;93:132–139. doi: 10.1161/01.RES.0000081596.90205.E2. [DOI] [PubMed] [Google Scholar]
- 49.Katanosaka Y, Iwata Y, Kobayashi Y, Shibasaki F, Wakabayashi S, Shigekawa M. J Biol Chem. 2005;280:5764–5772. doi: 10.1074/jbc.M410240200. [DOI] [PubMed] [Google Scholar]
- 50.Iwamoto T, Pan Y, Nakamura TY, Wakabayashi S, Shigekawa M. Biochemistry. 1998;37:17230–17238. doi: 10.1021/bi981521q. [DOI] [PubMed] [Google Scholar]
- 51.McDonough A, Zhang Y, Shin V, Frank JS. Am J Physiol Cell Physiol. 1996;270:C1221–C1227. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- 52.Sweadner KJ, Herrera V, Amato S, Moellmann A, Gibbons D, Repke K. Circ Res. 1994;74:669–678. doi: 10.1161/01.res.74.4.669. [DOI] [PubMed] [Google Scholar]
- 53.Scriven D, Dan P, Moore E. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dostanic I, Schultz Jel J, Lorenz JN, Lingrel JB. J Biol Chem. 2004;279:54053–54061. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]