

Abstract
Background:
Whole-genome comparisons can provide great insight into many aspects of biology. Until recently, however, comparisons were mainly possible only between distantly related species. Complete genome sequences are now becoming available from multiple sets of closely related strains or species.
Results:
By comparing the recently completed genome sequences of Vibrio cholerae, Streptococcus pneumoniae and Mycobacterium tuberculosis to those of closely related species - Escherichia coli, Streptococcus pyogenes and Mycobacterium leprae, respectively - we have identified an unusual and previously unobserved feature of bacterial genome structure. Scatterplots of the conserved sequences (both DNA and protein) between each pair of species produce a distinct X-shaped pattern, which we call an X-alignment. The key feature of these alignments is that they have symmetry around the replication origin and terminus; that is, the distance of a particular conserved feature (DNA or protein) from the replication origin (or terminus) is conserved between closely related pairs of species. Statistically significant X-alignments are also found within some genomes, indicating that there is symmetry about the replication origin for paralogous features as well.
Conclusions:
The most likely mechanism of generation of X-alignments involves large chromosomal inversions that reverse the genomic sequence symmetrically around the origin of replication. The finding of these X-alignments between many pairs of species suggests that chromosomal inversions around the origin are a common feature of bacterial genome evolution.
Background
Large-scale genomic rearrangements and duplications are important in the evolution of species. Previously, these large-scale genome-changing events were studied through genetic or cytological studies. With the availability of many complete genome sequences it is now possible to study such events through comparative genomics. The publication of the yeast genome has led to much better insight into the duplication events that have occurred in fungal and eukaryotic evolution (for example, see [1]). Large chromosomal duplications have also been found from analysis of completed chromosomes of Arabidopsis thaliana [2,3]. The ability to detect large-scale genomic changes is dependent in large part on which genomes are available. Such studies in bacteria, for example, have been limited by the availability of genomes only from distantly related sets of species. Recently, however, the genomes of sets of closely related bacterial species have become available. We have compared these closely related bacterial genomes and have discovered an unusual phenomenon - alignments of whole genomes that show an X-shaped pattern (which we refer to as X-alignments). Here we present the evidence for these X-alignments and discuss mechanisms that might have produced them.
Results and discussion
Whole-genome X-alignments between species at the DNA level
We compared the DNA sequences of the two chromosomes of Vibrio cholerae [4] with the sequence of the Escherichia coli chromosome [5] using a suffix tree alignment algorithm [6]. The analysis revealed a significant alignment at the DNA level between the V. cholerae large chromosome (chrI) [4] and the E. coli chromosome [5] spanning the entire length of these chromosomes (Figure 1a). Analysis of the reverse complement of V. cholerae chrI with E. coli also produced a significant alignment (Figure 1b). When superimposed, the two alignments produce a clear 'X' shape (Figure 1c) that is symmetric about the origin of replication of both genomes. This symmetry indicates that matching sequences tend to occur at the same distance from the origin but not necessarily on the same side of the origin. The X-alignment between V. cholerae and E. coli was found to be statistically significant using a test based on the number of matches found in diagonal strips in the alignment (see the Materials and methods section). Specifically, when V. cholerae chrI is aligned in the forward direction against E. coli, there are 459 maximal unique matching subsequences (MUMs; see the Materials and methods section), of which 177 occurred in a diagonal strip covering 10% of the total area (compared to the expected value of 46). The probability of observing this high a number of MUMs by chance is 4.7 × 10-59. The alignment of V. cholerae chrI in the reverse direction against E. coli (which corresponds to the MUMs on the anti-diagonal) has a probability of 1.8 × 10-90. As a control, we compared the genomes of distantly related species, such as E. coli and Mycobacterium tuberculosis. These do not show a significant X-alignment (Table 1).
Figure 1.
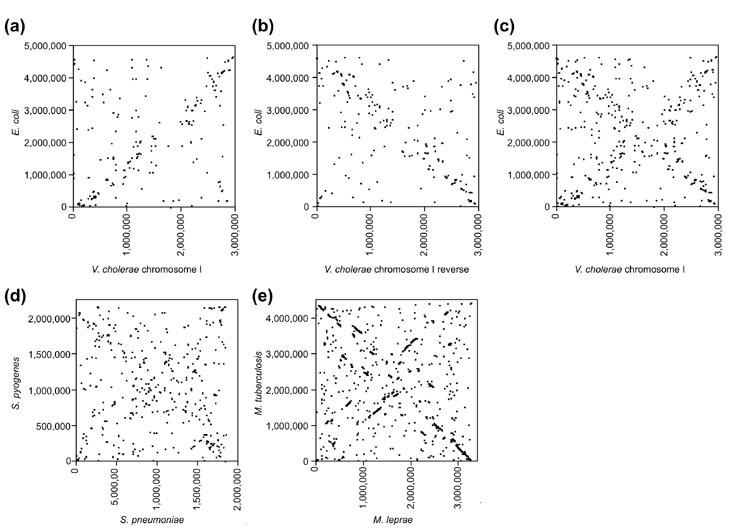
Between-species whole-genome DNA alignments. Plots of maximally unique matching subsequences (MUMs) between genomes as identified by the MUMmer program. (a)V. cholerae chrI forward strand versus E. coli forward strand. (b)E. coli forward versus V. cholerae chrI reverse. (c)V. cholerae chrI versus E. coli, forward and reverse overlaid. (d)S. pneumoniae forward versus S. pyogenes forward and reverse overlaid. (e)M. tuberculosis forward versus M. leprae forward and reverse overlaid. A point (x, y) indicates a DNA sequence that occurs once within each genome, at location x in one genome and at location y in the other genome. The matching sequences may occur on either the forward or the reverse strand; in either case, the locations indicate the 5' end of the sequences. The point (0,0) corresponds to the origin of replication for each genome.
Table 1.
Whole-genome DNA alignments using MUMmer
| Organism 1 | Organism 2 | Total | Matches | Probability* |
| MUMs | within | |||
| diagonal | ||||
| (10%) | ||||
| V. cholerae | E. coli | 459 | 177 | 4.7 ± 10-59 |
| V. cholerae (rev) | E. coli | 467 | 217 | 1.8 ± 10-90 |
| V. cholerae (rev) | V. cholerae | 342 | 86 | 8.2 ± 10-16 |
| E. coli (rev) | E. coli | 1,128 | 225 | 1.5 ± 10-23 |
| S. pyogenes | S. pneumoniae | 706 | 259 | 4.5 ± 10-80 |
| S. pyogenes (rev) | S. pneumoniae | 626 | 255 | 2.3 ± 10-90 |
| S. pyogenes (rev) | S. pyogenes | 367 | 96 | 1.1 ± 10-18 |
| S. pneumoniae (rev) | S. pneumoniae | 1,054 | 154 | 1.5 ± 10-6 |
| M. leprae (rev) | M. leprae | 449 | 89 | 3.5 ± 10-10 |
| M. tuberculosis (rev) | M. tuberculosis | 2,476 | 268 | 0.092 |
| E. coli | M. tuberculosis | 81 | 13 | 0.06 |
| E. coli (rev) | M. tuberculosis | 70 | 5 | 0.84 |
*Statistical significance was estimated as described in the text; rev, reverse complement sequence.
We have found that X-alignments of whole genomes are not limited to the V. cholerae versus E. coli comparison. For example, a whole-genome comparison of two bacteria in the genus Streptococcus - S. pyogenes [7] and S. pneumoniae (H. Tettelin, personal communication) - reveals a global X-alignment similar to that of V. cholerae versus E. coli (Figure 1d) which is also statistically significant (Table 1). In addition, an X-alignment is found between two species in the genus Mycobacterium - M. tuberculosis [8] and M. leprae [9] (Figure 1e) - as well as between two strains of Helicobacter pylori (data not shown). The X-alignments observed between any two pairs of genomes are not identical in every aspect. For example, in the alignment between the two Mycobacterium species, each conserved region is much longer than in the other genome pairs. We believe this is due to different numbers of evolutionary events between the species (see below). Whole-genome X-alignments were not found between any other pairs of species, although a related pattern was seen between some of the chlamydial species (see below).
Whole-genome X-alignments between species are also found at the proteome level
To test whether the X-alignments found in the DNA analysis could also be found at the level of whole proteomes, we conducted comparisons of homologous proteins between species (see the Materials and methods section). Figure 2a shows a scatterplot of chromosome positions of all proteins homologous between V. cholerae chrI and E. coli. The presence of many large gene families causes a great deal of noise in this comparison. This noise can be reduced by considering only the best matching homolog for each open reading frame (ORF), rather than all protein homologs (Figure 2b). This filtered protein comparison results in an X-alignment that is statistically significant (Table 2).
Figure 2.
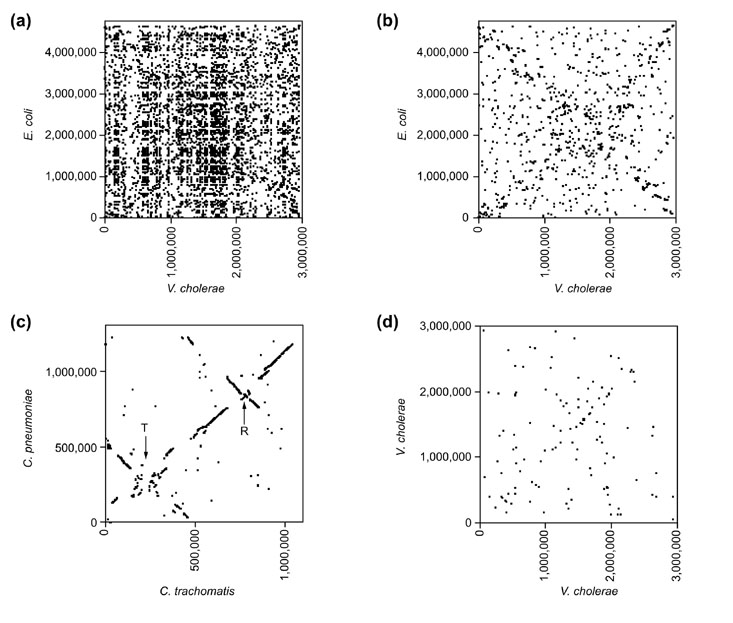
Whole-genome proteome alignments. Plots show the chromosome locations of pairs of predicted proteins that have significant similarity (on the basis of fasta3 comparisons). (a)V. cholerae chrI versus E. coli. All significant matches for each V. cholerae ORF are shown. (b)V. cholerae chrI versus E. coli, top matches. Only the best match for each V. cholerae ORF is shown. The filtering for top matches for each ORF removes noise due to the presence of many large multigene families. This X-alignment is highly statistically significant (Table 2). (c)Chlamydia trachomatis versus C. pneumoniae, top matches. Only the best match for each C. trachomatis ORF is shown. The position of the origins (R) and termini (T) of replication are slightly shifted to see the inversions better. This pattern is consistent with the occurrence a small number of inversions around the origin and terminus in the two lineages since their divergence from a common ancestor (see Figure 4). (d)V. cholerae chrI versus V. cholerae chrI, self-alignment. Only recently duplicated pairs of genes are shown. Recent duplications were operationally defined as those genes that were more similar to another gene in V. cholerae than to any gene in any other complete genome sequence. The faint X-alignment is statistically significant (Table 2). No significant X-alignment was detected when all pairs of paralogs were included.
Table 2.
Whole-genome protein-level comparisons
| Organism 1 | Organism 2 | Total | Matches | Probability* |
| matches | within | |||
| 10% | ||||
| diagonal | ||||
| Top matches | ||||
| V. cholerae | E. coli | 1,797 | 369 | 3.2 ± 10-40 |
| V. cholerae (rev) | E. coli | 1,797 | 441 | 2.3 ± 10-70 |
| V. cholerae | V. cholerae | 701 | 145 | 3.6 ± 10-17 |
| V. cholerae (rev) | V. cholerae | 701 | 70 | 0.52 |
| E. coli | E. coli | 1,985 | 286 | 3.6 ± 10-10 |
| E. coli (rev) | E. coli | 1,985 | 210 | 0.20 |
| Recent duplications† | ||||
| V. cholerae | V. cholerae | 195 | 60 | 1.0 ± 10-15 |
| V. cholerae (rev) | V. cholerae | 195 | 26 | 8.4 ± 10-4 |
*Statistical significance was estimated as described in the text. †Best match to another V. cholerae ORF versus any other complete genome.
Whole-genome X-alignments within species
The finding of the X-alignment pattern between species led us to search for similar patterns within species; that is, global alignments of a genome with its own reverse complement. Of the genomes for which we found between-species X-alignments (M. tuberculosis, M. leprae, S. pyogenes, S. pneumoniae, E. coli and V. cholerae), statistically significant self-alignments are detected for all except M. tuberculosis (Figure 3; probabilities shown in Table 1). Interestingly, these self-alignments are not as strong as those between species. Proteome analysis also shows an X-alignment within species (shown for V. cholerae chrI in Figure 2d; probabilities shown in Table 2). The X-alignment of proteins within V. cholerae chrI is statistically significant only for recently duplicated-genes, but disappears when all paralogs are included. The importance of filtering for recent duplications is discussed below.
Figure 3.
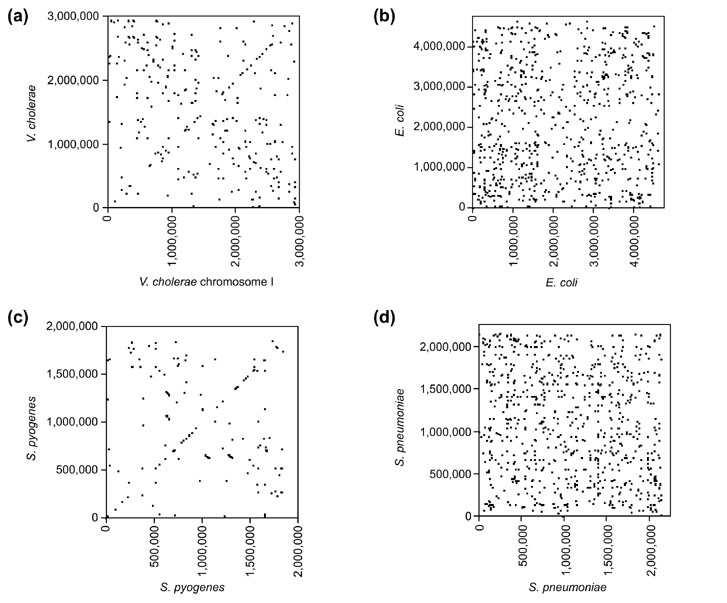
Within-genome DNA alignments. Plots of exactly matching sequences within four genomes as identified by the MUMmer program: (a)V. cholerae; (b)E. coli; (c)S. pyogenes; (d)S. pneumoniae. A point (x,y) indicates a DNA sequence that is repeated within the genome, occurring once at location x and again at location y. Points near the diagonal (y = x) correspond to tandem repeats. Points near the anti-diagonal (y = L - x, where L is genome length) correspond to repeats that occur at symmetric locations about the origin of location. The point (0,0) corresponds to the origin of replication in each plot. Statistically significant X-alignments occur in all four species (Table 1).
Model I: whole-genome inverted duplications
One possible explanation for an X-alignment within and between species is an ancestral inverted duplication of the whole genome, as has been suggested for E. coli [10]. The weak or missing X-alignment within species could be explained by gene loss of one of the two duplicates of many of the pairs of genes in the different lineages. Gene loss has been found to follow large chromosomal or genome duplications [11,12,13]. This gene loss is thought to stabilize large duplications by preventing recombination events between duplicate genes. If gene loss is responsible for the weak X-alignment within species, then to maintain the X-alignments between species, the member of the gene pair lost in a particular lineage should be essentially random. If an ancient inverted duplication followed by differential gene loss is the correct explanation for the observed X-alignments, one would expect the genes along one diagonal to be orthologous between species (related to each other by the speciation event), while the genes along the other diagonal should be paralogous (related to each other by the genome duplication event before the speciation of the two lineages). However, the evidence appears to contradict this model: likely orthologous gene pairs are equally distributed on each diagonal (data not shown).
Model II: chromosomal inversions about the origin and/or terminus
A second possible explanation for the X-alignments is that an underlying mechanism allows sections of DNA to move within the genome but maintains the distance of these sections from the origin and/or terminus. There are a variety of possible mechanisms for such movement, but we believe the most likely explanation is the occurrence of large chromosomal inversions that pivot around the replication origin and/or terminus. Large chromosomal inversions, including those that occur around the replication origin and terminus, have been shown to occur in E. coli and Salmonella typhimurium in the laboratory (see, for example, [14,15,16,17,18]). The occurrence of such inversions over evolutionary time scales was first suggested by comparative analysis of the complete genomes of four strains in the genus Chlamydia [19]. In that study, we found that the major chromosomal differences between C. pneumoniae and C. trachomatis (shown in Figure 2c) were consistent with the occurrence of large inversions that pivoted around the origin and terminus (including multiple inversions of different sizes). In Figure 4 we present a hypothetical model showing how a small number of inversions centered around the origin or terminus could produce patterns very similar to those seen in the Chlamydia, Mycobacterium and Helicobacter comparisons. The continued occurrence of such inversion over longer time scales would result in an X-alignment similar to that seen in the V. cholerae versus E. coli and S. pneumoniae versus S. pyogenes comparisons. Thus the different between-species X-alignments could be the result of different numbers of inversions between particular pairs of species.
Figure 4.
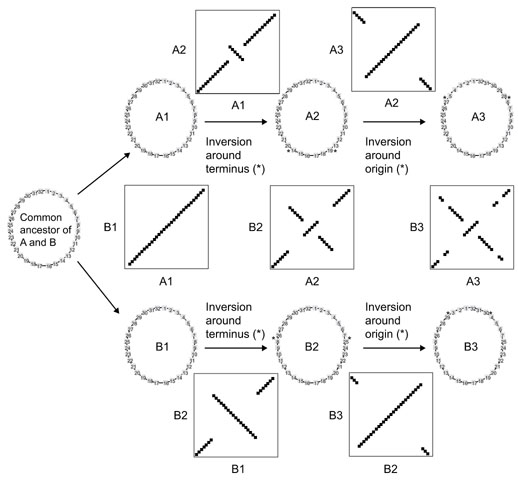
Schematic model of genome inversions. The model shows an initial speciation event, followed by a series of inversions in the different lineages (A and B). Inversions occur between the asterisks (*). Numbers on the chromosome refer to hypothetical genes 1-32. At time point 1, the genomes of the two species are still co-linear (as indicated in the scatterplot of A1 versus B1). Between time point 1 and time point 2, each species (A and B) undergoes a large inversion about the terminus (as indicated in the scatterplots of A1 versus A2 and B1 versus B2). This results in the between-species scatterplot looking as if there have been two nested inversions (A2 versus B2), similar to that seen for C. trachomatis versus C. pneumoniae (see Figure 2). Between time point 2 and time point 3 each species undergoes an additional inversion (as indicated in the scatterplots of B2 versus B3 and A2 versus A3). This results in the between-species scatterplots beginning to resemble an X-alignment, similar to that seen in M. tuberculosis versus M. leprae (see Figure 2).
Inversions about the origin and terminus could also produce an X-alignment within species, through the splitting of tandemly duplicated sequence. Many sets of tandemly duplicated genes are found in most bacterial genomes [19,20] (also see Figure 3a,c). As tandem duplications are inherently unstable (one of the duplicates can be rapidly eliminated by slippage and/or recombination events [21]), the fact that many tandem pairs are present within each genome suggests that tandem duplications occur frequently. Thus, it is reasonable to assume that occasionally a large inversion will split a pair of tandemly duplicated genes. An inversion that pivots about the origin and also splits a tandem duplication will result in a pair of paralogous genes spaced symmetrically on opposite sides of the origin.
If our inversion model is correct, then the genes along both diagonals in the between-species alignments should be orthologous, which is the case (see above). In contrast, genes along the anti-diagonal in the within-species X-alignments should be recent tandem duplicates that have been separated by inversions. This also appears to be the case - in the within-species analysis of V. cholerae chrI ORFs, the X-alignment shows up best when only recent duplicates are analyzed (Figure 2d). The splitting of tandem duplicates by inversions may be a general mechanism to stabilize the coexistence of duplicated genes, as it will prevent their elimination by unequal crossing-over or replication slippage events.
What could cause inversions that pivot around the origin and terminus of the genome to occur more frequently than other inversions? One possibility is that many inversions occur, but there is selection against those that change the distance of a gene from the origin or terminus. Such a possibility has been suggested by experimental work in E. coli [14,15]. Additional studies have, however, suggested that there is little selective difference between inversions and that instead there may be certain regions that are more prone to inversion than others [16,17,18,22,23]. Alternatively, the inversion events could be linked to replication, as has been suggested for small local inversion events [24]. Whatever the mechanisms, the fact that we find evidence for such inversions between many pairs of species suggests that they are a common feature of bacterial evolution. Many aspects of the X-alignments require further exploration. For example, to split a tandem duplication, an inversion must fall precisely on the boundary between two duplicated genes. This would appear to be unlikely, requiring a large number of inversions in order to generate a sufficient number of split gene pairs. If the mechanisms of gene duplication are somehow related to the mechanisms of inversion, however, then this model is more plausible. The process of duplicating a gene, if it occurs during replication, might promote a recombination event within the bacterial chromosome that inverts the sequence from the origin up to that point. As with inversion events, recombination and replication have been found to be tightly coupled [25].
Conclusions
We present here a novel observation regarding the conservation between bacterial species of the distance of particular genes from the replication origin or terminus. The initial observation was only possible due to the availability of complete genome sequences from pairs of moderately closely related species (for example, V. cholerae and E. coli). This shows the importance of having genome pairs from many levels of evolutionary relatedness. Comparisons of distantly related species enable the determination of universal features of life as well as of events that occur very rarely. Comparison of very closely related species allows the identification of frequent events such as transitional changes at third codon positions or tandem duplications. To elucidate all other events in the history of life, genome pairs covering all the intermediate levels of evolutionary relatedness will be needed.
Materials and methods
Genomes analyzed
Complete published genome sequences were obtained from the National Center for Biotechnology Information website [26] or from the TIGR Comprehensive Microbial Resource [27]. These included Aeropyrum pernix [28], Aquifex aeolicus [29], Archaeoglobus fulgidus [30], Bacillus subtilis [31], Borrelia burgdorferi [32], Campylobacter jejuni [33], Chlamydia pneumoniae AR39 [19], Chlamydia pneumoniae CWL029 [34], Chlamydia trachomatis (D/UW-3/Cx) [35], Chlamydia trachomatis MoPn [19], Deinococcus radiodurans [36], Escherichia coli [5], Haemophilus influenzae [37], Helicobacter pylori [38], Helicobacter pylori J99 [39], Methanobacterium thermoautotrophicum [40], Methanococcus jannaschii [41], Mycobacterium tuberculosis [8], Mycoplasma genitalium [42], Mycoplasma pneumoniae [43], Neisseria meningitidis MC58 [20], Neisseria meningitidis serogroup A strain Z2491 [44], Pyrococcus horikoshii [45], Rickettsia prowazekii [46], Synechocystis sp. [47], Thermotoga maritima [48], Treponema pallidum [49], and Vibrio cholerae [4]. In addition, a few unpublished genomes were analyzed: Streptococcus pyogenes (obtained from the Oklahoma University Genome Center website [7]), Streptococcus pneumoniae (H. Tettelin, personal communication), and Mycobacterium leprae (obtained from the Sanger Centre Pathogen Sequencing Group website [9]).
Whole-genome DNA alignments
DNA alignments of the complete genomic sequences of all bacteria used in this study were accomplished with the MUMmer program [6]. This program uses an efficient suffix tree construction algorithm to rapidly compute alignments of entire genomes. The algorithm identifies all exact matches of nucleotide subsequences that are contained in both input sequences; these exact matches must be longer than a specified minimum length, which was set to 20 base pairs for this comparison. To search for genome-scale alignments within species, complete bacterial and archaeal genomes (25 in total including all published genomes) were aligned with their own reverse complements. To search for between-species alignments, all genomes were aligned against all others in both orientations.
Whole-genome protein comparisons
The predicted proteome of each complete genome sequence (all predicted proteins in the genome) was compared to the proteomes of all complete genome sequences (including itself) using the fasta3 program [50]. Matches with an expected score (e-value) of 10-5 or less were considered significant.
Statistical significance of X-alignments
To calculate the statistical significance of the X-alignments, the maximal unique matching subsequences (MUMs) for unrelated genomes were examined and found to be uniformly distributed [6]. With a uniform background, the expected density of MUMs in any region of an alignment plot is a simple proportion of the area of that region to the entire plot. In particular, in an alignment with N total MUMs, the probability (Pr) of observing at least m matches in a region with area p can be computed using the binomial distribution in Equation 1:
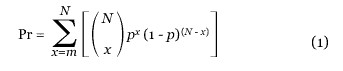
The alignment of V. cholerae chrI (both forward and reverse strands) versus E. coli contains 926 MUMs. The MUMs forming X-alignments appear along the diagonal (y = x) and the anti-diagonal (y = L -x, where L is the genome length). To estimate the significance of the alignments in both directions, diagonal strips were sampled along each of the diagonals. The width of each strip was set at 10% of the plot area and significance values were calculated (Table 1).
Identification of origins of replication
The origins of replication for the bacterial genomes have been characterized by a variety of methods. For E. coli, M. tuberculosis and M. leprae, the origins have been well-characterized by laboratory studies [51,52]. The origins and termini of C. trachomatis, C. pneumoniae and V. cholerae were identified by GC-skew [53] and by characteristic genes in the region of the origin [4,19]. GC-skew uses the function (G-C)/(G+C) computed on 2,000 bp windows across the genome, which exhibits a clear tendency in many bacterial genomes to be positive for the leading strand and negative for the lagging strand. The origin of H. pylori was determined by oligomer skew [54] and confirmed by GC-skew. The origins and termini of S. pneumoniae and S. pyogenes were determined by the authors of the present study using GC-skew analysis and the locations of characteristic genes, particularly the chromosome replication initiator gene dnaA.
Acknowledgments
Acknowledgements
We thank S. Eddy, M.A. Riley, T. Read, A. Stoltzfus, M-I Benito and I. Paulsen for helpful comments, suggestions and discussions. S.L.S. was supported in part by NSF grant IIS-9902923 and NIH grant R01 LM06845. S.L.S. and J.A.E were supported in part by NSF grant KDI-9980088. Data for all published complete genome sequences were obtained from the NCBI genomes database [26] or from The Institute for Genomic Research (TIGR) Microbial Genome Database [27]. The sequences of V. cholerae, S. pneumoniae, and M. tuberculosis (CDC 1551) were determined at TIGR with support from NIH and the NIAID. The M. leprae sequence data were produced by the Pathogen Sequencing Group at the Sanger Centre. Sequencing of M. leprae is funded by the Heiser Program for Research in Leprosy and Tuberculosis of The New York Community Trust and by L'Association Raoul Follereau. The M. tuberculosis CDC 1551 genome sequence was obtained from TIGR. The source of the S. pyogenes genome sequence was the Streptococcal Genome Sequencing Project funded by USPHS/NIH grant AI38406, and was kindly made available by B. A. Roe, S.P. Linn, L. Song, X. Yuan, S. Clifton, R.E. McLaughlin, M. McShan and J. Ferretti, and can be obtained from the website of the Oklahoma University Genome Center [7].
References
- Seoighe C, Wolfe KH. Updated map of duplicated regions in the yeast genome. Gene. 1999;238:253–261. doi: 10.1016/s0378-1119(99)00319-4. [DOI] [PubMed] [Google Scholar]
- Lin X, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Mayer K, Schuller C, Wambutt R, Murphy G, Volckaert G, Pohl T, Dusterhoft A, Stiekema W, Entian KD, Terryn N, et al. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature. 1999;402:769–777. doi: 10.1038/47134. [DOI] [PubMed] [Google Scholar]
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, et al. The genome sequence of Vibrio cholerae, the aetiologic agent of cholera. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett GI, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL. Alignment of whole genomes. Nucleic Acids Res. 1999;27:2369–2376. doi: 10.1093/nar/27.11.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oklahoma University Genome Center http://www.genome.ou.edu/strep.html
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Sanger Centre Pathogen Sequencing Group ftp://ftp.sanger.ac.uk/pub/pathogens/leprae
- Zipkas D, Riley M. Proposal concerning mechanism of evolution of the genome of Escherichia coli. Proc Natl Acad Sci USA. 1975;72:1354–1358. doi: 10.1073/pnas.72.4.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. The fate of duplicated genes: loss or new function? BioEssays. 1998;20:785–788. doi: 10.1002/(SICI)1521-1878(199810)20:10<785::AID-BIES2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Sankoff D. Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics. 1997;147:1259–1266. doi: 10.1093/genetics/147.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois V, Louarn J, Patte J, Rebollo JE, Louarn JM. Constraints in chromosomal inversions in Escherichia coli are not explained by replication pausing at inverted terminator-like sequences. Mol Microbiol. 1990;4:537–542. doi: 10.1111/j.1365-2958.1990.tb00621.x. [DOI] [PubMed] [Google Scholar]
- Rebollo JE, Francois V, Louarn JM. Detection and possible role of two large nondivisible zones on the Escherichia coli chromosome. Proc Natl Acad Sci USA. 1988;85:9391–9395. doi: 10.1073/pnas.85.24.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall A, Mahan MJ, Roth JR. Rearrangement of the bacterial chromosome: forbidden inversions. Science. 1988;241:1314–1318. doi: 10.1126/science.3045970. [DOI] [PubMed] [Google Scholar]
- Mahan MJ, Roth JR. Ability of a bacterial chromosome segment to invert is dictated by included material rather than flanking sequence. Genetics. 1991;129:1021–1032. doi: 10.1093/genetics/129.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall AM, Roth JR. Recombination between homologies in direct and inverse orientation in the chromosome of Salmonella : intervals which are nonpermissive for inversion formation. Genetics. 1989;122:737–747. doi: 10.1093/genetics/122.4.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, Ketchum KA, Hood DW, Peden JF, Dodson RJ, et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science. 2000;287:1809–1815. doi: 10.1126/science.287.5459.1809. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MB, Roth JR. Selection and endpoint distribution of bacterial inversion mutations. Genetics. 1983;105:539–557. doi: 10.1093/genetics/105.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan MJ, Roth JR. Reciprocality of recombination events that rearrange the chromosome. Genetics. 1988;120:23–35. doi: 10.1093/genetics/120.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AJ, Halliday JA. Inversions with deletions and duplications. Genetics. 1995;140:411–414. doi: 10.1093/genetics/140.1.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Morales E, Romero D. Recombination enhancement by replication (RER) in Rhizobium etli. Genetics. 2000;154:971–983. doi: 10.1093/genetics/154.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information, Entrez Genomes http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Genome
- The Institute for Genomic Research Microbial Genome Database http://www.tigr.org/tdb/mdb/mdb.html
- Kawarabayasi Y, Hino Y, Horikawa H, Yamazaki S, Haikawa Y, Jin-no K, Takahashi M, Sekine M, Baba S, Ankai A, et al. Complete genome sequence of an aerobic hyper-thermophilic crenarchaeon, Aeropyrum pernix K1. DNA Res. 1999;6:83–101. doi: 10.1093/dnares/6.2.83. [DOI] [PubMed] [Google Scholar]
- Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Grahams DE, Overbeek R, Snead MA, Keller M, Aujay M, et al. The complete genome of the hyperthemophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- Klenk H-P, Clayton RA, Tomb J-F, White O, Nelsen KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, et al. The complete genomic sequence of the hyperthermophilic, sulfate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- Kunst A, Ogasawara N, Moszer I, Albertini A, Alloni G, Azevedo V, Bertero M, Bessieres P, Bolotin A, Borchert S, et al. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, et al. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, et al. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman RW, Olinger L, Grimwood J, Davis RW, Stephens RS. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet. 1999;21:385–389. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, et al. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Alm RA, Ling LS, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, deJonge BL, et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature. 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- Smith DR, Doucette-Stamm LA, Deloughery C, Lee H, Dubois J, Aldredge T, Bashirzadeh R, Blakely D, Cook R, Gilbert K, et al. Complete genome sequence of Methanobacterium thermoautotrophicum DH: functional analysis and comparative genomics. J Bacteriol. 1996;179:7135–7155. doi: 10.1128/jb.179.22.7135-7155.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, Fitzgerald LM, Clayton RA, Gocayne JD, et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, Herrmann R. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Achtman M, James KD, Bentley SD, Churcher C, Klee SR, Morelli G, Basham D, Brown D, Chillingworth T, et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature. 2000;404:502–506. doi: 10.1038/35006655. [DOI] [PubMed] [Google Scholar]
- Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, et al. Complete sequence and gene organization of the genome of a hyperthermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281:375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Marsh RC, Worcel A. A DNA fragment containing the origin of replication of the Escherichia coli chromosome. Proc Natl Acad Sci USA. 1977;74:2720–2724. doi: 10.1073/pnas.74.7.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar L, Fsihi H, de Rossi E, Riccardi G, Rios C, Cole ST, Takiff HE. Organization of the origins of replication of the chromosomes of Mycobacterium smegmatis, Mycobacterium leprae and Mycobacterium tuberculosis and isolation of a functional origin from M. smegmatis. Mol Microbiol. 1996;20:283–293. doi: 10.1111/j.1365-2958.1996.tb02617.x. [DOI] [PubMed] [Google Scholar]
- Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Salzberg AJ, Kerlavage AR, Tomb JF. Skewed oligomers and origins of replication. Gene. 1998;217:57–67. doi: 10.1016/s0378-1119(98)00374-6. [DOI] [PubMed] [Google Scholar]