

Abstract
In a strategy to specifically target complement inhibitors to sites of complement activation and disease, recombinant fusion proteins consisting of a complement inhibitor linked to a C3 binding region of complement receptor (CR) 2 were prepared and characterized. Natural ligands for CR2 are C3 breakdown products deposited at sites of complement activation. Fusion proteins were prepared consisting of a human CR2 fragment linked to either the N terminus or C terminus of soluble forms of the membrane complement inhibitors decay accelerating factor (DAF) or CD59. The targeted complement inhibitors bound to C3-opsonized cells, and all were significantly more effective (up to 20-fold) than corresponding untargeted inhibitors at protecting target cells from complement. CR2 fusion proteins also inhibited CR3-dependent adhesion of U937 cells to C3 opsonized erythrocytes, indicating a second potential anti-inflammatory mechanism of CR2 fusion proteins, since CR3 is involved in endothelial adhesion and diapedesis of leukocytes at inflammatory sites. Finally, the in vivo validity of the targeting strategy was confirmed by the demonstration that CR2-DAF, but not soluble DAF, targets to the kidney in mouse models of lupus nephritis that are associated with renal complement deposition.
Introduction
Complement is an important component of immunity, but inappropriate and excessive activation of the complement system is involved in numerous pathological conditions. Complement activation products that mediate tissue injury are generated at various points in the complement pathway. Complement activation on a cell surface results in the cleavage of serum C3 and the covalent attachment of C3 fragments that serve as opsonins for immune effector cells. C3 cleavage also results in the generation of C3a, a soluble anaphylatoxic peptide. Later in the pathway, serum C5 is cleaved to release soluble C5a, a potent anaphylatoxin and chemoattractant with a wide range of bioactive properties. Cleavage of C5 initiates formation of the membrane attack complex (MAC), a cytolytic protein complex that assembles in cell membranes (for a detailed description of the complement system and activation pathways, see Rother et al. [ref. 1]).
Various types of complement-inhibitory proteins are currently under investigation for therapy of inflammatory disease and disease states associated with bioincompatibility (2). Two of the best therapeutically characterized inhibitors of human complement are a soluble form of complement receptor 1 (sCR1) and an anti-C5 monoclonal antibody. These systemically active inhibitory proteins have shown efficacy in various animal models of disease and more recently in clinical trials (3–9). Anti-C5 mAb inhibits the generation of C5a and the MAC, whereas sCR1 is an inhibitor of complement activation and also blocks the generation of C3 activation products. Soluble forms of human decay accelerating factor (DAF) and membrane cofactor protein (MCP), membrane inhibitors of complement activation, have also been shown to be protective in animal models of inflammation and bioincompatibility (10–14). CD59 is a membrane inhibitor of complement that blocks assembly of the MAC by binding to C8 and C9 but does not affect generation of complement opsonins or C3a and C5a. Soluble forms of CD59 (sCD59) have been produced, but low functional activity in vitro, particularly in the presence of serum, suggests that unmodified sCD59 will have little or no therapeutic efficacy (15–18).
Targeting complement inhibitors to sites of complement activation and disease is likely to improve their efficacy. Since complement plays an important role in host defense and immune complex catabolism, targeted complement inhibitors may also reduce potentially serious side effects resulting from systemic complement inhibition, particularly long-term complement inhibition. Recently, a modified form of sCR1 decorated with sialyl Lewisx (sLex) was prepared and shown to bind to endothelial cells expressing P and E selectin. sCR1sLex was shown to be a more potent therapeutic agent than sCR1 in rodent models of inflammatory disease (19–21). Specific targeting of complement inhibitors to a cell surface has been achieved by linking complement inhibitors to antibody fragments containing an antigen binding site. In feasibility studies, antibody-DAF (22) and antibody-CD59 (23) fusion proteins were more effective in vitro at protecting targeted cells than untargeted cells from complement. Nonspecific membrane targeting of recombinant complement inhibitors has also been achieved by coupling inhibitors to membrane-inserting peptides (24, 25). Here, we describe a novel means to target complement-inhibitory proteins that may have much broader therapeutic potential than previously described targeting strategies.
C3 activation fragments are abundant complement opsonins found at sites of complement activation, and they serve as ligands for various C3 receptors. One such receptor, complement receptor 2 (CR2), plays an important role in humoral immunity by way of its expression on mature B cells and follicular dendritic cells (26, 27). CR2 is a member of the C3 binding protein family and consists of 15 or 16 short consensus repeat (SCR) domains, structural units that are characteristic of these proteins (28, 29). Natural ligands for CR2 are iC3b, C3dg, and C3d, cell-bound breakdown fragments of C3 that bind to the two N-terminal SCR domains of CR2 (30, 31). Cleavage of C3 results initially in the generation and deposition of C3b on the activating cell surface. The C3b fragment is involved in the generation of enzymatic complexes that amplify the complement cascade. On a cell surface, C3b is rapidly converted to inactive iC3b, particularly when deposited on a host surface containing regulators of complement activation (i.e., most host tissue). Even in the absence of membrane-bound complement regulators, substantial levels of iC3b are formed because of the action of serum factor H. iC3b is subsequently digested to the membrane-bound fragments C3dg and then C3d by factor I and other proteases, but this process is relatively slow (32, 33). Thus, the C3 ligands for CR2 are relatively long lived once they are generated and will be present in high concentrations at sites of complement activation. For example, these C3 activation fragments have been shown to be present in the glomerulus in several forms of human glomerulonephritis (34). Because of the presence of these ligands, we considered soluble CR2 a rational choice as a targeting moiety for delivery of complement inhibitors to sites of complement-associated disease. We demonstrate herein that CR2-targeted DAF and CD59 bind to C3-coated targets and are significantly more potent than their untargeted counterparts at providing protection from complement. We corroborate previous data showing that sCD59 is not an effective inhibitor of complement-mediated lysis but show that CR2-mediated targeting of CD59 significantly increases its complement-inhibitory activity. Data are also presented that indicate CR2 fusion proteins act as antagonists of CR3 (Mac-1, CD11b/CD18), a leukocyte receptor involved in leukocyte adhesion and activation. Finally, using a mouse model of lupus nephritis that is associated with renal complement deposition, we show that a CR2-linked inhibitor is preferentially targeted to the kidney.
Methods
Cell lines and DNA.
All DNA manipulations were carried out in the mammalian expression vector PBM, derived from p118-mIgG1 (35) by deletion of mouse IgG1 Fc coding region. CHO cells used for protein expression were maintained in DMEM (GIBCO; Invitrogen Corp., Carlsbad, California, USA) supplemented with 10% FCS. Stably transfected CHO cell clones were cultivated in the presence of G418, and for recombinant protein expression, cells were cultured in suspension in serum-free medium for growth of CHO cells in suspension culture (CHO-S-SFM II) without FCS (Invitrogen Corp.). U937 cells were cultured in RPMI (Invitrogen Corp.) and 10% FCS.
Antibodies, reagents, and serum.
Rabbit antiserum to CHO cell membranes, human DAF, and CD59 were prepared by standard techniques (36). Mouse anti-DAF mAbs 1H4 (37) and 1A10 (38), rat anti-CD59 mAb YTH53.1 (39), and mouse anti human CR2 mAb 171 (binds to SCR 1-2) (40) are described. Anti-sheep erythrocyte IgM was from Research Diagnostic Inc. (Flanders, New Jersey, USA). All secondary antibodies were purchased from Sigma-Aldrich (St. Louis, Missouri, USA). Purified recombinant sCD59 was a gift from B.P. Morgan (University of Wales, Cardiff, United Kingdom). C6-depleted human serum was purchased from Quidel (San Diego, California, USA) and normal human serum (NHS) was obtained from the blood of healthy volunteers in the laboratory.
Construction of expression plasmids and protein expression.
The recombinant fusion proteins and soluble complement inhibitors prepared are depicted in Figure 1. cDNA constructs were prepared by joining the CR2 sequence encoding the four N-terminal SCR units (residues 1–250 of mature protein, Swissprot accession number P20023) to sequences encoding extracellular regions of DAF or CD59. The complement inhibitor sequences used encoded residues 1–249 of mature DAF protein sequence (Swissprot accession number P08174) and residues 1–77 of mature CD59 protein sequence (Swissprot accession number P13987). To join CR2 to complement inhibitor sequences, linking sequences encoding SS(GGGGS)3 and (GGGS)2 were used for fusion proteins containing CR2 at the C terminus and N terminus, respectively. Gene constructs were prepared by standard PCR methods (41). All cloning steps were performed in the PBM vector that was also used for protein expression (35). For expression, plasmids were transfected into CHO cells using lipofectamine according to the manufacturer’s instructions (Invitrogen Corp.). Stably transfected clones were selected by limiting dilution as described (35), and protein expression of clones was quantitated by ELISA.
Figure 1.

Diagram of the various CR2 complement-inhibitor fusion proteins.
ELISA and protein assays.
Detection of recombinant proteins and determination of relative protein concentration in culture supernatants was achieved using a standard ELISA technique (36). Depending on which type of recombinant protein was being assayed, the capture antibody was either anti-DAF mAb 1H4 or anti-CD59 mAb YTH53.1. Primary detection antibodies were either anti-DAF or anti-CD59 rabbit polyclonal antibody. In some ELISAs, anti-CR2 mAb 171 was also used as primary detection antibody, and although the assay was less sensitive with this antibody, similar data were obtained. The protein concentration of purified recombinant proteins was determined using a bicinchoninic acid protein assay kit (Pierce Chemical Co., Rockford, Illinois, USA).
Protein purification.
Recombinant proteins were purified from culture supernatant by affinity chromatography. Affinity columns were prepared by coupling either anti-DAF 1H4 mAb or anti-CD59 YTH53.1 mAb to HiTrap NHS-activated affinity columns (Pharmacia Biotech, Piscataway, New Jersey, USA) according to the manufacturer’s instructions. Culture supernatants containing recombinant proteins were adjusted to pH 8.0 and applied to affinity columns at a flow rate of 0.5 ml per minute. The column was washed with 6–8 vol of PBS, and recombinant proteins were eluted with 2–3 column vol of 0.1 M glycine (pH 2.4). The fractions containing fusion protein were collected into tubes containing 1 M Tris buffer (pH 8.0) and dialyzed against PBS.
SDS-PAGE and Western blotting.
Purified recombinant proteins were separated in SDS-PAGE 10% acrylamide gels (Bio-Rad Life Science, Hercules, California, USA) under nonreducing conditions. Gels were stained with Coomassie blue. For Western blotting, standard procedures were followed (36). Briefly, separated proteins were transferred to a polyvinylidene fluoride membrane, and the transferred proteins were detected by means of either anti-DAF mAb 1H4 or anti-CD59 mAb YTH53.1. Membranes were developed with an ECL detection kit (Amersham Biosciences, Piscataway, New Jersey, USA). CR2-CD59 was also analyzed by SDS-PAGE after glycanase treatment. CR2-CD59 (2 μg) was heated at 95°C for 3 minutes in 15 mM sodium phosphate buffer (pH 7.5) containing 0.1% SDS, 10 mM 2-mercaptoethanol, and 5 mM EDTA. After cooling, CR2-CD59 was incubated with 3 U of Flavobacterium meningosepticum N-glycanase (EC 3.5.1.52, Sigma-Aldrich) for 20 hours at 37°C in the presence of 1% Nonidet P40 and 0.3 mM PMSF.
Flow cytometry.
Binding of recombinant fusion proteins to C3-opsonized cells was determined by flow cytometry. CHO cells were incubated in 10% anti-CHO antiserum (30 minutes at 4°C), washed, and opsonized with C3 by incubation in 10% C6-depleted NHS (45 minutes at 37°C). The C3-opsonized cells were then washed and incubated with 1 μM recombinant protein (60 minutes at 4°C). After washing, cells were incubated with 10 μg/ml of either anti-DAF mAb 1H4 or anti-CD59 mAb YTH53.1 as appropriate (30 minutes at 4°C), followed by FITC-conjugated secondary antibody (1:100, 30 minutes at 4°C). Cells were then washed, fixed with 2% paraformaldehyde in PBS, and analyzed using a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, California, USA). All incubations and washes were performed in DMEM.
Analysis of CR2 fusion protein binding to C3 ligand.
Kinetic analysis of the interaction of the CR2 fusion proteins with C3dg-biotin was performed using surface plasmon resonance (SPR) measurements made on a BIAcore 3000 instrument (Biacore AB, Uppsala, Sweden). Human C3dg-biotin, prepared as described (42), was bound to the surface of BIAcore streptavidin sensor chips by injecting C3dg-biotin at 50 μg/ml over the surface of one flow cell of the chip at 2 μl per minute for 20 minutes. The flow buffer was 0.5× PBS (pH 7.4) plus 0.05% Tween 20. The SPR signal from captured C3dg generated BIAcore response units ranging from 250 to 500. Control streptavidin-coated flow cells were run in the absence of protein. Binding was evaluated over a range of CR2 fusion protein concentrations (15.6–500 nM) in 0.5× PBS and 0.05% Tween 20 at 25°C at a flow rate of 25 μl per minute. CR2 fusion protein samples were injected in 50 μl aliquots using the kinject command. Association of the fusion proteins with the ligand was monitored for 120 seconds, after which the complex was allowed to dissociate in the presence of buffer only for an additional 120 seconds. The binding surface was regenerated between analyses of different fusion protein concentrations by a 10-second pulse of 200 mM sodium carbonate (pH 9.5) at 50 μl per minute. Binding of CR2 fusion protein fragments to C3dg-immobilized flow cells was corrected for binding to control flow cells. Binding data were fitted to a 1:1 Langmuir binding model using BIAevaluation Version 3.1 software (BIAcore) and evaluated for best fit by low residual and χ2 values. The kinetic dissociation profiles obtained were used to calculate on and off rates (ka and kd) and affinity constants (KD) using the BIAevaluation Version 3.1 program. Between experiments, the streptavidin surface was regenerated with a 60-second pulse of 50 mM sodium hydroxide (pH 12.5) at 50 μl per minute, and C3dg-biotin was reapplied as described above.
Complement lysis assays.
CHO cells at 60–80% confluence were detached with versene (Invitrogen Corp.), washed twice, and resuspended to 106 cells per milliliter in DMEM. Cells were sensitized to complement by adding 10% rabbit anti-CHO cell membrane antiserum to cells (30 minutes at 4°C). Antiserum was then removed, and cells were resuspended in NHS diluted in DMEM. Final assay volumes were either 50 or 100 μl. After 60 minutes at 37°C, cell viability was determined either by trypan blue exclusion (both live and dead cells counted) or 51Cr release (43). Both assays gave similar results. To assay complement-inhibitory activity of recombinant proteins, the proteins were diluted in DMEM and added to NHS before addition to CHO cells. A final concentration of 10% NHS was used that resulted in approximately 90% lysis of unprotected antibody-sensitized CHO cells. Inhibition of complement-mediated hemolysis was determined using antibody-sensitized sheep erythrocytes (EAs) (Advanced Research Technologies, San Diego, California, USA). Hemolytic assays were carried out in gelatin veronal buffer (GVB++) (Advanced Research Technologies) in a final volume of 300 μl containing 2.5 × 107 EAs, NHS at a final dilution of 1:300, and incremental concentrations of fusion protein. Reaction mixtures were incubated at 37°C for 60 minutes, and reactions were stopped by addition of 300 μl of PBS containing 10 mM EDTA. Cells were removed by centrifugation, and cell lysis was assayed by spectrophotometric quantitation of hemoglobin in the supernatant at 413 nm.
Adhesion of U937 cells to erythrocytes.
Assays of CR3-dependent adhesion to C3-opsonized erythrocytes were performed essentially as described (44). Briefly, fresh sheep erythrocytes (SRBCs) were sensitized with a predetermined subagglutinating amount of rabbit anti-SRBC IgM for 30 minutes at 37°C in GVB (Advanced Research Technologies). After washing twice, C3b-opsonized SRBCs were prepared by incubating IgM-sensitized SRBCs with an equal volume of a 1:2 dilution of C6-deficient human serum in GVB++ (120 minutes at 37°C). Cells were washed twice and pellets resuspended in GVB. Most of the C3 bound to erythrocytes after this treatment is in the form of iC3b or C3d degradation products (CR2 ligands) because of the short half-life of C3b in serum. U937 cells (4 × 105 cells in 200 μl) were added to 50 μl of C3-opsonized SRBCs (2 × 106 cells), and the mixture was centrifuged (4 minutes per 40 g) and left at room temperature for 90 minutes. Cells were then examined by phase-contrast microscopy, and the number of U937 cells adherent to erythrocytes was determined. At least 100 erythrocytes were scored per sample, and the average number of U937 cells bound per erythrocyte was calculated. Triplicate determinations were made for each experiment performed. In some experiments, U937 cells were cultured for 3 days in the presence of 50 ng/ml PMA before harvest, a treatment that results in upregulation of CR3 (45, 46). Cells incubated with IgM-coated SRBCs alone or SRBCs incubated directly with C6-deficient human serum were used as controls.
Biodistribution studies.
Standard procedures for determining tissue distribution of injected radiolabeled proteins were followed (47, 48). Briefly, 1.7 μg of 125I-labeled CR2-DAF or soluble DAF (sDAF) was injected into the tail vein of 34-week-old (CR2-DAF at 4.20 × 106 cpm/μg and sDAF at 4.84 × 106 cpm/μg) or 8-week-old (CR2-DAF at 3.75 × 106 cpm/μg and sDAF at 3.58 × 106 cpm/μg) female NZB/NZW F1 mice (Jackson Labs, Bar Harbor, Maine, USA). Twenty-four or forty-eight hours after injection, mice were sacrificed, a blood sample was taken, and major organs were removed, shredded, and washed in PBS containing 10 mM EDTA and then weighed and counted. Targeting specificity was evaluated as percent injected dose per gram of tissue. Proteins were iodinated using the iodogen method according to the manufacturer’s instructions (Pierce Chemical Co.).
Immunofluorescence microscopy.
CR2-DAF or sDAF (270 μg) was injected into the tail vein of 24-week-old MRL/lpr mice. Twenty-four hours later, kidneys were removed and snap frozen. Cryostat sections (5 μM) prepared from frozen kidneys were fixed in acetone and processed for indirect immunofluorescence microscopy. An equimolar mixture of mouse anti-human DAF 1A10 and 1H6 mAbs were used as primary detection antibodies (final concentration, 10 μg/ml) with an anti-mouse IgG Fc-specific FITC-conjugated secondary antibody (F4143, Sigma-Aldrich). Standard procedures were followed (49), except that to reduce background staining, most likely caused by deposited immune complexes in the mouse kidney, the secondary FITC-labeled antibody was diluted 1:800 (10 times the recommended dilution). Digital images were acquired and optimized with Adobe Photoshop using identical settings.
Results
Construct design, expression, and purification.
Recombinant fusion proteins contained the four N-terminal SCR units of human CR2 linked to either the N terminus or the C terminus of soluble forms of human CD59 or DAF (constructs depicted in Figure 1). Recombinant proteins were purified from the culture supernatant of stably transfected CHO cell clones with yields of between 100 and 200 μg/l. Analysis of purified recombinant proteins by SDS-PAGE and Western blot revealed proteins within the expected molecular weight range (Figure 2), and except for CR2-CD59, all proteins migrated as a single band. The two bands seen for CR2-CD59 were due to differences in glycosylation, since CR2-CD59 migrated as a single band after glycanase treatment (data not shown).
Figure 2.

SDS-PAGE and Western blot analysis of purified recombinant fusion proteins and soluble complement inhibitors. Gels (10% acrylamide) were stained with Coomassie blue. Western blots were developed using antibodies to complement inhibitors as the primary antibody. MW, molecular weight; stds, standards.
Targeting of fusion proteins to complement opsonized cells.
C3 ligand for CR2 was deposited on CHO cells by incubation of CHO cells with complement-activating antibody and C6-depleted serum (to prevent MAC formation and cell lysis). C3 opsonization was confirmed by flow cytometric analysis using an antibody specific for human iC3b (50). All CR2-containing fusion proteins, but not sCD59 or sDAF, bound to C3-coated CHO cells (Figure 3).
Figure 3.

Binding of recombinant fusion proteins to C3-opsonized CHO cells. Antibody-sensitized CHO cells were incubated in C6-deficient human serum, washed, and incubated with soluble complement inhibitor (thin black trace) or fusion protein with CR2 at the N terminus (light gray trace) or C terminus (dark gray trace) at 20 μg/ml. Cell binding of recombinant proteins was detected by flow cytometry using anti-DAF or anti-CD59 mAbs followed by a FITC-labeled secondary antibody. Incubation of CHO cells with PBS instead of complement inhibitor is shown in the right-hand panel. Data are representative of three separate experiments.
Kinetic analysis of interaction between fusion proteins and C3dg ligand.
A comparison of the affinity of the different recombinant fusion proteins for the CR2 ligand C3dg was determined by SPR measurements. The experiments were performed by passing varying concentrations of the fusion proteins over Biacore streptavidin chips containing captured C3dg-biotin. Kinetic analysis of the data showed the best fit to a 1:1 (Langmuir) binding interaction model using global fitting parameters (Figure 4). Both of the fusion proteins with CR2 at the N terminus (CR2-DAF and CR2-CD59) showed similar binding profiles, with fast association and dissociation rates. In contrast, binding of fusion proteins with CR2 at the C terminus (DAF-CR2 and CD59-CR2) showed slow association and dissociation rates (Figure 4 and Table 1). The N-terminal CR2 fusion proteins, however, bound with the highest affinity (Table 1). CD59 fusion proteins bound with a higher affinity than DAF fusion proteins. Affinity constants of the CR2 fusion proteins for C3dg were between the ranges previously published for the interaction between CR2 and C3dg (42, 51). Soluble DAF and sCD59 did not bind to immobilized C3dg (data not shown). Although N-terminal CR2 fusion proteins bind C3dg with a higher affinity than C-terminal counterparts, flow cytometry indicated that a larger amount of the C-terminal CR2 fusion proteins bound to C3-opsonized CHO cells than did the corresponding N terminus CR2 fusion proteins (Figure 3). The reason for this may be due to different geometries and accessible surface domains of bound fusion proteins that may affect the interaction of anti-DAF or anti-CD59 detection antibodies used for flow cytometry or, alternatively, to differences in the form(s) of C3 activation fragments on the cell surface as compared with C3dg on Biacore chips. Further analysis is needed to resolve this issue, but the results of functional studies reported below support the former interpretation.
Figure 4.

Analysis of interaction between CR2 fusion proteins and C3dg by SPR. Solid lines indicate various concentrations of CR2 fusion proteins. Dashed lines show curves fitting to a 1:1 Langmuir binding model.
Table 1.
Kinetic parameters for recombinant fusion protein binding to immobilized C3dg-biotin

Complement-inhibitory activity of fusion proteins.
Complement-inhibitory activity of the targeted and untargeted complement inhibitors was analyzed by measuring their effect on complement-mediated lysis of both CHO cells and erythrocytes. In these experiments, antibody-sensitized cells and recombinant proteins were incubated in human serum at a concentration that resulted in 90–100% lysis of unprotected cells. For both cell types, the targeted complement inhibitors were significantly more effective than their respective untargeted proteins at inhibiting complement-mediated lysis. Targeted DAF proteins were more effective inhibitors than targeted CD59 (Figures 5 and 6). Fusion proteins containing CR2 linked to the N terminus of either DAF or CD59 were more effective inhibitors than C-terminal CR2 fusion proteins. The most potent inhibitor of complement lysis was CR2-DAF, requiring a concentration of 18 nM for 50% inhibition of CHO cell lysis. In contrast, untargeted sDAF required a concentration of 375 nM for 50% inhibition of CHO cell lysis, a 20-fold difference (Figure 5a). sCD59 was a particularly poor inhibitor of complement and provided only 25% protection from CHO cell lysis at 500 nM, the highest concentration tested. CR2-CD59, however, provided 50% inhibition of CHO cell lysis at 102 nM and was more effective than untargeted sDAF (Figure 6a). Table 2 compares the inhibitory activities of the various recombinant complement inhibitors. The higher complement-inhibitory activity of the N-terminal CR2 fusion proteins correlated with the higher affinity these proteins exhibited for C3dg ligand (Table 1).
Figure 5.

Inhibition of complement-mediated lysis by recombinant sDAF and DAF fusion proteins. Antibody-sensitized CHO cells (a) or SRBCs (b) were incubated with recombinant protein and 10% human serum (CHO cells) or 0.33% human serum (erythrocytes). These concentrations resulted in approximately 90% lysis of unprotected cells. Lysis was determined after 60 minutes at 37°C. Background lysis determined by incubating cells in heat-inactivated serum was less than 5% and was subtracted (mean ± SD, n = 4).
Figure 6.

Inhibition of complement-mediated lysis by recombinant sCD59 and CD59 fusion proteins. Antibody-sensitized CHO cells (a) or SRBCs (b) were incubated with recombinant protein and 10% human serum (CHO cells) or 0.33% human serum (erythrocytes). These concentrations resulted in approximately 90% lysis of unprotected cells. Lysis was determined after 60 minutes at 37°C. Background lysis determined by incubating cells in heat-inactivated serum was less than 5% and was subtracted (mean ± SD, n = 4).
Table 2.
Concentration of complement inhibitor providing 50% inhibition of lysis

There were some differences between the relative effectiveness of the complement inhibitors at protecting CHO cells and erythrocytes from complement-mediated lysis. This was particularly true for the DAF inhibitors; sDAF was significantly more effective at protecting erythrocytes than CHO cells from complement, although targeted DAF was still more effective. There was also little difference in the inhibitory activity of CR2-DAF and DAF-CR2 when erythrocytes were the target cells for complement lysis.
Effect of CR2 fusion proteins on cell adhesion.
CR3 is a leukocyte receptor involved in endothelial adhesion, diapedesis, and the activation of cell cytolytic mechanisms (phagocytosis and degranulation). Since CR2 and CR3 share the same iC3b complement ligand, we determined whether CR2 fusion proteins interfered with CR3-mediated cell binding. For these experiments, we used U937, a well-characterized promonocytic cell line (CR2–, CR3+) that binds to iC3b-coated erythrocytes in a CR3-dependent mechanism (46). All of the CR2 fusion proteins, but not sDAF or sCD59, significantly inhibited the binding of U937 cells to C3-opsonized SRBCs (P < 0.01). For this in vitro feasibility experiment, we used a relatively high 500-nM concentration of CR2 fusion protein, and each fusion protein inhibited U937 binding to a similar extent (Figure 7). Similar data were obtained in an experiment using U937 cells that were stimulated with PMA (data not shown), a treatment that results in upregulation of CR3 (45, 46). For complement opsonization of erythrocytes, IgM was used to activate complement, since IgG deposited on the erythrocytes would engage Fc-γ receptors expressed on U937 cells. In control experiments, U937 cells did not bind to SRBC preincubated with either IgM or C6-deficient human serum alone (data not shown). U937 cells also express CR4 (p150, 95, CD11c/CD18), a third complement receptor recognizing the iC3b ligand. However, binding of U937 cells to C3-opsonized erythrocytes is CR4-independent and appears to be related to the association of CR4 with the cytoskeleton and its immobility in the membrane (46).
Figure 7.

Effect of recombinant fusion proteins on U937 cell adhesion. SRBCs were sensitized with IgM antibody and incubated in C6-deficient serum. C3-opsonized erythrocytes were coincubated with U937 cells in the presence of 500 nM recombinant fusion protein or PBS. After incubation, the average number of U937 cells bound per erythrocyte was determined by microscopy (mean ± SD, n = 3).
Targeting of CR2-DAF to the kidneys of nephritic mice.
To determine whether a CR2 fusion protein will target a site of complement activation and disease in vivo, a biodistribution study of CR2-DAF and sDAF in female NZB/W F1 mice was performed. NZB/W F1 mice develop a spontaneous autoimmune disease that is very similar to human systemic lupus erythematosus (SLE), with the production of autoantibodies and the development of severe immune complex–mediated glomerulonephritis that is associated with complement deposition from 26 to 28 weeks of age (4, 52). Biodistribution of [125I]CR2-DAF and [125I]sDAF in 34-week-old NZB/W F1 mice was determined at 24 hours and 48 hours after injection. Twenty-four hours after tail-vein injection of [125I]CR2-DAF, a significantly higher proportion of radioactivity was localized to the kidney than to the other organs that were examined (Figure 8a). At 48 hours after injection of [125I]CR2-DAF, there was a similar level of radioactivity in the kidney as at 24 hours, but radioactivity in the liver and spleen was increased and blood radioactivity decreased (Figure 8b). The liver and spleen are sites of immune complex clearance and likely account for increased targeting of [125I]CR2-DAF to these organs at the later time point. [125I]sDAF showed no preferential binding in the kidney or any other organ (Figure 8, a and b). In 8-week-old prenephritic NZB/W F1 mice, there was no evidence of [125I]CR2-DAF targeting to the kidney (Figure 8c). Of further interest, [125I]sDAF was cleared much more rapidly from the circulation than [125I]CR2-DAF, suggesting that the CR2 moiety is functioning to prolong the circulatory half-life of the fusion protein. However, the level of [125I]CR2-DAF in the blood of younger mice at 24 hours was about half that recorded in the older mice, and the long circulatory half-life of [125I]CR2-DAF may be a consequence, at least in part, of it binding to circulating immune complexes. Targeting of CR2-DAF to complement deposited in the kidney was also examined in another murine model of SLE by direct examination of kidney sections. Similar to female NZB/W F1 mice, MRL/lpr mice develop severe proliferative glomerulonephritis with the deposition of complement in association with glomerular immune deposits by 24 weeks of age (53). CR2-DAF and sDAF were injected into the tail vein of 24-week-old MRL/lpr mice, and kidney sections were analyzed 24 hours later for human DAF immunoreactivity by fluorescence microscopy. Kidney sections from a mouse injected with CR2-DAF displayed a high level of DAF staining, with preferential localization in glomeruli in a pattern identical to that seen for immune complexes. No DAF staining was evident in glomeruli from a mouse injected with sDAF (Figure 9).
Figure 8.

Biodistribution of [125I]CR2-DAF and [125I]sDAF in nephritic 34-week-old and prenephretic 8-week-old NZB/W F1 mice. Radiolabeled proteins were injected into the tail vein, and biodistribution of radiolabel was determined after 24 hours or 48 hours. Each protein was injected into two mice.
Figure 9.
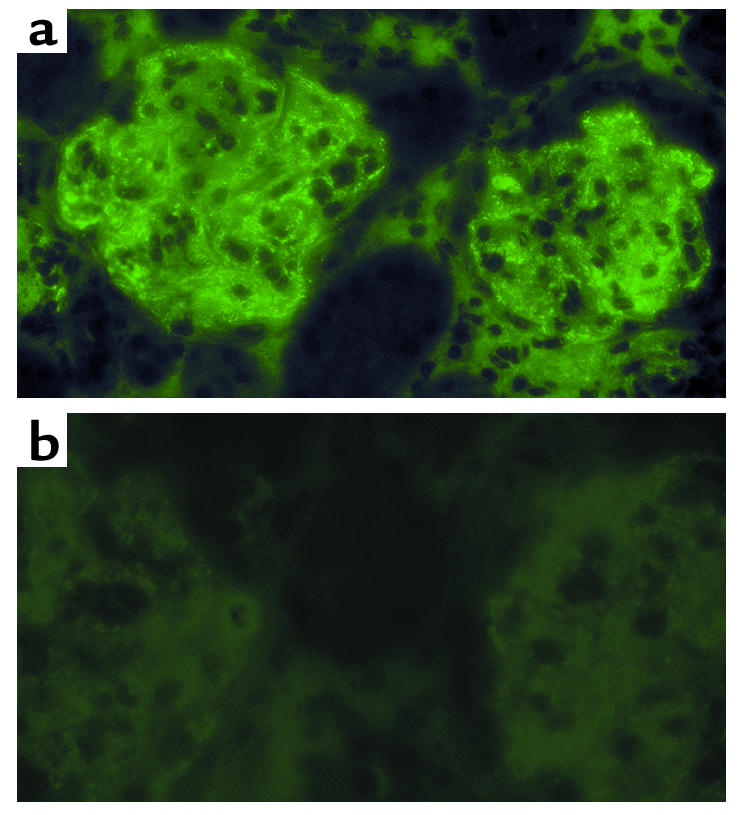
Imaging of CR2-DAF bound to glomeruli of 24-week-old MRL/lpr mice. Glomerular binding of CR2-DAF (a) and sDAF (b) was analyzed 24 hours after tail-vein injection of each protein. The figure shows immunofluorescence staining of kidney sections.
Discussion
This study describes the generation and characterization of soluble human DAF and CD59-containing proteins that are targeted to a site of complement activation. Targeting of CD59 and DAF was achieved by linking the inhibitors to a fragment of human CR2 that binds complement C3 activation products. The targeted proteins were significantly more potent than their untargeted counterparts at inhibiting complement. The C3 ligands for CR2 are relatively long lived and are covalently bound, often in large quantities, at sites of complement activation. Thus, CR2-mediated targeting of complement inhibition has the potential to be of therapeutic benefit for numerous complement-associated diseases or disease states. Consistent with this hypothesis, CR2-DAF was shown to target to the kidneys of nephritic NZB/W F1 mice. These mice produce autoantibodies with consequent formation and deposition of immune complexes in the kidney, resulting in complement activation and deposition (4, 52). Human CR2 binds human and mouse C3 ligands with similar affinities (54), and the biodistribution and microscopy studies establish that a CR2 fusion protein retains targeting function in vivo. However, human CD59 and soluble human DAF are poor inhibitors of rodent complement relative to their activity against human complement (with the exception of human DAF activity against the rat alternative pathway of activation) (43, 55, 56), and so the complement-inhibitory function of these proteins was not evaluated in a mouse model. Meaningful validation of the use of CR2-targeted complement inhibitors in a rodent model of disease will require the generation and characterization of rodent CR2 complement-inhibitor fusion proteins. The targeting approach is also likely to be effective for other inhibitors of complement such as soluble CR1, an inhibitor currently in clinical trials.
It is tempting to speculate that differences between the complement-inhibitory activity of the N- and C-terminal fusion proteins are due to the different C3dg ligand binding characteristics of the proteins. N-terminal CR2 fusion proteins were the most potent complement inhibitors, and these proteins bound C3dg with a higher affinity and with faster association and dissociation rates than C-terminal CR2 fusions. However, the difference in C3dg ligand affinity between the two CD59 fusion proteins and between the two DAF proteins was not great. One conceivable reason that a fast on-off rate may be advantageous could be related to cell surface “mobility” or the frequent release of the complement inhibitor from a fixed orientation, allowing frequent opportunities for more favorable positioning of the inhibitor for interaction with C3b or nascent MAC ligand (depending on the type of inhibitor). For CD59 in particular, previous data suggest that positioning relative to the site of MAC formation is important for its function (23, 57). Other possible reasons for the differences in the complement-inhibitory activity of N- and C-terminal CR2 fusion proteins include different positioning of the inhibitor moiety relative to its ligand at the cell surface and steric hindrance of the inhibitor’s ligand binding site. The relative affinities of the different CR2 fusion proteins for C3dg are reminiscent of the affinities of CR2-SCR 1–2 and CR2 SCR 1–15 for C3dg. The KD values for CR2 SCR1–2 and CR2 SCR 1–15 interactions with C3dg were similar, but CR2 SCR 1–2 associated and dissociated much faster, suggesting a contribution of the additional SCR domains to overall affinity (42). Analysis of the solution structure of another SCR-containing protein, factor H, indicated that SCR domains are folded back on themselves and interactions between SCR domains may modulate C3 ligand binding characteristics (58). Conformational variability between SCR domains is predicted to result from different (native) linker lengths, with longer linkers providing greater conformational flexibility. In this context, the CR2 and DAF SCR domains are linked with a relatively long Ser-Gly linker, and this may permit the fusion partners to fold back on one another, resulting in SCR-SCR interactions that may modulate CR2 binding affinity. This is a possible explanation for the lower KD of the DAF fusion proteins for C3dg as compared with the CD59 fusion proteins (CD59 is a single-domain compact globular protein with no SCR homology).
Complement-mediated lysis assays were performed using antibody-sensitized CHO cells or SRBCs as targets. There were marked differences in the relative activities of some of the complement inhibitors for protecting the different cells from complement-mediated lysis. sDAF, DAF-CR2, and CD59-CR2 were significantly more effective at protecting sheep erythrocytes than CHO cells from complement-mediated lysis. The reason for these differences is not clear, but it may be related to the multihit characteristics of nucleated cell lysis. Unlike erythrocytes, complement-mediated lysis of nucleated cells is not entirely due to colloid osmotic deregulation, and the deposition of multiple MACs in the plasma membrane is required (59–61). Most of the previous studies investigating the inhibitory activity of soluble (untargeted) complement inhibitors have been performed using erythrocytes as target cells for complement-mediated lysis. However, CHO cells most likely represent a more physiologically relevant target for in vitro experiments. Human cells were not used as targets in our experiments, since the expression of endogenous complement-inhibitory proteins would make interpretation of data difficult, and human erythrocytes and most human cell lines are resistant to lysis by human complement in vitro unless the function of endogenously expressed complement inhibitors is blocked.
Various mechanisms of complement-mediated damage are implicated in different disease conditions, and different diseases will most likely benefit from inhibition strategies acting at various points in the complement pathway. For example, if applicable for the disease, a particular benefit of blocking complement at a late step in the pathway would be that host defense functions and immune homeostasis mechanisms of complement would remain intact. Thus, a CD59-based inhibitor would provide advantages over inhibitors of complement activation (such as DAF and sCR1) in diseases in which the terminal cytolytic pathway is primarily implicated in pathogenesis. Soluble CD59 is unlikely to have therapeutic benefit because of its very poor activity in vitro, but we have shown that CR2-mediated targeting of CD59 significantly increased its complement-inhibitory activity. In fact, CR2-CD59 was more effective at inhibiting complement-mediated lysis than sDAF, and sDAF has shown therapeutic efficacy in vivo (11). Rodent analogues of CR2-CD59 may also be useful tools for dissecting the relative roles of early complement activation products versus MAC formation in disease pathogenesis. The relative contributions of the various complement activation products to tissue injury in many disease states is poorly understood and controversial.
The CR2 fusion proteins inhibited the binding of U937 cells to C3-opsonized erythrocytes. CR2 and CR3 both bind iC3b, and our data indicate that CR2 fusion proteins act as CR3 antagonists, since U937 binding to C3-opsonized erythrocytes is CR3 dependent (46). As an adhesion molecule, CR3 mediates endothelial adhesion and diapedesis at sites of inflammation through its high-affinity interaction with ICAM-1. As a complement receptor, CR3 promotes and enhances phagocytosis and degranulation through its interaction with iC3b. Both ICAM-1 and iC3b bind to overlapping epitopes on CR3 (62). CR3 can thus be an important determinant in promoting cell-mediated tissue damage at sites of inflammation, and antibodies that block CR3 have shown effectiveness in several inflammatory conditions (62). The antagonistic effect of CR2 on CR3 binding therefore indicates a second potential anti-inflammatory mechanism of action of the CR2 complement-inhibitor fusion proteins that may act synergistically with complement inhibition. Of further relevance, soluble CR2 has been shown to suppress antibody responses in mice (54), and CR2 complement-inhibitor fusion proteins may have additional immunologic effects that will need to be investigated.
In summary, targeting complement inhibitors to sites of complement activation and disease is likely to considerably enhance their efficacy. Indeed, the data indicate that for disease states that would benefit from CD59-based therapy, the targeting of CD59 to the site of complement activation will be a requirement. An advantage of CR2-mediated targeting over other targeting approaches, such as antibody-mediated targeting or sLex-mediated targeting to adhesion molecules, is that the CR2 moiety will target any accessible site of complement activation and will have broad therapeutic potential. CR2 fusion proteins can also act as CR3 (and CR2) antagonists, and this may represent a second and potentially important therapeutic benefit. Human CR2 complement-inhibitor fusion proteins are also less likely to be immunogenic than recombinant inhibitors containing antibody variable regions. The predicted ability of targeted inhibitors of complement activation to provide an effective local concentration with low levels of systemic inhibition also diminishes the possibility of compromising host defense mechanisms, particularly with long-term systemic complement inhibition (this is a less important consideration for CD59-based inhibitors). CR2-targeted inhibitors may also target infectious agents that activate complement, but whether this would seriously interfere with complement-dependent defense pathways is difficult to predict. This would depend on such considerations as the rate of pathogen multiplication relative to effective inhibitor levels both in the circulation and retained on host tissue at sites of complement activation.
Acknowledgments
We thank Masaki Imai for assistance with some experimental procedures. This work was supported by grants from the S.L.E. Foundation Inc., the American Heart Association, and the NIH (AI 34451 to S. Tomlinson and CA53615 to V.M. Holers).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: complement receptor (CR); decay accelerating factor (DAF); membrane attack complex (MAC); soluble form of complement receptor 1 (sCR1); membrane cofactor protein (MCP); soluble form of CD59 (sCD59); sialyl Lewisx (sLex); short consensus repeat (SCR); normal human serum (NHS); surface plasmon resonance (SPR); antibody-sensitized sheep erythrocytes (EAs); gelatin veronal buffer (GVB++); sheep erythrocytes (SRBCs); soluble DAF (sDAF); systemic lupus erythematosus (SLE).
References
- 1.Rother, K., Till, G.O., and Haensch, G.M., editors. 1998. The complement system. Springer-Verlag. Berlin, Germany. 564 pp.
- 2.Lambris, J.D., and Holers, V.M. 2000. Therapeutic interventions in the complement system. Humana Press. Totowa, New Jersey, USA. 258 pp.
- 3.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc. Natl. Acad. Sci. U. S. A. 1995;92:8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, et al. Amelioration of lupus-like autoimmune disease in NZB/W F1 mice after treatment with a blocking monoclonal antibody specific for complement component C5. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8563–8568. doi: 10.1073/pnas.93.16.8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplan M. Eculizumab (Alexion) Curr. Opin. Investig. Drugs. 2002;3:1017–1023. [PubMed] [Google Scholar]
- 6.Whiss PA. Pexelizumab Alexion. Curr. Opin. Investig. Drugs. 2002;3:870–877. [PubMed] [Google Scholar]
- 7.Weisman HF, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 8.Rioux P. TP-10 (AVANT Immunotherapeutics) Curr. Opin. Investig. Drugs. 2001;2:364–371. [PubMed] [Google Scholar]
- 9.Zamora MR, et al. Complement inhibition attenuates human lung transplant reperfusion injury: a multicenter trial. Chest. 1999;116(Suppl. 1):46S. doi: 10.1378/chest.116.suppl_1.46s. [DOI] [PubMed] [Google Scholar]
- 10.Higgins PJ, et al. A soluble chimeric complement activating inhibitory protein that possesses both decay-accelerating and factor I cofactor activities. J. Immunol. 1997;158:2872–2881. [PubMed] [Google Scholar]
- 11.Moran P, et al. Human recombinant soluble decay accelerating factor inhibits complement activation in vitro and in vivo. J. Immunol. 1992;149:1736–1743. [PubMed] [Google Scholar]
- 12.Christiansen D, Milland J, Thorley BR, McKenzie IF, Loveland BE. A functional analysis of recombinant soluble CD46 in vivo and a comparison with recombinant soluble forms of CD55 and CD35 in vitro. Eur. J. Immunol. 1996;26:578–585. doi: 10.1002/eji.1830260312. [DOI] [PubMed] [Google Scholar]
- 13.Salerno CT, et al. A soluble chimeric inhibitor of C3 and C5 convertases, complement activation blocker-2, prolongs graft survival in pig-to-rhesus monkey heart transplantation. Xenotransplantation. 2002;9:125–134. doi: 10.1034/j.1399-3089.2002.1o042.x. [DOI] [PubMed] [Google Scholar]
- 14.Kroshus TJ, et al. A recombinant soluble chimeric complement inhibitor composed of human CD46 and CD55 reduces acute cardiac tissue injury in models of pig-to-human heart transplantation. Transplantation. 2000;69:2282–2289. doi: 10.1097/00007890-200006150-00011. [DOI] [PubMed] [Google Scholar]
- 15.Rushmere NK, Van Den Berg CW, Morgan BP. Production and functional characterization of a soluble recombinant form of mouse CD59. Immunology. 2000;99:326–332. doi: 10.1046/j.1365-2567.2000.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Quigg RJ, He C, Hack BK, Alexander JJ, Morgan BP. Production and functional analysis of rat CD59 and chimeric CD59-Crry as active soluble proteins in Pichia pastoris. Immunology. 2000;99:46–53. doi: 10.1046/j.1365-2567.2000.00945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugita Y, et al. Recombinant soluble CD59 inhibits reactive haemolysis with complement. Immunology. 1994;82:34–41. [PMC free article] [PubMed] [Google Scholar]
- 18.Meri S, Lehto T, Sutton CW, Tyynela J, Baumann M. Structural composition and functional characterization of soluble CD59: heterogeneity of the oligosaccharide and glycophosphoinositol (GPI) anchor revealed by laser-desorption mass spectrometric analysis. Biochem. J. 1996;316:923–935. doi: 10.1042/bj3160923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulligan MS, et al. Endothelial targeting and enhanced antiinflammatory effects of complement inhibitors possessing sialyl lewisX moieties. J. Immunol. 1999;162:4952–4959. [PubMed] [Google Scholar]
- 20.Ritterhaus CW, et al. Recombinant glycoproteins that inhibit complement activation and also bind the selectin adhesion molecules. J. Biol. Chem. 1999;274:11237–11244. doi: 10.1074/jbc.274.16.11237. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, et al. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Lu S, Morrison SL, Tomlinson S. Targeting of functional antibody-decay accelerating factor (DAF) fusion proteins to a cell surface. J. Biol. Chem. 2001;276:27290–27295. doi: 10.1074/jbc.M100436200. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H-F, Yu J, Bajwa E, Morrison SL, Tomlinson S. Targeting of functional antibody-CD59 fusion proteins to a cell surface. J. Clin. Invest. 1999;103:55–66. doi: 10.1172/JCI4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linton SM, et al. Therapeutic efficacy of a novel membrane-targeted complement regulator in antigen-induced arthritis in the rat. Arthritis Rheum. 2000;43:2590–2597. doi: 10.1002/1529-0131(200011)43:11<2590::AID-ANR29>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 25.Smith GP, Smith RA. Membrane-targeted complement inhibitors. Mol. Immunol. 2001;38:249–255. doi: 10.1016/s0161-5890(01)00047-5. [DOI] [PubMed] [Google Scholar]
- 26.Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- 27.Carroll MC. The role of complement in B cell activation and tolerance. Adv. Immunol. 2000;74:61–88. doi: 10.1016/s0065-2776(08)60908-6. [DOI] [PubMed] [Google Scholar]
- 28.Holers VM. Complement receptors. Year Immunol. 1989;4:231–240. [PubMed] [Google Scholar]
- 29.Dierich MP, Schulz TF, Eigentler A, Huemer H, Schwable W. Structural and functional relationships among receptors and regulators of the complement system. Mol. Immunol. 1988;25:1043–1051. doi: 10.1016/0161-5890(88)90136-8. [DOI] [PubMed] [Google Scholar]
- 30.Lowell CA, et al. Mapping of the Epstein-Barr virus and C3dg binding sites to a common domain on complement receptor type 2. J. Exp. Med. 1989;170:1931–1946. doi: 10.1084/jem.170.6.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szakonyi G, et al. Structure of complement receptor 2 in complex with its C3d ligand. Science. 2001;292:1725–1728. doi: 10.1126/science.1059118. [DOI] [PubMed] [Google Scholar]
- 32.Law SK, Fearon DT, Levine RP. Action of the C3b-inactivator on the cell-bound C3b. J. Immunol. 1979;122:759–765. [PubMed] [Google Scholar]
- 33.Seya T, Nagasawa S. Limited proteolysis of complement protein C3b by regulatory enzyme C3b inactivator: isolation and characterization of a biologically active fragment, C3d,g. J. Biochem. (Tokyo) 1985;97:373–382. doi: 10.1093/oxfordjournals.jbchem.a135064. [DOI] [PubMed] [Google Scholar]
- 34.Holers, V.M. 2001. Complement in clinical immunology. Mosby. Philadelphia, Pennsylvania, USA. 289 pp.
- 35.Quigg RA, et al. Blockade of antibody-induced glomerulonephritis with Crry-Ig, a soluble murine complement inhibitor. J. Immunol. 1998;160:4553–4560. [PubMed] [Google Scholar]
- 36.Harlow, E., and Lane, D. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory. Cold Spring Harbor, New York, USA. 726 pp.
- 37.Coyne KE, et al. Mapping of epitopes, glycosylation sites, and complement regulatory domains in human decay accelerating factor. J. Immunol. 1992;149:2906–2913. [PubMed] [Google Scholar]
- 38.Kinoshita T, Medof ME, Silber R, Nussenzweig V. Distribution of decay-accelerating factor in peripheral blood of normal individuals and patients with paroxysmal nocturnal hemoglobinuria. J. Exp. Med. 1985;162:75–92. doi: 10.1084/jem.162.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies A, et al. CD59, an LY-6-like protein expressed in human lymphoid cells, regulates the action of the complement membrane attack complex of homologous cells. J. Exp. Med. 1989;170:637–654. doi: 10.1084/jem.170.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guthridge JM, et al. Epitope mapping using the X-ray crystallographic structure of complement receptor type 2 (CR2)/CD21: identification of a highly inhibitory monoclonal antibody that directly recognizes the CR2-C3d interface. J. Immunol. 2001;167:5758–5766. doi: 10.4049/jimmunol.167.10.5758. [DOI] [PubMed] [Google Scholar]
- 41.Dieffenbach, C.W., and Dveksler, G.S., editors. 1995. PCR primer: a laboratory manual. Cold Spring Harbor Laboratory Press. Cold Spring Harbor, New York, USA. 714 pp.
- 42.Guthridge JM, et al. Structural studies in solution of the recombinant N-terminal pair of short consensus/complement repeat domains of complement receptor type 2 (CR2/CD21) and interactions with its ligand C3dg. Biochemistry. 2001;40:5931–5941. doi: 10.1021/bi0101749. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, et al. Protection of human breast cancer cells from complement-mediated lysis by expression of heterologous CD59. Clin. Exp. Immunol. 1999;115:13–18. doi: 10.1046/j.1365-2249.1999.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duits AJ, Jainandunsing SM, van de Winkel JG, Capel PJ. Selective enhancement of Leu-CAM expression by interleukin 6 during differentiation of human promonocytic U937 cells. Scand. J. Immunol. 1991;33:151–159. doi: 10.1111/j.1365-3083.1991.tb03745.x. [DOI] [PubMed] [Google Scholar]
- 45.Rothlein R, Springer TA. The requirement for lymphocyte function-associated antigen 1 in homotypic leukocyte adhesion stimulated by phorbol ester. J. Exp. Med. 1986;163:1132–1149. doi: 10.1084/jem.163.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ross GD, Reed W, Dalzell JG, Becker SE, Hogg N. Macrophage cytoskeleton association with CR3 and CR4 regulates receptor mobility and phagocytosis of iC3b-opsonized erythrocytes. J. Leukoc. Biol. 1992;51:109–117. doi: 10.1002/jlb.51.2.109. [DOI] [PubMed] [Google Scholar]
- 47.Sharkey RM, Motta-Hennessy C, Pawlyk D, Siegel JA, Goldenberg DM. Biodistribution and dose estimates for yttrium- and iodine-labeled monoclonal antibody IgG and fragments in nude mice. Cancer Res. 1990;50:2330–2336. [PubMed] [Google Scholar]
- 48.Sharkey RM, Natale A, Goldenberg DM, Mattes MJ. Rapid blood clearance of immunoglobulin G2a and immunoglobulin G2b in nude mice. Cancer Res. 1991;51:3102–3107. [PubMed] [Google Scholar]
- 49.Coligan, J.E., Kruisbeek, A.M., Margulies, D.H., Shevach, E.M., and Strober, W., editors. 2002. Current protocols in immunology [book on CD-ROM]. John Wiley and Sons Inc. Hoboken, New Jersey, USA.
- 50.Iida K, Mitomo K, Fujita K, Tamura N. Characterization of three monoclonal antibodies against C3 with selective specificities. Immunology. 1987;62:413–417. [PMC free article] [PubMed] [Google Scholar]
- 51.Sarrias MR, et al. Kinetic analysis of the interactions of complement receptor 2 (CR2, CD21) with its ligands C3d, iC3b, and the EBV glycoprotein gp350/220. J. Immunol. 2001;167:1490–1499. doi: 10.4049/jimmunol.167.3.1490. [DOI] [PubMed] [Google Scholar]
- 52.Andrews BS, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J. Exp. Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biesecker G, Katz S, Koffler D. Renal localization of the membrane attack complex in systemic lupus erythematosus nephritis. J. Exp. Med. 1981;154:1779–1794. doi: 10.1084/jem.154.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hebell T, Ahearn JM, Fearon DT. Suppression of the immune response by a soluble complement receptor of B lymphocytes. Science. 1991;254:102–105. doi: 10.1126/science.1718035. [DOI] [PubMed] [Google Scholar]
- 55.Harris CL, Spiller OB, Morgan BP. Human and rodent decay-accelerating factors (CD55) are not species restricted in their complement-inhibiting activities. Immunology. 2000;100:462–470. doi: 10.1046/j.1365-2567.2000.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu J, et al. Mapping the regions of the complement inhibitor CD59 responsible for its species selectivity. Biochemistry. 1997;36:9423–9428. doi: 10.1021/bi970832i. [DOI] [PubMed] [Google Scholar]
- 57.Fodor WL, Rollins SA, Guilmette ER, Setter E, Squinto SP. A novel bifunctional chimeric complement inhibitor that regulates C3 convertase and formation of the membrane attack complex. J. Immunol. 1995;155:4135–4138. [PubMed] [Google Scholar]
- 58.Aslam M, Perkins SJ. Folded-back solution structure of monomeric factor H of human complement by synchrotron X-ray and neutron scattering, analytical ultracentrifugation and constrained molecular modelling. J. Mol. Biol. 2001;309:1117–1138. doi: 10.1006/jmbi.2001.4720. [DOI] [PubMed] [Google Scholar]
- 59.Koski CL, Ramm LE, Hammer CH, Mayer MM, Shin ML. Cytolysis of nucleated cells by complement: cell death displays multi-hit characteristics. Proc. Natl. Acad. Sci. U. S. A. 1983;80:3816–3820. doi: 10.1073/pnas.80.12.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramm LE, Whitlow MB, Mayer MM. Transmembrane channel formation by complement: functional analysis of the number of C5b6, C7, C8, and C9 molecules required for a single channel. Proc. Natl. Acad. Sci. U. S. A. 1982;79:4751–4755. doi: 10.1073/pnas.79.15.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takeda J, et al. Number of hits necessary for complement-mediated hemolysis. Microbiol. Immunol. 1986;30:461–468. doi: 10.1111/j.1348-0421.1986.tb02972.x. [DOI] [PubMed] [Google Scholar]
- 62.Ross GD. Regulation of the adhesion versus cytotoxic functions of the Mac-1/CR3/αMβ2-integrin glycoprotein. Crit. Rev. Immunol. 2000;20:197–222. [PubMed] [Google Scholar]