

Abstract
Background:
Genome comparisons have revealed major lateral gene transfer between the three primary kingdoms of life - Bacteria, Archaea, and Eukarya. Another important evolutionary phenomenon involves the evolutionary mobility of protein domains that form versatile multidomain architectures. We were interested in investigating the possibility of a combination of these phenomena, with an invading gene merging with a pre-existing gene in the recipient genome.
Results:
Complete genomes of fifteen bacteria, four archaea and one eukaryote were searched for interkingdom gene fusions (IKFs); that is, genes coding for proteins that apparently consist of domains originating from different primary kingdoms. Phylogenetic analysis supported 37 cases of IKF, each of which includes a 'native' domain and a horizontally acquired 'alien' domain. IKFs could have evolved via lateral transfer of a gene coding for the alien domain (or a larger protein containing this domain) followed by recombination with a native gene. For several IKFs, this scenario is supported by the presence of a gene coding for a second, stand-alone version of the alien domain in the recipient genome. Among the genomes investigated, the greatest number of IKFs has been detected in Mycobacterium tuberculosis, where they are almost always accompanied by a stand-alone alien domain. For most of the IKF cases detected in other genomes, the stand-alone counterpart is missing.
Conclusions:
The results of comparative genome analysis show that IKF formation is a real, but relatively rare, evolutionary phenomenon. We hypothesize that IKFs are formed primarily via the proposed two-stage mechanism, but other than in the Actinomycetes, in which IKF generation seems to be an active, ongoing process, most of the stand-alone intermediates have been eliminated, perhaps because of functional redundancy.
Background
Comparative genome analysis has revealed major lateral gene transfer between the three primary kingdoms of life, Bacteria, Archaea, and Eukarya [1,2,3,4]. The best recognized form of lateral gene flux is the transfer of numerous genes from mitochondria and chloroplasts to eukaryotic nuclear genomes [5]. Far beyond that, however, the role of lateral gene exchange, along with lineage-specific gene loss, as one of the principal factors of evolution, at least among prokaryotes, is obvious from the fact that the great majority of conserved families of orthologous genes show a 'patchy' phyletic distribution [6,7]. In many cases, such families are shared by phylogenetically distant species (for example, bacteria and archaea), while they are missing in some of the more closely related species (for example, bacteria from the same lineage). Correlations have been noticed between the preferred routes of gene transfer and the lifestyles of the organisms involved. Thus, massive gene exchange seems to have occurred between archaeal and bacterial hyperthermophiles [8,9], whereas certain parasitic bacteria, for example, chlamydia and spirochetes, appear to have acquired significantly more eukaryotic genes than free-living bacteria [10,11,12].
Another evolutionary trend that is predominant in eukaryotes, but is important also in bacteria and archaea, involves the evolutionary mobility of protein domains that combine to form variable multidomain architectures [13,14,15,16,18]. Domain fusion is one of the foundations of most forms of regulation and signal transduction in the cell. Examples include prokaryotic transcriptional regulators, most of which consist of the DNA-binding helix-turn-helix domain fused to a variety of small-molecule-binding domains [19], the two-component signal transduction system that is based on fusions of histidine kinases with sensor domains and of receiver domains with DNA-binding domains [20], and the sugar phosphotransferase (PTS) systems that include complex fusions of several enzymes [21]. In the evolution of eukaryotes, domain fusion takes the form of domain accretion, whereby proteins from complex organisms (such as animals) that are involved in various forms of regulation and signal transduction tend to accrue multiple domains that facilitate the formation of complex networks of interactions [22].
We were interested in exploring the possibility of a meeting between these two major evolutionary phenomena - lateral gene exchange and gene fusion - which would result in the formation of multidomain proteins in which different domains display distinct evolutionary provenance. In particular, we sought to identify fusions between domains originating from different primary kingdoms - Bacteria, Archaea and Eukarya - which we term interkingdom gene (domain) fusions (IKFs), and obtain clues to the pathways of IKF origin through comparative genome analysis. We show that, although IKF in general is a rare phenomenon, one bacterial lineage, the Actinomycetes, displays a significantly increased frequency of such events; we also propose a probable mechanism for IKF formation.
Results and discussion
To identify IKFs, all protein sequences encoded in the analyzed genomes were compared to the non-redundant protein database, and those proteins in which distinct parts showed the greatest similarity to homologs from different primary kingdoms were identified (see the Materials and methods section). In most cases, the reported alignments were highly statistically significant, leaving no doubt that true homologs were detected (Table 1). On the few occasions when the database search statistics in themselves were not fully convincing (for example, the OB-fold nucleic acid-binding domain in the Bacillus subtilis protein YhcN and the methyltransferase domain in the YabN protein, also from B. subtilis), the homologous relationship was validated by detection of the salient sequence motifs known to be involved in the corresponding protein functions (data not shown). Such motif analysis was performed for all analyzed domains in order not only to validate homology, but also to distinguish between active and inactivated forms of enzymes. Figure 1 shows multiple alignments of two domains involved in an IKF, illustrating the conservation of the characteristic functional motifs and the specific similarity between each of the domains of the IKF protein (in this case from Aquifex aeolicus) and their archaeal and bacterial homologs, respectively.
Table 1.
Interkingdom domain fusions and their probable origins
| IKF gene | Best 'native' hit | Best 'alien' hit | Protein function | Stand-alone | Comment |
| (GI number and gene | (E-value, amino acid | (E-value, amino | paralog of the | ||
| name) and origin | residue range, | acid residue range, | alien domain | ||
| of domains | species)/domain | species)/domain | |||
| function | function | ||||
| Archaea | |||||
| Aeropyrum pernix | |||||
| 5106104_ | 2621953_Mth | 2633525_Bs | Hydroxymethyl- | None | Pyrococci encode proteins with |
| APE2400 | 5e-27; | 4e-54; | pyrimidine | the same domain organization | |
| Archaeal-bacterial | 282-445; | 16-272; | phosphate kinase | andclosest similarity to A. pernix; | |
| uncharacterized domain | hydroxymethyl- | involved in thiamine | M. jannaschii encodes a protein | ||
| conserved among | pyrimidine phosphate | biosynthesis | with the same domain | ||
| archaea (homolog | kinase | (additional function?) | organization but low similarity; | ||
| of the amino-terminal | Mt encodes a HMP-kinase with | ||||
| domain of sialic acid | moderate similarity | ||||
| synthase) | |||||
| Methanococcus jannaschii | |||||
| 1591138_ | 2128140_Mj; | 7270033_At; | Unknown; | None | The amino-terminal domain is |
| MJ0434 | 1e-19; | 0.003; | possible role | present in several stand-alone | |
| Archaeal- | 2-94; | 120-222; | in stress response | copies in M. jannaschii, but | |
| bacterial-eukaryotic | uncharacterized | AIG2-like | otherwise, is seen mostly in | ||
| domain | stress-related | bacteria; the possibility of | |||
| protein | acquisition of a bacterial gene | ||||
| by the Methanococcus lineage | |||||
| is conceivable | |||||
| Methanobacterium thermoautotrophicum | |||||
| 2621249_ | 5103547_Ap; | 1651798_Ssp; | Membrane-associated | None | In Ssp, the amino-terminal |
| MTH204 | 1e-34; | 0.002; | 5-formyl- | domain is fused to another | |
| Archaeal- | 137-326; | 8-139; | tetrahydrofolate | uncharacterized domain. An | |
| eukaryotic/ | 5-formyl- | uncharacterized | cyclo-ligase(?); | ortholog with conserved | |
| bacterial | tetrahydrofolate | membrane-associated | exact function | domain organization is seen | |
| cyclo-ligase | domain | unknown | in Mycobacterium, but many | ||
| other bacteria encode stand- | |||||
| alone versions of this domain, | |||||
| which could be the actual sources | |||||
| of horizontal gene transfer | |||||
| 2621673_ | 3256572_Ph; | 2984130_Aa; | GTPase, possible | 2621855 | |
| MTH594 | 3e-10; | 6e-19; | role in signal | ||
| Archaeal-bacterial | 5-137; | 233-390; | transduction | ||
| inactivated RecA | GTPase | ||||
| domain | |||||
| 2622642_ | 5105992_Ap; | 2569943_Axy; | Glucose-1-phosphate | None | |
| MTH1523 | 3e-36; | 2e-05; | thymidylyl transferase/ | ||
| Archaeal-bacterial | 5-226; | 226-334; | glucose-6-phosphate | ||
| glucose-1-phosphate | mannose-6- | isomerase | |||
| thymidylyl transferase | phosphate isomerase | ||||
| Bacteria | |||||
| Aquifex aeolicus | |||||
| 2983622_ | 2633696_Bs; | 2650176_Af; | Signal | None | |
| aq_1151 | 5e-65; | 0.005; | transduction | ||
| Bacterial-archaeal | 325-795; | 116-279; | c-di-GMP | ||
| c-di-GMP phospho- | PAS/PAC | phospho-diesterase | |||
| diesterase | domain | ||||
| 2984285_ | 586875_Bs; | 3915955_Mj; | Molybdenum | None | |
| aq_2060 | 4e-63 | 3e-09; | cofactor | ||
| Bacterial-archaeal | 1-252; | 270-441; | bisynthesis enzyme(?) | ||
| PHP superfamily | pyruvate | ||||
| hydrolase | formate-lyase | ||||
| activating enzyme | |||||
| (Fe-S cluster | |||||
| oxidoreductase) | |||||
| Bacillus subtilis | |||||
| 2632283_yaaH, | 4980914_Tm | 399377_Rn | Chitinase | 2635915 | B. subtilis encodes two |
| 1945087_ydhD | 1e-06 | 2e-11 | paralogous proteins with the | ||
| Bacterial-eukaryotic | 2-92; | 221-402; | same domain architecture | ||
| LysM repeat domain | chitinase | ||||
| 2633242_yhcR | 645819_Dr; | 2622704_Mth; | Nuclease-nucleotidase | None | |
| Bacterial-archaeal | 1e-64; | 0.008 | (probable repair | ||
| 584-1068; | 151-257; | enzyme) | |||
| 5'-nucleotidase; | nucleic acid-binding | ||||
| 1175987_ | domain (OB-fold) | ||||
| ECR100; | |||||
| 2e-09; | |||||
| 377-521; | |||||
| thermonuclease | |||||
| 2632325_yabN | 4981449_Tm; | 3873806_Ce; | Methyl-transferase/ | None | Other than in chlamydiae, |
| Bacterial-eukaryotic | 2e-62; | 0.003; | pyro-phosphatase | the SWI domain is seen | |
| 223-483; | 7-125; | (metabolic enzyme | in eukaryotic chromatin- | ||
| MazG (predicted pyro- | SAM-dependent | of an unknown | associated proteins, leading | ||
| phosphatase) | methyl-transferase | pathway?) | to the suggestion that | ||
| chlamydial topoisomerase | |||||
| is involved in chromosome | |||||
| condensation | |||||
| Chlamydophyla pneumoniae | |||||
| 4377077_ | 730965_Bs; | 3581917_Sp; | DNA topoisomerase I, | 7189103 | SWI is a typical eukaryotic |
| CPn0769 | e-148; | 3e-10; | possibly involved in | domain not found in | |
| Bacterial-eukaryotic | 1-727; | 792-866; | chromatin | prokaryotes other than | |
| DNA topoisomerase I | SWI domain | condensation | chlamydia (the ortholog | ||
| in Chlamydia trachomatis has the | |||||
| same domain architecture) | |||||
| Deinococcus radiodurans | |||||
| 6459294_ | 7248325_Sco; | 6754878_Mm; | DNase | None | The G9a domain is not |
| DR1533 | 0.001; | 9e-28; | detectable in other prokaryotes. | ||
| Bacterial-eukaryotic | 171-265; | 4-148; | In eukaryotes, this domain so | ||
| McrA family | G9a domain (DNA- | far has been found only as part | |||
| endonuclease | binding?) | of multidomain nuclear proteins, | |||
| including transcription factors | |||||
| Escherichia coli | |||||
| 1787179_ | 94933_Ppu; | 3747107_Rn; | Oxidoreductase | None | The eukaryotic domain is present |
| b0947 | 3e-10; | 3e-32; | (as a partial sequence) also in the | ||
| Bacterial-eukaryotic | 287-367; | 4-261; | beta-proteobacterium Vogesella. | ||
| ferredoxin | uncharacterized | This domain contains a conserved | |||
| domain (thiol | pair of cysteines, which together | ||||
| oxidoreductase?) | with the ferredoxin fusion, may | ||||
| suggest a thiol oxidoreductase | |||||
| activity. Most of the eukaryotic | |||||
| proteins containing this domain | |||||
| appear to be mitochondrial, | |||||
| suggesting the possibility of an | |||||
| alternative evolutionary scenario | |||||
| 1787678_ | 487713_Sli; | 5459012_Pab; | Methyl-transferase/ | None | |
| b1410 | 3e-05; | 1e-17; | Lipase (exact function | ||
| Bacterial-archaeal | 408-522; | 33-274; | unclear) | ||
| SAM-dependent | lyso-phospholipase | ||||
| methyl-transferase | |||||
| 1787679_ynbD | 1591375_Mj; | 7160233_Sp; | Membrane-associated | None | An unusual case of fusion |
| Archaeal-eukaryotic | 4e-04; | 1e-06; | bifunctional | between an apparently archaeal | |
| 50-218; | 346-415; | phosphatase | and a typical eukaryotic domain | ||
| membrane-associated | tyrosine phosphatase | in a bacterium | |||
| acid phosphatase | |||||
| 1788589_ | 5763950_Sco; | 3860247_At; | Bifunctional enzyme; | None | |
| b2255 | 4e-35; | 1e-55; | exact function unclear | ||
| Bacterial-eukaryotic | 1-259; | 318-652; | |||
| methionyl-tRNA | dTDP-glucose 4-6- | ||||
| formyl-transferase | dehydratase | ||||
| 1788938_yfiQ | 929735_Nsp; | 2649370_Af; | acetyl-CoA synthetase/ | None | |
| bacterial-Archaeal/ | 8e-32; | 4e-85; | acetyl-transferase; exact | ||
| eukaryotic | 637-874; | 6-689; | function unclear | ||
| acetyl-transferase | acetyl-CoA synthetase | ||||
| Mycobacterium tuberculosis | |||||
| 2909507_ | 6469244_Sco; | 4151109_Tbr; | Adenylate cyclase/ | 7476546, | M. tuberculosis encodes three |
| Rv2488c, | 5e-64; | 6e-04; | ATPase; probable | 7476738 | paralogous proteins that consist |
| 2791528_Rv1358, | 19-603; | 6-167; | transcription regulator | of three domains, the eukaryotic- | |
| 1419061_ | 4726088_Rer; | adenylate cyclase | type adenylate cyclase, AP | ||
| Rv1358 | 2e-12; | (apoptotic) ATPase and DNA- | |||
| Bacterial-eukaryotic | 818-1073 | binding response regulator, and | |||
| two stand-alone versions of | |||||
| adenylate cyclase, which show the | |||||
| closest similarity to the cyclase | |||||
| domain of the multidomain | |||||
| proteins | |||||
| 1314025_ | 120037_Tt; | 178213_Hs; | Ferredoxin/ | 2076681 | D. radiodurans also encodes the |
| Rv0886 | 1e-11; | 4e-65; | ferredoxin reductase | eukaryotic-type ferredoxin | |
| Bacterial-eukaryotic | 2-79; | 93-543; | reductase, but the ferredoxin | ||
| ferredoxin | ferredoxin reductase | fusion is unique to mycobacteria | |||
| 3261732_ | 2661695_Sco; | 279520_Dd; | cAMP-dependent | 4455714 | |
| Rv0998 | 3e-13; | 7e-07; | acetyl-transferase(?) | (M. leprae) | |
| Bacterial-eukaryotic | 148-328; | 30-105; | |||
| acetyl-transferase | cAMP-binding domain | ||||
| 2326726_ | 421331_Cvi; | 2645721_Mm; | Bifunctional enzyme of | 1929080 | |
| Rv1683 | 1e-24; | 6e-26; | poly (3-hydroxy-butyrate) | ||
| Bacterial-eukaryotic | 23-359; | 456-972; | synthesis | ||
| poly (3-hydroxy- | very-long-chain | ||||
| butyrate) synthase | acyl-CoA synthetase | ||||
| 1403447_ | 6752338_Sco; | 3892714_At; | Polyfunctional enzyme | 2661651 | In this protein, the domain of |
| Rv2006 | 2e-27; | 8e-27; | of trehalose metabolism | apparent eukaryotic origin | |
| Bacterial-eukaryotic | 23-240; | 264-521; | is flanked by bacterial domains | ||
| phosphatase; | trehalose-6-phosphate | from both sides | |||
| 6448751_Sco; | phosphatase | ||||
| 0.0; | |||||
| 534-1320; | |||||
| trehalose hydrolase | |||||
| 2896788_ | 117648_Ec; | 3073773_Mm; | Polyfunctional enzyme | 2337823 | The presence of the stand-alone |
| Rv2051c | 1e-16; | 4e-31; | of lipid metabolism | (M. leprae); | version of the eukaryotic |
| Bacterial-eukaryotic | 94-514; | 588-829; | 6468712 | domain in Streptomyces suggests | |
| apolipoprotein | dolichol-phosphate- | (Streptomyces | an ancient horizontal transfer | ||
| N-acyltransferase | mannose synthase | coelicolor) | |||
| 2791523_ | 6225563_Scy; | 1098605_Cnu; | Multifunctional enzyme | None | |
| Rv2483c | 7e-16; | 5e-22; | of phospholipid | ||
| Bacterial-eukaryotic | 36-253; | 289-492; | metabolism | ||
| phosphoserine | 1-acyl-sn- | ||||
| phosphatase | glycerol-3-phosphate | ||||
| acyltransferase | |||||
| 2894233_ | 2633801_Bs; | 4538974_At; | Molybdopterin synthase | 2076687 | The same domain organization |
| Rv3323c | 3e-19; | 7e-06; | is seen in D. radiodurans, but in | ||
| Bacterial-eukaryotic | 89-208; | 2-82; | this case, both components | ||
| molybdopterin | molybdopterin | appear to be of bacterial origin | |||
| synthase large subunit | synthase small subunit | ||||
| (MoaE) | (MoaD) | ||||
| 2960152_ | 4753872_Sco; | 466119_Ce; | cAMP-regulated | 2501688 | M. tuberculosis encodes two |
| Rv3728, | 1e-35; | 7e-20; | efflux pump(?) | strongly similar paralogs with | |
| 7477551_ | 56-428; | 549-964; | the same domain architecture | ||
| Rv3239c | transmembrane | cAMP-binding domain- | |||
| Bacterial-eukaryotic | efflux protein | phosphoesterase | |||
| 2960153_ | 4731342_Sl; | 1591330_Mj; | Bifunctional enzyme | 1806159 | The amino-terminal domain |
| Rv3729 | 3e-14; | 3e-58; | of molybdenum | stand-alone paralog is more | |
| Bacterial-archaeal | 510-776; | molybdenum | cofactor biosynthesis | similar to archaeal homologs | |
| C5-O-methyl- | cofactor biosynthesis | than to the stand-alone paralog, | |||
| Transferase | protein MoaA | but nevertheless, the latter | |||
| (mitomycin | (Fe-S oxidoreductase) | appears to be of archaeal origin | |||
| biosynthesis) | |||||
| 3261806_ | 40487_Cg; | 7304009_Dm; | Secreted protein | 7649504 | The stand-alone version of the |
| Rv3811 | 3e-12; | 2e-12; | (S. coelicolor) | eukaryotic domain is present | |
| Bacterial-eukaryotic | 404-494; | 198-384; | only in Streptomyces | ||
| major secreted | peptidoglycan | ||||
| protein | recognition protein | ||||
| Treponema pallidum | |||||
| 3322964_ | 7225946_Nm; | 320868_Sc; | Uridine kinase | None | A co-linear ortholog is present |
| TP0667 | 9e-04; | 2e-13; | in Thermotoga | ||
| Bacterial-eukaryotic | 10-154; | 290-488; | |||
| threonyl-tRNA | uridine kinase | ||||
| synthetase (TGS and | |||||
| H3H domains) | |||||
| Thermotoga maritima | |||||
| 4981276_ | 68516_Bs; | 3218401_Sp; | Uridine kinase | None | A co-linear ortholog is present |
| TM0751 | 3e-07; | 2e-11; | in Treponema | ||
| Bacterial-eukaryotic | 11-200; | 288-475; | |||
| threonyl-tRNA | uridine kinase | ||||
| synthetase (TGS and | |||||
| H3H domains) | |||||
| Eukaryotes | |||||
| Saccharomyces cerevisiae | |||||
| 536367_ | 586134_Bt; | 7450047_Aa; | Bifunctional signal- | 5249 | SurE homologs are not |
| Ybr094w | 9e-10; | 8e-09; | transduction protein | (Yarrowia | detectable in eukaryotes other |
| Eukaryotic/ | tubulin-tyrosine ligase | acid phosphatase | lipolytica) | than yeasts | |
| Bacterial-archaeal | (SurE) | ||||
| 1431219_ | 577625_Hs; | 3328426_Ct | |||
| YDL141w | 1e-39 | 5e-27; | |||
| Eukaryotic- | Biotin-[propionyl- | biotin protein ligase | Bifunctional biotin- | None | An ortholog with an identical |
| bacterial | CoA-carboxylase(ATP- | protein ligase | domain architecture is present | ||
| hydrolysing)] ligase | in S. pombe | ||||
| 458922_ | 477096_Gg; | 1653075_Ssp; | heat shock | NONE | An ortholog with an identical |
| YHR206W | 8e-18; | 7e-17; | transcription | domain architecture is present | |
| Eukaryotic-bacterial | 78-216 | 375-503; | factor | in S. pombe (3327019) | |
| heat shock | CheY domain | ||||
| transcription factor | |||||
| domain | 2983676_Aa; | Siroheme synthase | 2330809 | S. pombe also encodes a co-linear | |
| 486539_ | 1146165_At; | 1e-04; | (S. pombe) | ortholog (3581882); apparent | |
| YKR069w | 3e-34; | 22-188; | displacement of the bacterial | ||
| Eukaryotic-bacterial | 249-556; | precorrin-2 oxidase | precorrin-2 oxidase by a distinct | ||
| urophorphyrin III | Rossmann fold domain | ||||
| methylase | |||||
| 1302305_ | 4938476_At; | 3212189_Hi; | Multifunctional enzyme | None | Co-linear orthologs in S. pombe |
| YNL256w | 5e-65; | 5e-05; | of folate biosynthesis | (7490442) and Pneumocystis | |
| Eukaryotic-bacterial | 324-861 | 62-148; | carinii (283062) | ||
| 7,8-dihydro-6- | 187-297; | ||||
| hydroxymethylpterin- | dihydro-neopterin | ||||
| pyro-phosphokinase+ | aldolase | ||||
| Dihydro-pteroate | |||||
| synthase | |||||
| 1419887_ | 7297709_Dm; | 5918510_Sco; | Bifunctional RNA | 2213559 | The known bacterial homologs |
| YOL066c | 2e-72; | 2e-10; | modification enzyme | (S. pombe) | have a two-domain organization; |
| Eukaryotic-bacterial | 42-408; | 436-574; | the evolutionary scenario could | ||
| large ribosomal | pyrimidine deaminase | have included domain | |||
| subunit pseudoU | rearrangements | ||||
| synthase | |||||
| 1419865_ | 2462827_At; | 1075360_Hi; | Transcriptional regulator None | Yeast encodes three strongly | |
| YOL055c, | 1e-39; | 6e-24; | of thiamine biosynthesis | similar paralogs with identical | |
| 2132251_ | 22-390; | 342-549; | genes(?) | domain organization; co-linear | |
| YPL258c, | phosphomethyl | transcriptional | orthologs are present in other | ||
| 2132289_ | pyrimidinekinase | activator | ascomycetes | ||
| YPR121w | (thiamine biosynthesis) | ||||
| Eukaryotic-bacterial | |||||
| 1370444_ YPL214c | 2746079_Bn; | 2648451_Af; | Bifunctional thiamine | None | Except for the one from |
| Eukaryotic-archaeal/ | 1e-27; | 9e-27; | biosynthesis enzyme | A. fulgidus, all highly conserved | |
| Bacterial | 9-233; | 251-531; | homologs of the kinase domain | ||
| thiamin-phosphate | hydroxyethyl-thiazole | of this protein are bacterial; it | |||
| pyro-phosphorylase | kinase | appears likely that the A. fulgidus | |||
| gene is the result of horizontal | |||||
| transfer |
The following complete genomes were analyzed. Archaea: Aeropyrum pernix (Ap); Archaeoglobus fulgidus (Af); Methanococcus jannaschii (Mj); Methanobacterium thermoautotrophicum (Mth); Pyrococcus horikoshii (Ph); Bacteria: Aquifex aeolicus (Aa); Borrelia burgdorferi (Bb); Bacillus subtilis (Bs); Chlamydophila pneumoniae (Cp); Deinococcus radiodurans (Dr); Escherichia coli (Ec); Haemophilus influenzae (Hi); Helicobacter pylori (Hp); Mycobacterium tuberculosis (Mt); Mycoplasma pneumoniae (Mp); Rickettsia prowazekii (Rp); Synechocystis sp (Ssp); Thermotoga maritima (Tm); Treponema pallidum (Tp). No IKFs were detected in the genomes that are not shown in the table. Additional species name abbreviations: At, Arabidopsis thaliana; Axy, Acetobacter xylinus; Bn, Brassica napus; Ce, Caenorhabditis elegans; Cvi, Chromatium vinosum; Gg, Gallus gallus; Hs, Homo sapiens; Mm, Mus musculus; Rn, Rattus norvegicus; Sco, Streptomyces coelicolor; Sl, Streptomyces lavendulae.
Figure 1.
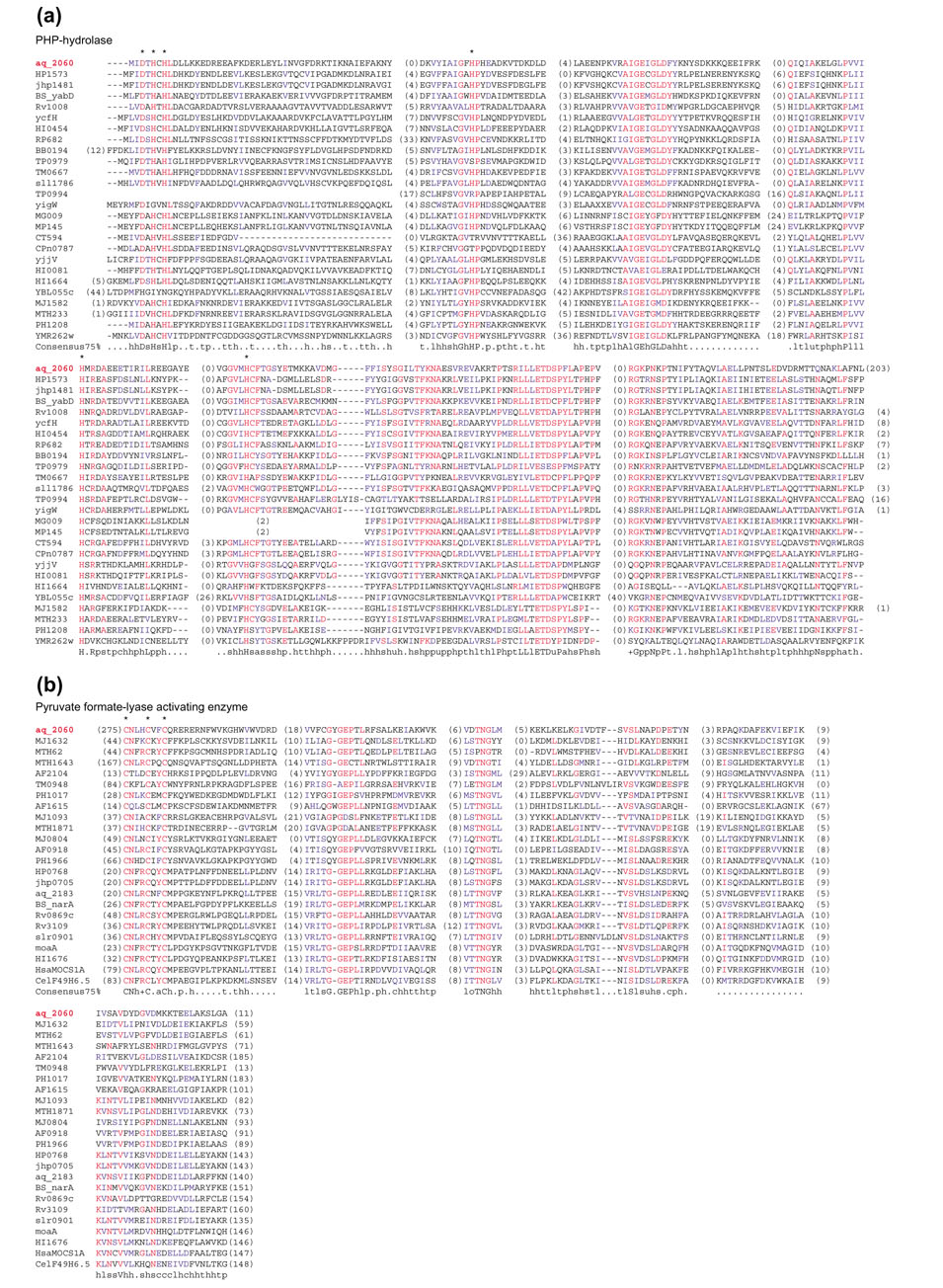
Multiple alignments of two domains comprising an interkingdom domian fusion. Alignments of (a) the PHP-hydrolase domain [4] and (b) the pyruvate formate lyase activating enzyme domain of the IKF protein aq_2060 from A. aeolicus. The sequences of the aq_2060 domains are placed with the most similar sequences of the corresponding stand-alone enzymes, bacterial ones in the case of PHP-hydrolase and archaeal ones in the case of the pyruvate formate lyase activating enzyme. The phylogenetic trees produced form these alignments are shown in Figure 2c. The numbers in parentheses show the lengths of regions between the aligned blocks that are not shown. The consensus includes amino acid residues and residue classes that are conserved in 75% of the aligned sequences; the residue classes are as follows: h, hydrophobic; l, aliphatic; a, aromatic; s, small; u, tiny; p, polar; b, big; t, residues with high turn-forming propensity. Asterisks show the predicted active site residues; note the replacements in some of the sequences that are predicted to be inactivated versions of the respective enzymes (see text). The alignments were colored using the BOXSHADE program [30]; individual residues conserved in at least 50% of the aligned sequences are in red; residues similar to the conserved ones and groups of conserved similar residues are in blue.
In several cases, the chimeric origin of a gene was obvious at a qualitative level because no homolog of the 'alien' domain with comparable sequence similarity was detected in the recipient superkingdom (Table 1, Figure 2a,b). For the rest of the candidate IKFs, phylogenetic tree analysis was performed to corroborate the origin of the invading domain by horizontal transfer; statistically significant grouping of a candidate IKF domain with homologs from the donor superkingdom provides such evidence (Figure 2c,d). The overall number of confirmed IKFs is relatively small - 37 in 21 compared genomes (about 0.1% of the genes) - compared to the total number of likely interkingdom gene transfers. For completely sequenced bacterial genomes this has been conservatively estimated as 1-2% of the genes, with a greater fraction (2-10%) detected in archaea and hyperthermophilic bacteria ([23], and K.S. Makarova, L. Aravind and E.V.K., unpublished observations). Examination of the clusters of orthologous groups (COGs) of proteins from complete genomes [6], in which multidomain proteins are split into the constituent domains if the orthologs of the latter are present as stand-alone forms in some of the genomes, shows that IKFs constitute only a small fraction of all fusions of evolutionarily mobile domains (Figure 3). Generally, the small number of identified IKFs compared to the total number of inferred horizontal transfer events and the total number of domain fusions could be compatible with a random model of domain fusion subsequent to lateral gene transfer.
Figure 2.
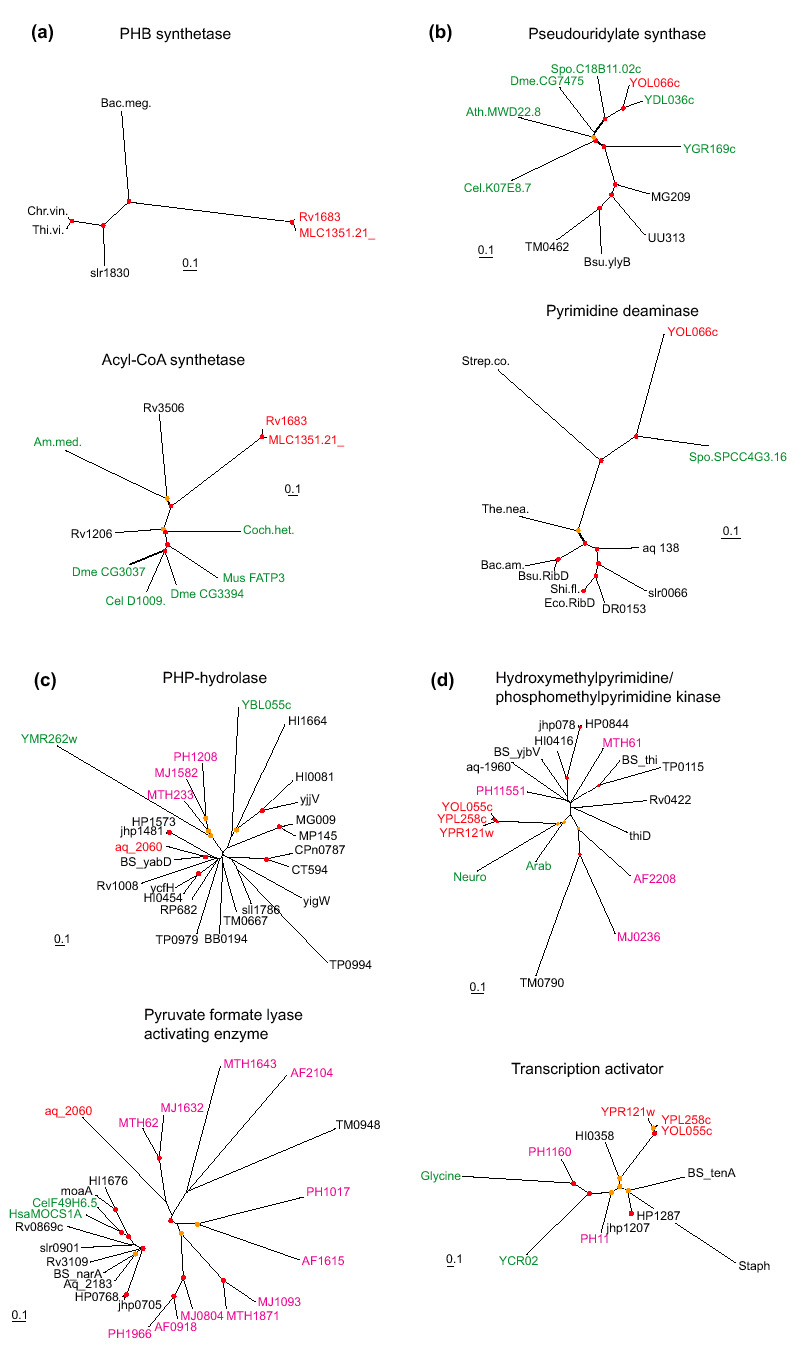
Examples of phylogenetic trees supporting the contribution of interkingdom horizontal gene transfer to the emergence of interkingdom domain fusions. The names of proteins from different primary kingdoms are color-coded: black, bacterial; pink, archaeal; green, eukaryotic; the domains involved in the apparent IKF are shown in red. Red circles show nodes with bootstrap support >70%, and yellow circles show nodes with 50-70% support. The bar unit corresponds to 0.1 substitutions per site (10 PAM). (a) IKF: Rv1683 (gi| 7476858) from M. tuberculosis. Fusion of a bacterial poly(3-hydroxy-butyrate) (PHB) synthase and eukaryotic very long chain acyl-CoA synthetase. Note the absence of eukaryotic homologs in the PHB synthase tree and of bacterial homologs other than the two from M. leprae in the acyl-CoA synthetase tree. (b) IKF: yeast YOL066c (gi|6324506). Fusion of a eukaryotic pesudouridylate synthetase with a bacterial pyrimidine deaminase. Note the absence of eukaryotic homologs, other than that from S. pombe, in the pyrimidine deaminase tree. (c) IKF: aq_2060 (gi|2984285) from Aquifex aeolicus. This protein is a fusion of a PHP superfamily hydrolase of apparent bacterial origin and a pyruvate formate-lyase activating enzyme of archaeal origin. (d) IKF: yeast YOL055c (gi|1419865), YPL258c (gi|2132251) and YPR121w (gi|2132289) from S. cerevisiae. Fusion of a eukaryotic phosphomethylpyrimidine kinase and a bacterial transcriptional activator. Species abbreviations: Bac.meg., Bacillus megaterium; Chr.vin., Chromatium vinosum; Thi.vi., Thiocystis violacea; Am.med., Amycolatopsis mediterranei; Coch.het., Cochliobolus heterostrophus; Dme, Drosophila melanogaster; Cel, Caenorhabditis elegans; Mus, Mus musculus; Spo, Schizosaccharomyces pombe; Ath, Arabidopsis thaliana; Strep.co., Streptomyces coelicolor; The.nea., Thermotoga neapolitana; Bac. am, Bacillus amyloliquefaciens; Shi.fl., Shigella flexneri; Hsa, Homo sapiens.
Figure 3.
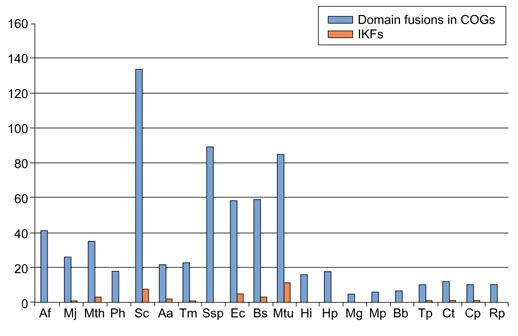
Overall numbers of domain fusions estimated using the COGs and interkingdom domain fusions encoded in completely sequenced genomes. The data for estimating the overall number of domain fusions were from the current COG release [6], which does not include several bacterial and archaeal species (for example, Aeropyrum pernix and Deinococcus radiodurans) that have been analyzed in the present work (Table 1). Accordingly, the data for these genomes are not shown in the figure. Species name abbreviations: Af, Archaeoglobus fulgidus; Mj, Methanococcus jannaschii; Mth, Methanobacterium thermoautotrophicum; Ph, Pyrococcus horikoshii; Sc, Saccharomyces cerevisiae; Aa, Aquifex aeolicus; Tm, Thermotoga maritima; Ssp, Synechocystis sp.; Ec, Escherichia coli; Bs, Bacillus subtilis; Mtu, Mycobacterium tuberculosis; Hi, Haemophilus influenzae; Hp, Helicobacter pylori; Mg; Mycoplasma genitalium; Mp, Mycoplasma pneumoniae; Bb, Borrelia burgdorferi; Tp, Treponema pallidum; Ct, Chlamydia trachomatis; Cp, Chlamydophila pneumoniae; Rp, Rickettsia prowazekii.
However, the distribution of IKFs among genomes is distinctly non-random, suggesting that such a simple model may be incorrect. Specifically, 12 IKFs were detected in Mycobacterium tuberculosis and 10 were found in the yeast Saccharomyces cerevisiae, but only a small number or none was identified in each of the other bacterial and archaeal genomes (Figure 2, Table 1). The excess of IKFs in Mycobacterium is particularly notable, given that the fraction of genes horizontally transferred from archaea and eukaryotes in the mycobacterial genome is only slightly greater than that in most of the other bacteria, and considerably lower than that in the hyperthermophilic bacteria Aquifex and Thermotoga (K.S. Makarova, L. Aravind and E.V.K., unpublished observations). Similarly, whereas the overall number of domain fusions in M. tuberculosis is greater than in most other bacteria, the difference is insufficient to account for the over-representation of IKFs; furthermore, the cyanobacterium Synechocystis sp. has an even greater overall number of fusions but does not have any detectable IKFs (Figure 3). At present, we cannot provide a defendable biological explanation for the comparatively high frequency of IKF in Mycobacterium. It is tempting to interpret this trend in terms of adaptation of this bacterium to its relatively recently occupied parasitic niche, but examination of the individual IKF cases does not offer immediate clues in mycobacterial biology. The yeast IKFs clearly represent relatively recent horizontal transfers distinct from the gene influx from the mitochondria following the establishment of endosymbiosis because, under the protocol of IKF detection used here, only those alien domains were identified that have no counterparts in other eukaryotes.
Most of the IKFs are unique, but B. subtilis, M. tuberculosis and yeast each also encode families of two to three paralogous IKFs, which apparently have evolved by duplication subsequent to the respective fusion events (Table 1). Strikingly, the same IKF, the three-domain uridine kinase, is shared by Treponema pallidum and Thermotoga maritima (Table 1). Given that these two bacteria are not specifically related and that Borrelia burgdorferi, the second spirochete whose genome has been sequenced, encodes a typical bacterial uridine kinase, the presence of a common IKF in Treponema and Thermotoga cannot be realistically attributed to vertical inheritance of this gene from a common ancestor. It thus probably reflects horizontal transfer of the gene encoding the three-domain protein subsequent to its emergence in either the spirochetes or the Thermotogales.
Two evolutionary issues pertaining to IKFs need to be addressed, namely the mechanism(s) of their origin and the selective forces responsible for their preservation. From general considerations, it seems likely that IKFs have evolved via a two-step process, which involves lateral transfer of the complete gene coding for the IKF's alien portion, followed by domain fusion. This scenario rests on the assumption that the acquired foreign gene is selectively advantageous, because otherwise it would have been inactivated by mutations before recombination could take place. Under this mechanism, the alien portion of an IKF is likely to be present in the recipient genome also as a stand-alone gene. A clear-cut case of such a duplication of a horizontally transferred domain has been noticed in Chlamydia, whose genomes encode the SWI domain, implicated in chromatin condensation, both as a stand-alone protein and as the carboxy-terminal portion of topoisomerase I [10]. Apart from this case, the IKFs fall into two readily discernible classes, namely those from Mycobacterium and all the rest. M. tuberculosis (the only complete genome of an actinomycete available) possesses considerably more IKFs than any other bacterial or archaeal species (see above), and typically, the alien portions of these proteins show high level of similarity to the homologs from the donor superkingdom (eukaryotes). Most significantly, there is also, with a single exception, a stand-alone counterpart in the mycobacterial genome; in some cases, such a counterpart is seen only in a closely related species, M. leprae, and in one case, it is found in Streptomyces, a distantly related actinomycete (Table 1). In the other genomes, the IKFs are generally less similar to the apparent donor and, with a few exceptions, stand-alone versions of the alien domains are missing (Table 1). The hypothesis that seems to be most compatible with these observations is that IKFs indeed evolve via a stand-alone, horizontally transferred intermediate, but in the case of ancient IKFs, these intermediates are typically eliminated during evolution, perhaps because their function becomes redundant with the formation of the IKF. The IKFs identified in actinomycetes appear to result from relatively recent gene fusion events so that the original, stand-alone transferred genes are still present in the genome.
The IKFs include a variety of protein functions. Only some of these are well understood such as, for example, those of the bifunctional nucleotide and coenzyme metabolism enzymes that are particularly abundant in yeast (Table 1). In other cases, the function of an IKF-encoded protein could be predicted only tentatively on the basis of the functions of its constituent domains (Table 1). The selective advantage of the formation of multidomain proteins, at least as far as enzymes are involved, lies in the possibility of effective coupling of the reactions catalyzed by the different domains [16]; this may be generalized also for functional coordination of non-enzymatic domains. Fusion may result in the addition of a regulatory function to an enzymatic one. For example, it appears most likely that the RNA-binding TGS domain [24] in the uridine kinases of Treponema pallidum and Thermotoga maritima is involved in autoregulation of translation. The unusual aspect of the IKFs appears to be the compatibility of evolutionarily distant domains.
Examination of the phyletic distribution of the multidomain architectures of IKFs may help in pinpointing the evolutionary stage at which the fusion (but not necessarily the preceding horizontal gene transfer) has occurred. For example, the fusion of the SWI domain with topoisomerase belongs after the radiation of Chlamydia from other bacterial lineages, but before the radiation of Chlamydia pneumoniae and Chlamydia trachomatis (Table 1). The majority of IKFs detected in the yeast S. cerevisiae are also present in Schizosaccharomyces pombe and/or other ascomycetes (Table 1, and data not shown), but not in any other eukaryotes, and accordingly, they should have evolved at a relatively early stage of fungal evolution, but not before the fungal clade diverged from the rest of the eukaryotic crown group.
Finally, it should be noted that formation of some of the IKFs might have required more complex rearrangements of the contributing proteins than simple domain fusion. Figure 4 shows the domain architectures of proteins that contribute domains to two IKFs. In each case, a simple fusion between genes encoding the respective individual domains is insufficient to explain the emergence of the IKF. For example, the uridine kinase example mentioned above (Figure 4a) should have involved isolation of the TGS-HxxxH domains of threonyl-tRNA synthetase before or concomitantly with their fusion with the uridine kinase. The specific molecular mechanism could have involved selective duplication of the upstream portion of the threonyl-tRNA synthetase gene. Similarly, the sialic acid synthase homologous domain, which is fused to hydroxymethylpyrimidine phosphate kinase in A. pernix and pyrococci, appears to have been derived from two-domain proteins that additionally contain a helix-turn-helix DNA-binding domain (Figure 4b). These hypotheses of a complex mechanism of gene fusion involved in the emergence of IKFs are based on a limited sample of sequenced genomes. An alternative possibility is that, before the postulated horizontal transfer event, the recipient domain(s) has been encoded by a stand-alone gene; such genes that do not contain the fused alien domain may yet be discovered in newly sequenced genomes. In fact, a stand-alone version of the sialic acid synthase homologous domain is seen in Methanobacterium, although it is considerably less similar to the IKF than the version fused to the HTH domain (Figure 4b).
Figure 4.
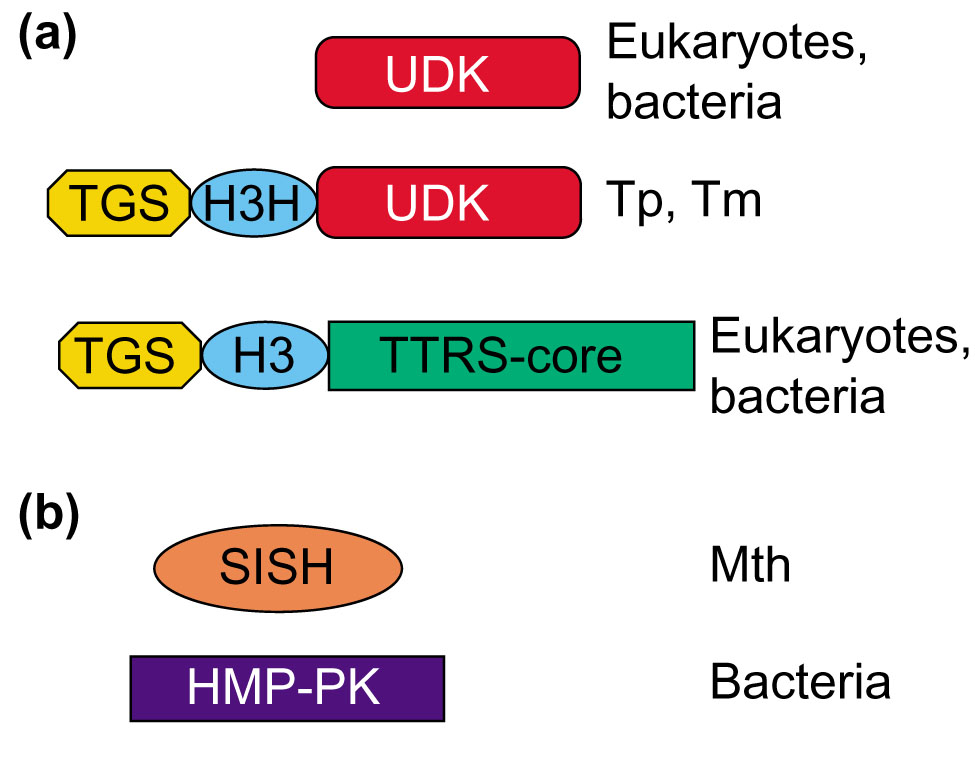
Multidomain architectures of interkingdom fusion proteins and their homologs (examples). (a) The three-domain uridine kinase; (b) the sialic acid synthase homologous domain fused to hydroxymethylpyrimidine phosphate kinase. Domain name abbreviations: TTRS, threonyl-tRNA synthetase; UDK, uridine kinase; TGS and H3H, amino-terminal domains of TTRS; HMP-PK, hydroxymethylpyrimidine phosphate kinase; SISH, sialic acid synthase homologous domain; HTH, helix-turn-helix DNA-binding domain. Different shades represent distinct sequence families of each domain. Species name abbreviations: Tp, Treponema pallidum; Tm, Thermotoga maritima; Mth, Methanobacterium thermoautotrophicum; Mj, Methanococcus jannaschii; Ap, Aeropyrum pernix; Ph, Pyrococcus horikoshii; Pa, Pyrococcus abyssii.
The identification of IKFs underscores the complexity of the evolutionary process as revealed by comparison of multiple genomes. In and by itself, this phenomenon may not have a unique biological significance, but it reveals the overlap between two major evolutionary trends, horizontal gene transfer and protein domain rearrangement, and shows that domains, rather then entire proteins (genes), should be considered fundamental units of genetic material exchange.
Materials and methods
Protein sequences encoded in 21 complete genomes of archaea, bacteria and the yeast Saccharomyces cerevisiae were extracted from the Genome division of the Entrez retrieval system [25]. Each protein encoded in these genomes was used as the query in a comparison against the non-redundant protein sequence database (National Center for Biotechnology Information, NIH, Bethesda, USA) using the BLASTP program [26]. For each query, the set of local similarities detected by BLASTP was automatically (using a Perl script written for this purpose) screened for putative IKFs, that is situations in which the query did not have full-size homologs outside its immediate taxonomic group (for example, the Proteobacteria for Escherichia coli) and in which different regions of the query showed the greatest similarity to proteins from different primary kingdoms. The pseudocode for the script follows:

The script itself is available as an additional data file. The candidate IKF cases were further examined to detect situations where one or more distinct regions of the query could be classified as 'native' or 'alien' either on the basis of the lack of close homologs from the respective primary kingdom or using phylogenetic analysis. Multiple sequence alignments were generated using the ClustalW program [27], and when necessary, manually corrected to ensure the proper alignment of conserved motifs typical of the respective domains. Phylogenetic trees were constructed using the PROTDIST and FITCH programs of the PHYLIP package [28]. Trees were made separately for each domain of a putative IKF, and its mixed ancestry was considered confirmed if the affinities of the domains with different primary kingdoms were supported by bootstrap values of at least 50%. Additional iterative database searches were performed using the PSI-BLAST program [26,29] in order to predict functions of the individual domains of the identified IKFs in cases when these were not immediately clear.
Additional data
The following additional data are included with the online version of this paper: the Perl script used to screen local similarities for putative IKFs.
Supplementary Material
The Perl script used to screen local similarities for putative IKFs
References
- Doolittle WF. Lateral genomics. Trends Cell Biol. 1999;9:M5–M8. [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. . Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Mushegian AR, Galperin MY, Walker DR. Comparison of archaeal and bacterial genomes: computer analysis of protein sequences predicts novel functions and suggests a chimeric origin for the archaea. . Mol Microbiol. 1997;25:619–637. doi: 10.1046/j.1365-2958.1997.4821861.x. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Gray MW. Evolution of organellar genomes. Curr Opin Genet Dev. 1999;9:678–687. doi: 10.1016/s0959-437x(99)00030-1. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. . Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Aravind LR, Tatusov L, Wolf YI, Walker DR, Koonin EV. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. . Trends Genet. 1998;14:442–444. doi: 10.1016/s0168-9525(98)01553-4. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. . Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. . Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Subramanian G, Koonin EV, Aravind L. Comparative genome analysis of pathogenic spirochetes - Borrelia burgdorferi and Treponema pallidum. Infect Immun. 2000;68:1633–1648. doi: 10.1128/iai.68.3.1633-1648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Koonin EV. Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends Genet. 1999;15:173–175. doi: 10.1016/s0168-9525(99)01704-7. [DOI] [PubMed] [Google Scholar]
- Doolittle RF. The multiplicity of domains in proteins. . Annu Rev Biochem. 1995;64:287–314. doi: 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Ilipoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. . Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Who is your neighbor: new computational approaches in functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M., Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. . Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res. 1999;27:4658–4670. doi: 10.1093/nar/27.23.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebe TW, Stock JB. The histidine protein kinase superfamily. . Adv Microb Physiol. 1999;41:139–227. doi: 10.1016/s0065-2911(08)60167-8. [DOI] [PubMed] [Google Scholar]
- Saier MH, Jr, Reizer J. The bacterial phosphotransferase system: new frontiers 30 years later. Mol Microbiol. 1994;13:755–764. doi: 10.1111/j.1365-2958.1994.tb00468.x. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Aravind L, Kondrashov AS. The impact of comparative genomics on our understanding of evolution. Cell. 2000;101:573–576. doi: 10.1016/s0092-8674(00)80867-3. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV. Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-tRNA synthetases - analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710. [PubMed] [Google Scholar]
- National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/Entrez/Genome/org.html
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. . Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Koonin EV. PSI-BLAST - a tool for making discoveries in sequence databases. Trends Biochem Sci. 1998;23:444–447. doi: 10.1016/s0968-0004(98)01298-5. [DOI] [PubMed] [Google Scholar]
- BOXSHADE http://www.ch.embnet.org/software/BOX_form.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Perl script used to screen local similarities for putative IKFs