

Abstract
Site-directed mutagenesis was used to produce 27 single cysteine mutants of bacteriophage M13 major coat protein spanning the whole primary sequence of the protein. Single-cysteine mutants were labeled with nitroxide spin labels and incorporated into phospholipid bilayers with increasing acyl chain length. The SDSL is combined with ESR and CD spectroscopy. CD spectroscopy provided information about the overall protein conformation in different mismatching lipids. The spin label ESR spectra were analyzed in terms of a new spectral simulation approach based on hybrid evolutionary optimization and solution condensation. This method gives the residue-level free rotational space (i.e., the effective space within which the spin label can wobble) and the diffusion constant of the spin label attached to the protein. The results suggest that the coat protein has a large structural flexibility, which facilitates a stable protein-to-membrane association in lipid bilayers with various degrees of hydrophobic mismatch.
INTRODUCTION
Protein frustration arising from the presence of lipids that do not match the protein hydrophobic thickness has been suggested to be an important factor in determining a stable thermodynamic association of a membrane protein with the lipids (1–4). A minimal bilayer perturbation is achieved when the hydrophobic length of the protein matches that of the surrounding lipids. To avoid hydrophobic mismatch, the protein may i), change the conformational space of the amino acid side chains; ii), change backbone conformations, thereby modifying its secondary structure elements; iii), change the tilt angles of the transmembrane segments; iv), change its partitioning in the lipid bilayer; or v), change its aggregation state in the membrane. On the other hand, lipids also can reduce the free energy by a change of the order of the lipid acyl chains as well as by a phase transition to nonlamellar structures (3,5,6).
To study the effect of hydrophobic mismatch on protein structure, it is advisable to avoid complicating factors such as different electrostatic properties of the lipids, lipid miscibility, or preferential phase partitioning. It is therefore usual to work with a homolog series of PC lipid bilayers with an increasing acyl chain length (6–8). The major coat protein of bacteriophage M13, which is composed of 50 amino acid residues, is often used as a reference system for such experiments since during part of the phage life cycle, this protein is stored in the cell membrane (9,10). For example, the various effects of different hydrophobic thicknesses on the lipid selectivity, oligomerization, lateral segregation, and tilt angle of the coat protein have been addressed previously by fluorescence spectroscopy (7,8,11–14). From these studies it follows that the protein exhibits a large selectivity toward phospholipids that allows a good hydrophobic matching (7). Upon decreasing the hydrophobic thickness the tilt angle of the transmembrane α-helix increases, thereby enabling much of the hydrophobic part of the protein to be inside the lipid bilayer. In addition the transmembrane α-helix may rotate along its long axis to optimize the hydrophobic and electrostatic interactions of the C-terminal phenylalanines and lysines (8,15). Reconstitution of the protein in short chain lipid bilayers also results in a loss of helical structure and formation of a stretched conformation of the hinge region that connects the N-terminal domain and transmembrane α-helix (13). So far, however, no attempts were made to obtain a detailed overall structure of bacteriophage M13 major coat protein in mismatching lipid systems. In general, there are also only a few references to the detailed structure analysis of other membrane proteins in mismatching lipids (1).
SDSL has proven to be a powerful approach for the study of membrane protein structure, topology, and dynamics (16–19). In this approach, site-specifically placed cysteine residues are used to introduce nitroxide radicals at a well-defined position in a protein sequence. ESR spectroscopy then yields the rotational restrictions of the attached label that arise from structural constraints due to secondary, tertiary, or quaternary structure of the protein. In principle it should therefore be possible to resolve the membrane protein structure if one were able to analyze motional patterns of spin labels attached at various specific positions along the protein primary sequence. Recently, a new approach for the detailed analysis of spin label ESR spectra was developed that enables one to search for the most probable motional patterns by ESR spectral simulation and global parameter optimization, followed by a condensation algorithm that filters and groups the solutions found in the optimization runs (20). In particular, by using this analysis, it becomes possible to determine the distribution of the local conformational space of a protein spin labeled at specific sites along the protein sequence in different lipid environments.
In this work, site-directed mutagenesis was used to produce 27 single cysteine mutants of the 50-amino-acid major coat protein spanning the whole primary sequence of the protein. Following the approach of our previous work (21), single-cysteine mutants were labeled with nitroxide spin labels and incorporated into lipid bilayers with increasing acyl chain length. CD spectroscopy was applied to infer the overall protein conformation in different mismatching lipids. The ESR data were used to determine the free rotational space. The results suggest that the coat protein has a large structural flexibility, which facilitates a stable protein-to-membrane association in lipid bilayers with various degrees of hydrophobic mismatch.
MATERIALS AND METHODS
Sample preparation
Various site-specific single cysteine mutants of bacteriophage M13 major coat protein were prepared, purified, and labeled with 3-maleimido proxyl spin label (Aldrich, Milwaukee, WI) as described previously (15,22). Labeled mutants were reconstituted into 14:1 PC, 16:1 PC, 18:1 PC, 20:1 PC, and 22:1 PC bilayers (all lipids purchased from Avanti Polar Lipids, Alabaster, AL) at L/P 100 as reported earlier (15,23). For the purpose of ESR measurements, the proteoliposomes were then concentrated using lyophilization and subsequent rehydration and were collected by high-speed centrifugation (21).
CD spectroscopy
CD measurements were performed at room temperature on a Jasco J-715 Spectropolarimeter (Jasco, Tokyo, Japan) in the wavelength range 190–250 nm using a 1-mm path length. The CD settings were 100-s scan time, 1-nm bandwidth, 0.1-nm resolution, and 125-ms response time. Up to 50 spectra were accumulated to improve the signal/noise ratio. CD spectra of M13 coat protein reconstituted using the cholate dialysis reconstitution procedure (23) in each lipid bilayer (14:1 PC, 16:1 PC, 18:1 PC, 20:1 PC, and 22:1 PC) and the respective background spectra were recorded in 100 mM sodium phosphate buffer (pH 8.0) under the same experimental conditions. Difference spectra were obtained by subtracting the background spectra from the corresponding spectra. For the purpose of comparison, the different CD spectra were normalized to the same intensity at 222 nm.
ESR spectroscopy
Samples of reconstituted spin-labeled mutants in different lipid bilayers (14:1 PC, 18:1 PC, and 22:1 PC) were filled up to 5 mm in 50-μl glass capillaries that were accommodated within standard 4-mm diameter quartz tubes. ESR spectra were recorded at room temperature on a Bruker ESP 300E ESR spectrometer (Bruker Instruments, Billerica, MA) equipped with a 108TMH/9103 microwave cavity. The ESR settings were 6.38-mW microwave power, 0.1-mT modulation amplitude, 40-ms time constant, 80-s scan time, 10-mT scan width, and 338.9-mT center field (19,24–26). Up to 20 spectra were collected to improve the signal/noise ratio.
ESR spectral simulation, optimization, and solution condensation
The ESR spectra of spin-labeled mutants were simulated as published previously (21). In short, in accordance with the molecular dynamic simulations where picosecond to nanosecond motions of the attached spin label are observed (27), a fast motional averaging approximation was applied to analyze the local motion of the spin label attached to the protein. Typically two parameters, ϑ and φ, were used to describe the maximum amplitudes of restricted tilt (opening cone angle ϑ) and axial rotation (a cone asymmetry angle φ) in partial averaging of the magnetic properties of the spin Hamiltonian.
The magnetic interaction tensors g and A were linearly corrected with a parameter pA that describes the effects of polarity. Furthermore, a parameter prot that counts for effect of proticity on the g tensor was applied (28). Proticity describes the effect of proton binding to the spin label on its magnetic tensor components. It was found that the relative error for parameter prot was quite large, as expected for single frequency ESR measurements. Therefore this parameter will not be used further in our discussion. The magnetic tensor values used in simulation for the g tensor are 2.0091, 2.0051, and 2.0026; and for the A tensor, 0.700, 0.670, and 3.345 mT. The tensor components were corrected according to Steinhoff et al. (28) as follows: Axx:= Axx × pA; Ayy:= Ayy × pA; Azz:= Azz × pA; gxx:= gxx + (gxx − gfe) × (−0.77 × (pA − 1) − prot); gyy:= gyy + (gyy − gfe) × (−0.06 × (pA − 1)); gzz:= gzz + (gzz − gfe) × (−0.12 × (pA − 1)), where gfe is the free electron g. When calculating the convolution of the magnetic field distribution and the basic line shape, two line width parameters, τc and W, were applied. A Lorentzian line was used in the motional narrowing approximation with a single effective rotational correlation time τc (29). To describe an additional broadening of the spectral line arising from nonmotional effects, a constant W was applied. This parameter arises from unresolved hydrogen superhyperfine interactions and contributions from paramagnetic impurities (e.g., oxygen), in addition to external magnetic field inhomogeneities, field modulation effects, and intermolecular spin-spin interaction if present and applicable. Since the line broadening parameter W is not relevant for the local spin label motion, we will not discuss it further in this work.
To resolve coexisting motional patterns from the ESR spectra, the simulated spectra were composed from four independent components defined by four solution sets {ϑ, φ, τc, W, pA, prot} and appropriate weights, resulting in 27 independent spectral parameters (note that we get 27 parameters by 4 × 6 + 3 (for the relative weights)). Motional patterns, or a distribution of motional patterns, were resolved from the ESR spectra with the implementation of multi-run HEO and a condensation algorithm that filters and groups the solutions found in the optimization runs (called GHOST condensation) (20,21,30).
To guide the optimization for solving the inverse problem, a common fitness function was introduced: the reduced χ2. This function was calculated from the sum of the squared residuals between the experimental and simulated spectral points divided by the squared standard deviation of the experimental points and by the number of points in the experimental spectrum (in our case 1024). Due to the complex search space of the spectral parameters (27 in total), the HEO algorithm was enhanced with a special shaking operator to prevent crowding of solutions (30). For multi-run HEO optimization, 20 runs were applied. Typically 10 different best-fit parameter sets of each run were extracted from the multiple HEO runs. The large amount of information generated was finally condensed by the GHOST condensation algorithm (20) that filters the solutions according to their goodness of fit and solution density in the parameter space and performs group recognition by a slicing method (31).
RESULTS
Protein overall secondary structure
The overall structure of the major coat protein reconstituted into different lipid bilayers was determined with CD spectroscopy. As given in Fig. 1, the CD spectra of the spin-labeled T46C mutant protein are indicative of an overall helical conformation. However, small differences in the CD spectra of the protein in the individual lipid bilayers are observed. The negative ellipticity at 210 nm increases from 14:1 PC to 18:1 PC but decreases again from 18:1 PC to 22:1 PC. For the protein reconstituted in 22:1 PC, the zero crossing of the ellipticity is shifted to a slightly higher wavelength. There is, however, no indication of irreversible β-sheet aggregation for the protein in different PC bilayers.
FIGURE 1.

CD spectra of 3-maleimidoproxyl-labeled M13 T46C mutant coat protein reconstituted in unilamellar vesicles of 14:1 PC, 16:1 PC, 18:1 PC, 20:1 PC, and 22:1 PC at L/P 100 in 100 mM sodium phosphate buffer (pH 8.0) at room temperature. The CD spectra were normalized to the same intensity at 222 nm.
Protein local structure
To resolve the local conformation of the protein in different lipid bilayers, 27 out of 50 amino acid residues were replaced for a cysteine residue, spin labeled, reconstituted into lipid bilayers, and the ESR spectra were recorded. Based on the CD results, bilayers with the protein reconstituted in the two extreme lipid systems 14:1 PC and 22:1 PC were selected for further study (i.e., the thinnest and thickest lipid bilayer). As a rough indicator for the spin label mobility, the outer hyperfine splitting 2Azz for the various mutants reconstituted in 14:1 PC and 22:1 PC bilayers is shown in Fig. 2. In general the spin labels in 22:1 PC bilayers are more restricted as compared to 14:1 PC bilayers. However, the overall pattern of mobility for the different protein segments in 14:1 PC and 22:1 PC bilayers is about the same. Both the C- and N-termini of the protein are unrestricted in mobility, whereas the central part of the protein appears to be strongly immobilized. Although analysis of the outer hyperfine splitting 2Azz of the ESR spectra suggests some modifications of the conformation of the protein in the two lipid bilayers, it is not possible to relate this directly with the CD results.
FIGURE 2.

Outer hyperfine splitting 2Azz of ESR spectra of M13 mutant coat protein labeled with 3-maleimidoproxyl at various positions and reconstituted into 14:1 PC (solid circles) and 22:1 PC (triangles) multilamellar vesicles at L/P 100 in 150 mM NaCl, 10 mM Tris (pH 8.0), and 0.2 mM EDTA at room temperature.
To extract the local structural information, the ESR spectra were simulated with a model of asymmetric motional restriction (21) and characterized via a multi-run hybrid evolution optimization method (20). For all 27 mutants, the quality of the simulated ESR spectra is excellent—the reduced χ2 is between 3 and 5 at a signal/noise ratio between 250 and 400. As an example, the experimental and fitted ESR spectra of the spin-labeled mutants S13C, A25C, A35C, and T46C in the two lipid bilayers are shown in Fig. 3. For reference purposes, these mutants are selected to be well distributed along the primary sequence. Fig. 3 also contains the corresponding GHOST plots that provide the most significant and probable groups of solutions of spectral parameters. The ESR line shapes for the selected mutants in 14:1 PC along the protein primary sequence are different, ranging from isotropic (position 13), to moderately immobilized (position 46), to very anisotropic (positions 25 and 35). Different rotational restrictions of the spin labels as suggested by different spectral line shapes are well resolved by the GHOST plots. For example, the rotational space for position 13 is completely open as suggested by the large ϑ and φ values. On the other hand the rotational space for spin label at position 25 is very restricted as suggested by the green-colored component. As was discussed previously (21), only the “most restricted” component represents the transmembrane state of the protein and will be discussed further in the text. Other components resolved by this methodology can involve nonspecific labeling and other local conformations with lower probabilities. In the thicker lipid bilayer, 22:1 PC, all selected mutants show a more restricted rotational space. The most pronounced difference is observed at position 13, where the spin label experiences a significantly more restricted rotational space in 22:1 PC as compared to 14:1 PC.
FIGURE 3.

ESR spectra of 3-maleimidoproxyl spin-labeled M13 mutant coat proteins S13C, A25C, A35C, and T46C reconstituted into 14:1 PC and 22:1 PC multilamellar vesicles at L/P 100 in 150 mM NaCl, 10 mM Tris (pH 8.0), and 0.2 mM EDTA at room temperature (black line). Spectral line heights are normalized to the same central line height. Simulated spectra (red lines) were fitted to the experimental ESR spectra (black lines) using an HEO optimization. GHOST condensations plots are given where the parameter ϑ is the cone angle within the spin label can move, and parameter φ describes the asymmetry of the cone. The relative fractions of a group of solutions for the two spectral parameters are indicated for each spin-labeled mutant. The red, green, and blue colors of the solutions codes for the relative values of τc, W, and pA in their definition intervals {0–3 ns}, {0–4 G}, and {0.8–1.2}, respectively (40).
The GHOST condensation plots shown in Fig. 3 represent the distribution of the effective free rotational spaces, i.e., the cones defined by the maximal amplitudes of tilt and axial rotations, within which the spin label attached to the protein can wobble. Based on these parameters, the normalized free rotational space, Ω, of the spin label can be defined as
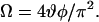 |
(1) |
This parameter measures the space angle, i.e., the surface of the cone left for spin label wobbling. It was deduced from the experimental data, which indicated that the product of the two cone angles is most sensitive for the protein local motion. Note that both angles in this product are normalized to the maximal amplitude, which is π/2. In Fig. 4 the normalized free rotational space Ω of the transmembrane state of the protein reconstituted into 14:1 PC and 22:1 PC bilayers is presented. In this figure, protein domains with differences in the free rotational space can be well identified, for example, segments with no restriction at both terminal ends or segments with large restrictions in the transmembrane α-helix.
FIGURE 4.

Positional dependence of the normalized free rotational space Ω (i.e., the effective space within which the spin label can wobble) of the solutions belonging to the transmembrane state of the M13 coat protein (21), obtained from ESR spectral simulation of spin-labeled protein when reconstituted into 14:1 PC (solid circles) and 22:1 PC (triangles) lipid bilayers. The data points are plotted on a logarithmic scale and represent the center of mass of the Ω distribution, and the error bars point to their second moments. The ESR spectra were recorded at room temperature above the gel-to-liquid crystalline phase transition temperature of 14:1 PC and 22:1 PC.
Protein-folding elements in mismatching lipid environments
By a close inspection of the data in Fig. 4, one can observe different folding elements along the protein sequence. For example, in 14:1 PC bilayers there is no restriction up to position 13, the notable exception, however, being position 10. Next, a relatively constant value for the normalized free rotational space Ω around 0.4 is found from positions 15 to 23 comprising the amphipathic helix. There is a decrease in the normalized free rotational space Ω going from amino acid residues 23–25. This segment represents a part of the so-called hinge region that connects the amphipathic and transmembrane α-helix. Spin-labeled site 25 is the most strongly immobilized one. The penetration of the protein through the lipid-water interface at the C-terminal side seems to be abrupt and associated with amino acid positions 44 and 46.
In the thicker 22:1 PC bilayers, the protein structure adapts to the membrane in a slightly different manner. Overall, the normalized free rotational space Ω is reduced as compared to 14:1 PC, indicating an increased restriction of the local spin label motion. An unstructured N-terminus is seen up to position 7. The restriction of the normalized free rotational space Ω associated with the N-terminal domain shifts toward the N-terminus and extends from amino acid residues 10–23. However, the membrane location at the C-terminus is not affected. Both in 22:1 PC and 14:1 PC, there is a relative strong restriction for positions 23–29.
Protein side-chain rotational diffusion
From a traditional point of view, it is reasonable to expect that the rotational moment of the spin label attached to the protein at different locations along the protein sequence and at constant temperature is approximately constant. From Fig. 4, however, it follows that the space in which the spin label can move differs along the protein sequence. As the rotational moment is approximately constant at a given temperature and environment, the rotational correlation time τc, which is related to the time lapse between subsequent molecular collisions that affect the rotational moment, should correlate with the free rotational space. This implies that the spin label in a more confined environment can keep its rotational moment for a shorter time, i.e., it has a shorter correlation time. On the other hand, a spin label that is spatially less restricted remembers its motional momentum for a longer time, i.e., it has a longer correlation time. To normalize the effect of spatial restrictions on the rotational correlation time, it is useful to recalculate the rotational correlation time taking into account the surface of the cone where the spin label moves. Similar to the classical lateral diffusion constant definition, we can therefore define the rotational diffusion constant D as the quotient of one-fourth of the mean-square displacement and rotational correlation time τc. Note that mean-square displacement in rotational diffusion is approximated with the product of wobble angles, which is proportional to the free rotational space Ω:
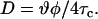 |
(2) |
The dependence of the diffusion constant D along the protein sequence is shown in Fig. 5. In comparing the data for 14:1 PC and 22:1 PC, it is striking to note that, except for positions 12 and 13, the differences in the diffusion constant are small. The spin label diffusion constant is ∼3–4 times lower for sites that are located in the amphipathic domain and transmembrane α-helix, as compared to sites that are located in the N- and C-terminus. Furthermore, there is a strong gradient in the rotational diffusion constant in both the N- and C-terminus. The spin label attached at the far end of the C-terminus (position 49) has a lower diffusion constant as compared to spin labels in the beginning of the N-terminus.
FIGURE 5.

Positional dependence of the rotational diffusion constant D obtained from ESR spectral simulation of spin-labeled M13 coat protein when reconstituted in 14:1 PC (solid circles) and 22:1 PC (triangles) bilayers.
DISCUSSION
The overall and local structure of a membrane protein reconstituted in lipid bilayers with varying degrees of hydrophobic mismatch were studied by CD and SDSL ESR spectroscopy. Site-specific protein cysteine mutants that were selected to be approximately regularly spaced along the primary sequence of the M13 major coat protein were reconstituted into 14:1 PC, 16:1 PC, 18:1 PC, 20:1 PC, and 22:1 PC bilayers. In this work we demonstrate that SDSL in combination with a new computational approach of ESR analysis is capable of providing detailed local molecular information in terms of protein local rotational restrictions that arise due to reconstitution in lipid bilayers with hydrophobic mismatch. The ultimate goal of this approach will be to combine this information to obtain the overall structure of the protein.
Under hydrophobic matching conditions, M13 major coat protein has been reported to be stably associated in a lipid bilayer (7,13,14). In case of hydrophobic mismatch, however, both lipids and protein molecules will tend to reorganize themselves to lower the free energy. As follows from the ratio of the CD intensities 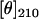 /
/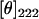 the overall secondary structure of the protein changes when going from a matching 18:1 PC bilayer to either thinner or thicker lipid bilayers. Although CD spectroscopy may enable one to obtain a global structure of the protein, provided the availability of a good reference data set, it is less suitable to determine local molecular details needed to explain relatively small structural rearrangements upon hydrophobic mismatch.
the overall secondary structure of the protein changes when going from a matching 18:1 PC bilayer to either thinner or thicker lipid bilayers. Although CD spectroscopy may enable one to obtain a global structure of the protein, provided the availability of a good reference data set, it is less suitable to determine local molecular details needed to explain relatively small structural rearrangements upon hydrophobic mismatch.
To resolve local conformations of the protein in either positively or negatively hydrophobic mismatching lipid bilayers, we will consider only the effect of the two lipid bilayer extremes (i.e., 14:1 PC and 22:1 PC) on the local structure of the protein. As the majority of the spin-labeled protein sites appeared to be moderately immobilized in both lipid systems, an attempt was made to explain the structural rearrangements on the basis of the outer hyperfine splitting 2Azz. The overall motion of the protein in lipid bilayers is assumed to be slow on the ESR timescale. When the slow tumbling motion of the protein in the lipid bilayer is decoupled from the fast side-chain motion, the main contribution to the ESR spectrum would be an increase of the outer hyperfine splitting. Apart from spin-labeled mutants 25 and 29, which have a relatively large outer hyperfine splitting, all the other spin-labeled mutants have much smaller outer hyperfine splittings. On the other hand, when the slow overall protein motion and fast amino acid side-chain motion are correlated on the ESR timescale, the magnetic tensor components would partially average out and the slow protein motion would result in a distribution of spectral solutions. The contribution of the slow protein motion, however, should be similar along the labeled sites in the protein primary sequence. Since we observed a typical segmental pattern of protein mobility (i.e., flexible N- and C-terminus and amphipathic and transmembrane parts), it is not very likely that a possible contribution of the slow protein motion significantly affects the interpretation of the results.
In the literature, the outer hyperfine splitting 2Azz of spin-labeled proteins is frequently used as a measure of the local constraints for the anisotropic rotational motion of the spin label (27,32). Such an approach was carried out previously on membrane-embedded M13 coat protein by Bashtovyy et al. (33), who used the outer hyperfine splitting 2Azz to search through the database of known micellar NMR structures (34) to find the best fitting conformation by a constrained geometry optimization in a phospholipid environment. However, in the absence of a detailed molecular model that resolves local constraints and free rotational space in nonmatching 14:1 PC and 22:1 PC lipid bilayers, such a general approach is not feasible. Furthermore, in complex multi-component spectra, the outer hyperfine splitting 2Azz may not represent the relevant site-specific information (i.e., a small immobilized fraction producing outer wings of the spectrum may dominate the value of 2Azz). Therefore, to extract the ESR spectral information that will be suitable for structural modeling, a new method for data analysis is introduced here that gives the most probable motional restriction patterns for the spin-labeled site (20,21). This method provides a new parameter: the normalized free rotational space Ω of the spin label while attached to the protein (Eq. 1).
The normalized free rotational space Ω data (Fig. 4) in both 14:1 PC and 22:1 PC are consistent with the generally accepted fold pattern of M13 major coat protein (10,34,35). In both lipid bilayers there is an unstructured N-terminus followed by an amphipathic helix extending up to position 23. The hinge region that connects the amphipathic helix and transmembrane α-helix is reflected by strongly decreased values of Ω. The transmembrane helix is ended with a flexible unstructured short C-terminus. The membrane location at the C-terminus is not affected by the lipid bilayer thickness, in agreement with previous work (8). This effect can be assigned to a unique concerted action of the C-terminal phenylalanines (Phe-42 and Phe-45) and lysines (Lys-40, Lys-43, and Lys-44) in membrane anchoring (9,10).
It is remarkable that given the range of the lipid bilayer thicknesses and differences of the physicochemical properties of 14:1 PC and 22:1 PC, the overall differences in the local structures of the protein in the two lipid systems are relatively small. This indicates that the local free rotational space of the spin label attached to the protein is dominated primarily by the conformation of the protein and affected only slightly by the lipid surroundings. The only strong difference that can be observed is related to the length of the unstructured N-terminal domain, which is seven amino acid residues long in 22:1 PC and 14 residues in 14:1 PC, an exception being position 10, which is severely restricted already in 14:1 PC. The observed length of the unstructured N-terminal domain is consistent with the low-resolution structure of the protein that is emerging from the analysis of site-directed FRET data (P. V. Nazarov, R. B. M. Koehorst, W. L. Vos, V. V. Apanasovich, and M. A. Hemminga, unpublished).
The resolution of the data presented in Fig. 4 further enables fine tuning of the local structure of the protein within a given structural element. It is interesting to note that in both lipid systems, there is a relative strong restriction for positions 23–29. The most pronounced effect is found for position 25, which is much more constrained than the average for the spin labels in the α-helical transmembrane domain. This is probably due to a cluster of bulky amino acid residues in the vicinity (i.e., Ile-22, Tyr-21, Tyr-24, and Trp-26) (33,37,38). These amino acid residues could restrict the local spin label motion, but also the aromatic residues could compact the helix by a sandwich effect due to the planar aromatic side chains. Based on these findings, the local spin label motion can be described in terms of structural hierarchy: 1), structural effects due to the primary structure of the protein, i.e., structural effects due to limitation of the rotational space by the side chains of neighboring amino acid residues; 2), structural effects due to secondary or higher structures of the protein, i.e., folding elements such as α-helix or an unstructured state; and 3), structural effects due to the surrounding lipid molecules.
Consistent with this classification, two rotational diffusion regimes of the spin labels attached to the protein (see Fig. 5) can be identified. The spin label rotational diffusion is either low in the structured part of the protein by the presence of an α-helical conformation in the amphipathic domain and transmembrane section or much higher at both unstructured protein termini. Clearly, the normalized free rotational space Ω is the most sensitive parameter to describe the local spin label environment.
The normalized free rotational space Ω in 14:1 PC in both the amphipathic helix and transmembrane α-helix is less constrained than in 22:1 PC. In addition, the smaller value of the outer splitting 2Azz (Fig. 2) and the larger value of the normalized free rotational space Ω in 14:1 PC bilayers may indicate a less stable and more loosely defined helix, i.e., hydrogen bonds break and close often. From molecular dynamics simulations, it follows that hydrogen bonds that keep the helix structure together can break and reform on the ESR timescale (39). Therefore, ESR spectroscopy should be sensitive enough to pick up this effect. It is presumed that making a hydrogen bond is energetically more favorable in the transmembrane protein domain than in the N-terminal domain. Therefore, it is expected that the transmembrane α-helix is stiffer and more rigid with a reduced rotational space as compared to the amphipathic helix. This is consistent with the normalized free rotational space Ω data (see Fig. 4) that show lower values in the transmembrane α-helix as compared to the amphipathic helix. Thus we can conclude that on average the transmembrane α-helix is more stable than the N-terminal helix. The transition from a more loosely defined amphipathic helix to a more rigid transmembrane helix is indicated by the large change in the normalized free rotational space Ω between position 22 and 25 in both lipid systems.
The SDSL approach developed in this work for membrane proteins provides an alternative structural view as compared to other biophysical techniques, such as NMR and x-ray crystallography. The residue-level information that one can extract from SDSL is useful for obtaining information about protein folding, protein topology, protein-lipid interactions, hydrophobic mismatch, and detection of function-related conformational changes of membrane proteins in different lipid systems. The key advantage of the SDSL approach, however, is that it is independent of the size of the proteolipid system and can be applied in different membrane model systems, i.e., micelles and liposomes. Nevertheless the major challenge that still lies ahead is how to exploit the information about the local free rotational space to obtain a detailed overall structure of membrane proteins with the resolution of one amino acid.
Acknowledgments
This work was partly supported by contract No. QLG-CT-2000-01801 of the European Commission (MIVase—New Therapeutic Approaches to Osteoporosis: targeting the osteoclast V-ATPase) and P1-0060 of the Slovenian Research Agency. D.S., J.Š., and M.A.H. are members of the COST D22 Action “Protein-Lipid Interactions” of the European Union.
Abbreviations used: SDSL, site-directed spin labeling; ESR, electron spin resonance; CD, circular dichroism; PC, phosphatidylcholine; 14:1 PC, 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine; 16:1 PC, 1,2-dipalmitoleoyl-sn-glycero-3-phosphocholine; 18:1 PC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; 20:1 PC, 1,2-dieicosenoyl-sn-glycero-3-phosphocholine; 22:1 PC, 1,2-dierucoyl-sn-glycero-3-phosphocholine; FRET, Förster (or fluorescence) resonance energy transfer; GHOST, condensation algorithm that filters and groups the solutions found in optimization runs; HEO, hybrid evolutionary optimization; L/P, lipid/protein molar ratio.
References
- 1.Lee, A. G. 2003. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 1612:1–40. [DOI] [PubMed] [Google Scholar]
- 2.Bechinger, B. 2001. Solid-state NMR investigations of interaction contributions that determine the alignment of helical polypeptides in biological membranes. FEBS Lett. 504:161–165. [DOI] [PubMed] [Google Scholar]
- 3.Killian, A. J. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–415. [DOI] [PubMed] [Google Scholar]
- 4.Lohner, K., and E. J. Prenner. 1999. Differential scanning calorimetry and x-ray diffraction studies of the specificity of the interaction of antimicrobial peptides with membrane-mimetic systems. Biochim. Biophys. Acta. 1462:141–156. [DOI] [PubMed] [Google Scholar]
- 5.De Planque, M. R., and J. A. Killian. 2003. Protein-lipid interactions studied with designed transmembrane peptides: role of hydrophobic matching and interfacial anchoring. Mol. Membr. Biol. 20:271–284. [DOI] [PubMed] [Google Scholar]
- 6.Balgavy, P., and F. Devinsky. 1996. Cut-off effects in biological activities of surfactants. Adv. Colloid Interface Sci. 12:23–63. [DOI] [PubMed] [Google Scholar]
- 7.Fernandes, F., L. M. Loura, R. Koehorst, R. B. Spruijt, M. A. Hemminga, A. Fedorov, and M. Prieto. 2004. Quantification of protein-lipid selectivity using FRET: application to the M13 major coat protein. Biophys. J. 87:344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehorst, R. B., R. B. Spruijt, F. J. Vergeldt, and M. A. Hemminga. 2004. Lipid bilayer topology of the transmembrane α-helix of M13 major coat protein and bilayer polarity profile by site-directed fluorescence spectroscopy. Biophys. J. 87:1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stopar, D., R. B. Spruijt, C. J. Wolfs, and M. A. Hemminga. 2003. Protein-lipid interactions of bacteriophage M13 major coat protein. Biochim. Biophys. Acta. 1611:5–15. [DOI] [PubMed] [Google Scholar]
- 10.Stopar, D., R. B. Spruijt, and M. A. Hemminga. 2006. Anchoring mechanisms of membrane-associated M13 major coat protein. Chem. Phys. Lipids. 141:83–93. [DOI] [PubMed] [Google Scholar]
- 11.Spruijt, R. B., C. J. A. M. Wolfs, J. W. G. Verver, and M. A. Hemminga. 1996. Accessibility and environmental probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 35:10383–10391. [DOI] [PubMed] [Google Scholar]
- 12.Meijer, A. B., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2001. Membrane-anchoring interactions of M13 major coat protein. Biochemistry. 40:8815–8820. [DOI] [PubMed] [Google Scholar]
- 13.Spruijt, R. B., C. J. M. Wolfs, and M. A. Hemminga. 2004. Membrane assembly of M13 major coat protein: evidence for a structural adaptation in the hinge region and a tilted transmembrane domain. Biochemistry. 43:13972–13980. [DOI] [PubMed] [Google Scholar]
- 14.Fernandes, F., L. M. Loura, M. Prieto, R. Koehorst, R. B. Spruijt, and M. A. Hemminga. 2003. Dependence of M13 major coat protein oligomerization and lateral segregation on bilayer composition. Biophys. J. 85:2430–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meijer, A. B., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2001. Configurations of the N-terminal amphipathic domain of the membrane-bound M13 major coat protein. Biochemistry. 40:5081–5086. [DOI] [PubMed] [Google Scholar]
- 16.Hubbell, W. L., and C. Altenbach. 1994. Investigation of structure and dynamics in membrane proteins using site-directed spin labeling. Curr. Opin. Struct. Biol. 4:566–573. [Google Scholar]
- 17.Fajer, P. G. 2000. Electron spin resonance spectroscopy labeling in peptide and protein analysis. In Encyclopedia of Analytical Chemistry. R. A. Mayers, editor. John Wiley & Sons, Chichester, UK. 1–37.
- 18.Hustedt, E. J., and A. H. Beth. 1999. Nitroxide spin-spin interactions: applications to protein structure and dynamics. Annu. Rev. Biophys. Biomol. Struct. 28:129–153. [DOI] [PubMed] [Google Scholar]
- 19.Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 1996. Local dynamics of the M13 major coat protein in different membrane-mimicking systems. Biochemistry. 35:15467–15473. [DOI] [PubMed] [Google Scholar]
- 20.Strancar, J., T. Koklic, Z. Arsov, B. Filipic, D. Stopar, and M. A. Hemminga. 2005. Spin label EPR-based characterization of biosystem complexity. J. Chem. Inf. Model. 45:394–406. [DOI] [PubMed] [Google Scholar]
- 21.Stopar, D., J. Strancar, R. B. Spruijt, and M. A. Hemminga. 2005. Exploring the local conformational space of a membrane protein by site-directed spin labeling. J. Chem. Inf. Model. 45:1621–1627. [DOI] [PubMed] [Google Scholar]
- 22.Spruijt, R. B., A. B. Meijer, C. J. A. M. Wolfs, and M. A. Hemminga. 2000. Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of bacteriophage M13. Biochim. Biophys. Acta. 1509:311–323. [DOI] [PubMed] [Google Scholar]
- 23.Spruijt, R. B., C. J. A. M. Wolfs, and M. A. Hemminga. 1989. Aggregation-related conformational change of membrane-associated coat protein of bacteriophage M13. Biochemistry. 28:9158–9165. [DOI] [PubMed] [Google Scholar]
- 24.Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 1998. Mimicking initial interactions of bacteriophage M13 coat protein disassembly in model membrane systems. Biochemistry. 37:10181–10187. [DOI] [PubMed] [Google Scholar]
- 25.Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 1997. In situ aggregational state of M13 bacteriophage major coat protein in sodium cholate and lipid bilayers. Biochemistry. 36:12268–12275. [DOI] [PubMed] [Google Scholar]
- 26.Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2002. Structural characterization of bacteriophage M13 solubilization by amphiphiles. Biochim. Biophys. Acta. 1594:54–63. [DOI] [PubMed] [Google Scholar]
- 27.LaConte, L. E. W., V. Voelz, W. Nelson, E. Michael, and D. Thomas. 2002. Molecular dynamics simulation of site-directed spin labeling: experimental validation in muscle fibers. Biophys. J. 83:1854–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinhoff, H. J., A. Savitsky, C. Wegener, M. Pfeiffer, M. Plato, and K. Möbius. 2000. High-field EPR studies of the structure and conformational changes of site-directed spin labeled bacteriorhodopsin. Biochim. Biophys. Acta. 1457:253–262. [DOI] [PubMed] [Google Scholar]
- 29.Strancar, J., M. Sentjurc, and M. Schara. 2000. Fast and accurate characterization of biological membranes by EPR spectral simulations of nitroxides. J. Magn. Reson. 142:254–265. [DOI] [PubMed] [Google Scholar]
- 30.Kavalenka, A. A., B. Filipic, M. A. Hemminga, and J. Strancar. 2005. Speeding up a genetic algorithm for EPR-based spin label characterization of biosystem complexity. J. Chem. Inf. Model. 45:1628–1635. [DOI] [PubMed] [Google Scholar]
- 31.Strancar, J., T. Koklic, and Z. Arsov. 2003. Soft picture of lateral heterogeneity in biomembranes. J. Membr. Biol. 196:135–146. [DOI] [PubMed] [Google Scholar]
- 32.Marsh, D. 1981. Membrane spectroscopy. Electron spin resonance: spin labels. In Membrane Spectroscopy. E. Grell, editor. Springer-Verlag, Berlin. 51–142.
- 33.Bashtovyy, D., D. Marsh, M. A. Hemminga, and T. Pali. 2001. Constrained modeling of spin-labeled major coat protein mutants from M13 bacteriophage in a phospholipid bilayer. Protein Sci. 5:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papavoine, C. H. M., B. E. C. Christiaans, R. H. A. Folmer, R. N. H. Konings, and C. W. Hilbers. 1998. Solution structure of the M13 major coat protein in detergent micelles: a basis for a model of phage assembly involving specific residues. J. Mol. Biol. 282:401–419. [DOI] [PubMed] [Google Scholar]
- 35.McDonnell, P. A., K. Shon, Y. Kim, and S. J. Opella. 1993. fd coat protein structure in membrane environments. J. Mol. Biol. 233:447–463. [DOI] [PubMed] [Google Scholar]
- 36.Reference deleted in proof.
- 37.Yuen, C. T., A. R. Davidson, and C. M. Deber. 2000. Role of aromatic residues at the lipid-water interface in micelle-bound bacteriophage M13 major coat protein. Biochemistry. 39:16155–16162. [DOI] [PubMed] [Google Scholar]
- 38.Wolkers, W. F., R. B. Spruijt, A. Kaan, R. N. H. Konings, and M. A. Hemminga. 1997. Conventional and saturation-transfer ESR of spin-labeled mutant bacteriophage M13 coat protein in phospholipid bilayers. Biochim. Biophys. Acta. 1327:5–16. [DOI] [PubMed] [Google Scholar]
- 39.Berjanskii, M., M. Riley, and S. R. Van Doren. 2002. Hsc70-interacting HPD loop of the J domain of polyomavirus T antigens fluctuates in ps to ns and micros to ms. J. Mol. Biol. 321:503–516. [DOI] [PubMed] [Google Scholar]
- 40.Štrancar, J., T. Koklič, Z. Arsov, B. Filipič, D. Stopar, and M. A. Hemminga. 2005. Spin label EPR-based characterization of biosystem complexity. J. Chem. Inf. Model. 45:394–406. [DOI] [PubMed] [Google Scholar]