

Abstract
Cell membranes have complex lipid compositions, including an asymmetric distribution of phospholipids between the opposing leaflets of the bilayer. Although it has been demonstrated that the lipid composition of the outer leaflet of the plasma membrane is sufficient for the formation of raft-like liquid-ordered (lo) phase domains, the influence that such domains may have on the lipids and proteins of the inner leaflet remains unknown. We used tethered polymer supports and a combined Langmuir-Blodgett/vesicle fusion (LB/VF) technique to build asymmetric planar bilayers that mimic plasma membrane asymmetry in many ways. We show that directly supported LB monolayers containing cholesterol-rich lo phases are inherently unstable when exposed to water or vesicle suspensions. However, tethering the LB monolayer to the solid support with the lipid-anchored polymer 1,2-dimyristoyl phophatidylethanolamine-N-[poly(ethylene glycol)-triethoxysilane] significantly improves stability and allows for the formation of complex planar-supported bilayers that retain >90% asymmetry for 1–2 h. We developed a single molecule tracking (SPT) system for the study of lipid diffusion in asymmetric bilayers with coexisting liquid phases. SPT allowed us to study in detail the diffusion of individual lipids inside, outside, or directly opposed to lo phase domains. We show here that lo phase domains in one monolayer of an asymmetric bilayer do not induce the formation of domains in the opposite leaflet when this leaflet is composed of palmitoyl-oleoyl phosphatidylcholine and cholesterol but do induce domains when this leaflet is composed of porcine brain phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and cholesterol. The diffusion of lipids is similar in lo and liquid-disordered phase domains and is not affected by transbilayer coupling, indicating that lateral and transverse lipid interactions that give rise to the domain structure are weak in the biological lipid mixtures that were employed in this work.
INTRODUCTION
Since the discovery that some cell membranes could be separated into soluble and insoluble fractions by cold extraction with a mild nonionic detergent like Triton X-100 and that these fractions are distinct in their lipid and protein compositions (1,2), experiments by numerous investigators have been performed to determine the origin and function of detergent resistant membranes (DRMs). Although many questions remain unanswered, most of the evidence from both cell and model membranes indicates that the solubility of lipids in Triton X-100 can be correlated to the phase in which they reside at the time of exposure and that the DRM fraction in fact forms liquid-ordered (lo) phase bilayers that separate in vitro from surrounding domains of liquid-disordered (ld) phase bilayers. Conceptual lo phase domains in cell membranes were given the name lipid rafts because of the belief that they serve as platforms for the transport of material from the Golgi to specific parts of the plasma membrane (PM) or for the sequestration of signaling components within the PM (3). Furthermore, rafts have been implicated as sites of actin polymerization (4), as well as viral assembly and budding (5–10).
A major problem with attempts to understand PM domain structure and function by means of correlation of the behavior of DRM fractions is that the contents of these fractions do not match what is currently known about the lipid distribution in the PM. The lipid composition of DRMs is high in sphingomyelin (SM) and glycosphingolipids (GSLs), which are the only lipid species in the PM that contain predominantly fully saturated hydrocarbon chains. Although some monounsaturated phospholipids (PLs) have been recovered from DRMs from cells (11) and can be forced into lo phase regions of model membranes with high cholesterol (12), saturated PLs or SM are required for the onset of ld-lo phase coexistence (12,13). However, the lipid composition of the mammalian PM is asymmetric (14), with nearly all of the SM residing in the extracellular leaflet. Phosphatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidylinositol (PI) exist overwhelmingly in the cytoplasmic leaflet, whereas phosphatidylcholine (PC) and cholesterol are relatively equally distributed between both leaflets (15–17). Therefore, the lipid fraction of the extracellular leaflet of the PM consists of mostly unsaturated PC, saturated SM, and cholesterol, i.e., the necessary components for the formation of lo phase domains. On the other hand, the cytoplasmic leaflet is mainly composed of unsaturated PC, PE, PS, and cholesterol. Model membrane studies in our laboratory and others (18) have so far failed to produce lo phase domains in mixtures mimicking the inner leaflet of the PM (see also Fig. 1), and inner-leaflet PLs are overwhelmingly soluble in detergent-treated cell extracts (2). Yet several groups have reported the recovery of inner leaflet-associated proteins in DRMs (19–21). The question therefore arises: how can rafts that exist only in the outer leaflet of the PM lead to the sequestration of proteins associated with the inner leaflet? In the absence of conclusive experimental data to resolve this question, the following speculative explanations are frequently cited in the literature: 1), lipid rafts in the outer leaflet induce the formation of ordered lipid domains in the inner leaflet, which in turn sequester acylated proteins; 2), transmembrane proteins that are associated with outer leaflet lipid rafts directly interact with proteins on the inner leaflet, resulting in their insolubility; or 3), there is no transbilayer effect, and the insoluble nature of some proteins is fortuitous and independent of lipid association.
FIGURE 1.

Fluorescence micrographs of planar-supported bilayers mimicking the inner leaflet lipid composition of the mammalian PM. Bilayers were composed of equimolar amounts of porcine brain PC and PE (A) or PC, PE, and PS (B) and cholesterol as indicated. Bilayers were formed by the LB/LS method on quartz slides at room temperature and stained with 0.5% Rh-DPPE in the LS monolayer only. The white bar represents 10 μm.
Fluorescence studies in giant vesicles have revealed much detail about the thermodynamics and dynamics of the lipid phase behavior in the liquid-liquid phase coexistence region (13,22–25), but such systems cannot be made with asymmetric lipid distributions. Planar-supported bilayers are attractive alternative systems for studying the formation, nature, and transbilayer effects of lo phase domains because 1), lipid composition and sometimes lipid asymmetry can be controlled, 2), proteins can be reconstituted in a biologically active state, and 3), the planar geometry of the bilayer facilitates the simple interpretation of structural and dynamical data obtained from fluorescence microscopy including total internal reflection fluorescence microscopy (TIRFM), fluorescence recovery after photobleaching (FRAP), single molecule fluorescence tracking (sometimes called single particle tracking, SPT), and fluorescence interference contrast (FLIC) microscopy.
FLIC microscopy is a relatively new method to measure distances in the z-direction with exquisite resolution. We have recently extended the method and developed a modified fitting procedure that allowed us to measure lipid asymmetry in planar-supported bilayers by FLIC microscopy (26). In that work, we also showed that asymmetric lipid bilayers could be made by a combined Langmuir-Blodgett/vesicle fusion (LB/VF) technique (27) that retained nearly 100% lipid asymmetry upon completion. The motivation behind making asymmetric planar-supported bilayers is to mimic the asymmetric nature and thus provide a good new model system of the PM. An interesting application of this system is to examine whether a transbilayer coupling exists between certain different lipid compositions in opposing leaflets. In particular, we are interested in addressing the question of whether lo phase domains in the outer leaflet induce changes in the phase behavior of the inner leaflet as postulated in one of the hypothetical raft coupling scenarios.
In this study, we present the development of a model system consisting of a polymer-supported asymmetric planar bilayer that contains stable lo phase domains in one leaflet and a homogeneous ld phase in the opposite leaflet. The possible transbilayer influence of asymmetric lo phase domains on lipids in the opposing leaflet is studied by epifluorescence imaging, FRAP, and SPT. The stability of the initial (LB) monolayer upon exposure to vesicle solutions during the formation of LB/VF bilayers and the effects of phase, composition, and tethered polymer supports on this process are examined by fluorescence imaging and TIRFM. The FLIC method is utilized to measure the retention of asymmetry as well as the time course of lipid flip-flop in fluid-phase-supported lipid bilayers.
MATERIALS AND METHODS
The following materials were purchased and used without further purification: 1-palmitoyl-2-oleoyl phosphatidylcholine (POPC), 1,2-dipalmitoyl phosphatidylcholine (DPPC), 1,2-dimyristoyl phosphatidylethanolamine (DMPE), 1,2-dimyristoyl phosphatidylethanolamine-N-[7-nitro-2-1,3-benzoxadiazol-4-yl] (NBD-DMPE), 1,2-dipalmitoyl phosphatidylethanolamine-N-[lissamine rhodamine B] (Rh-DPPE), porcine brain PC (bPC), porcine brain PE (bPE), porcine brain PS (bPS), and porcine brain SM (bSM) (Avanti Polar Lipids, Alabaster, AL); Alexa Fluor 647 (A647) succinimidyl ester (Molecular Probes, Eugene, OR); cholesterol, HEPES, and glycerol (Sigma Chemical, St. Louis, MO); chloroform, ethanol, methanol, ether, Contrad detergent, all inorganic salts, acids, bases, and peroxide (Fisher Scientific, Fair Lawn, NJ). 1,2-dimyristoyl phophatidylethanolamine-N-[poly(ethylene glycol)-triethoxysilane] (DPS) was custom synthesized by Nektar Therapeutics (Huntsville, AL).
Fluorescent labeling 500 μL of a 2 mg/mL solution of DMPE in methanol/chloroform (3:1) was mixed with 1 mg of A647 succinimidyl ester, stirred rapidly for 1 h at room temperature, then incubated at 4°C overnight. Fluorescently labeled lipid (A647-DMPE) was separated from unreacted DMPE and dye by preparative thin-layer chromatography as described by Kates (28) on 1-mm-thick silica gel plates (Whatman, Clifton, NJ) with a developing solvent of chloroform/methanol/water (65:25:4) and eluting solvent of chloroform/methanol/ether (1:1:1). A647-DMPE was stored at −20°C in chloroform.
Large unilamellar vesicles
The desired lipids were codissolved in chloroform or chloroform/methanol. Solvent was evaporated under a stream of N2 gas followed by vacuum for at least 1 h. The resulting residue was suspended in reconstitution buffer (RB: 25 mM HEPES, 100 mM KCl, 1 mM CaCl2, pH = 7.4), rapidly vortexed, freeze-thawed five times by submersion in liquid N2 followed by water at 40°C, and extruded by 15 passes through two polycarbonate membranes with a pore diameter of 100 nm (Avestin, Ottawa, ON). Vesicles could be stored at 4°C for up to 5 days before use.
Planar-supported bilayers
Planar bilayers were prepared on 40 mm × 25 mm × 1 mm quartz slides (Quartz Scientific, Fairport Harbor, OH) for epifluorescence microcopy, FRAP, SPT, or TIRFM. Slides were cleaned by boiling in Contrad detergent for 10 min, then hot bath sonicated while still in detergent for 30 min, followed by extensive rinsing with water, then methanol, then water again. Remaining organic residue was removed by immersion in three volumes of 95% H2SO4 to one volume of 30% H2O2, followed by extensive rinsing in water. Immediately before use, slides were further cleaned for 10 min in an argon plasma sterilizer (Harrick Scientific, Ossining, NY). Si/SiO2 chips were used as a substrate for FLIC experiments. Silicon wafers were oxidized at 1000°C until the SiO2 layer reached a thickness of 350 nm. The surface was then patterned by photolithography and HF etching to form four distinct levels of oxide with thicknesses ranging from 10 to 200 nm. Oxide thicknesses were measured by microellipsometry (Plasmos SD2000, Munich, Germany). Immediately before use, the chips were cleaned in three volumes of 95% H2SO4 to one volume of 30% H2O2, followed by extensive rinsing in water. Bilayers were formed by either the LB/LS (29) or LB/VF technique (27). The different methods for the formation of directly supported and polymer-supported planar bilayers were described in detail recently (26).
Fluorescence microscopy
For all but the SPT experiments, we used a Zeiss Axiovert 35 fluorescence microscope (Carl Zeiss, Thornwood, NY) with either a mercury lamp or an argon ion laser (Innova 300C, Coherent, Palo Alto, CA) as a light source. A mirror cube at the back of the microscope was used to switch between epiillumination by the laser and the lamp. Another removable mirror was used to switch between TIR- and epiillumination by the laser. For epifluorescence imaging and FLIC experiments, bilayers labeled with 0.5% Rh-DPPE were excited with the mercury lamp through a 546-nm band-pass filter (BP546/10, Schott Glaswerke, Mainz, Germany) and observed through a 610-nm band-pass filter (D610/60, Chroma, Brattleboro, VT). For TIRFM experiments, Rh-DPPE was excited by the laser at 514 nm and observed through the same 610-nm band-pass filter. For FRAP experiments, NBD-DMPE was excited by the laser at 488 nm and observed through a 535-nm band-pass filter (D535/40, Chroma). A 40× water immersion objective (Zeiss; numerical aperture (N.A.) 0.75) was used in all cases. The intensity of the laser beam was computer controlled through an acoustooptic modulator (AOM-40, IntraAction, Bellwood, IL) or could be blocked entirely by a computer-controlled shutter. Fluorescence emission intensity was measured using a photomultiplier tube (Thorn EMI 9658A, Ruislip, UK). Images were recorded by a charge-coupled device (CCD) cooled to −12°C (Sensicam QE, Cooke, Auburn Hills, MI). Image analysis and data acquisition was done using LabVIEW software (National Instruments, Austin, TX).
TIRFM
A focused laser beam was directed through a trapezoidal prism onto the quartz-buffer interface where the planar membrane was attached. The prism-quartz interface was lubricated with glycerol to allow easy translocation of the sample cell on the microscope stage. The beam was totally internally reflected at an angle of 72° from the surface normal, resulting in an evanescent wave that decays exponentially through the solution as follows:
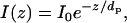 |
(1) |
where I0 is the incident intensity and dp is the total penetration depth. For our setup, dp was 90 nm. An elliptical area of ∼250 μm × 65 μm was illuminated and observed.
FRAP
Bilayers were bleached in a pattern of parallel stripes (30) and the data were fit to the model
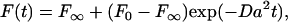 |
(2) |
where F0 and F∞ are the initial and final fluorescence intensities after bleaching, respectively, a = 2π/p, p is the stripe period (12.7 or 3.2 μm), and D is the lateral diffusion coefficient. The mobile fraction mf, which reflects the % of observed fluorescence recovery within the time frame of a FRAP experiment (<1 min), is given by
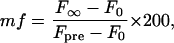 |
(3) |
where Fpre is the fluorescence intensity before photobleaching. Ten to fifteen regions on at least two independently prepared bilayers were sampled to determine the reported average values.
FLIC microscopy
FLIC microscopy (31–33) was used to measure the average distance of all fluorescent dyes from a reflective interface. When directly supported on a SiO2 substrate, a 2-nm cleft of water separates the bilayer from the surface (34). The average distance 〈d〉 of all dyes from the top of the cleft and the fraction of dyes in the distal monolayer,
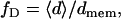 |
(4) |
where dmem is the membrane thickness, were determined as described (26). Using our data collection and fitting methods, it is possible to determine 〈d〉 with an accuracy of ±5 Å.
Single molecule tracking
SPT experiments were carried out with a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss) using a dye laser (599, Coherent) pumped by an argon ion laser (Innova 90C-5, Coherent) as excitation source. The laser intensity was modulated by an acoustooptic modulator (Isomet, Springfield, VA), and the first-order diffracted beam was directed through a shutter and a focusing lens (f = 300 mm, Newport, Irvine, CA) into the fluorescence illuminator of the microscope. The total power of this beam was typically set to 20–70 mW. To track single A647-DMPE lipids, the laser beam was reflected by a dichroic mirror (660dclp, Chroma) and focused onto the back focal plane of a 63× water immersion objective (Zeiss, N.A. = 0.95). This produced a circular field of view with a diameter of ∼30 μm. In some experiments, the sample was illuminated by total internal reflection. This was achieved by directing the laser beam by means of a quartz prism at an angle of 72° to the normal on the sample slide, which was optically coupled to the prism using glycerol. Fluorescent light passing the dichroic mirror was observed through a 665-nm long-pass filter (HQ665lp, Chroma) by an electron multiplying CCD (iXon DV887AC-FI, Andor, Belfast, UK). The CCD was cooled to −50°C and the gain was typically set to 165, which corresponds to an electron gain factor of ∼60 according to the data sheet of the manufacturer. Images of 128 × 128 pixel2 were acquired in two different modes. In the frame transfer mode used for fast diffusing species, a series of 20 images was taken with an exposure time of 30 ms and a cycle time of 32 ms. For slowly diffusing samples, individual images (30 ms exposure) were acquired every 1 to 2 s with the laser illumination set to zero and the shutter closed between each image. The laser intensity, the shutter, and the camera were controlled by a homemade program written in LabVIEW (National Instruments).
To observe Rh-DPPE fluorescence in the same area where single fluorescent A647-DMPE molecules were tracked, a mirror at the rear of the microscope was switched to direct the light of a mercury lamp through a 540-nm band-pass filter (D540/25, Chroma) and via a dichroic mirror (565dclp, Chroma) through the objective. The fluorescent light was observed through a 605-nm band-pass filter (D605/55, Chroma) with the same Andor camera that was also used to detect the single molecules.
Analysis of SPT data
Single molecule recognition and trajectory reconstructions were performed with a homemade image analysis program written in LabVIEW (National Instruments). Images were cross correlated with a Gaussian representation of the expected point spread function for particles at the diffraction limit emitting at ∼700 nm (35). By applying a threshold to the cross correlated image, fluorescent spots were recognized and the center of mass for each spot could be determined from a region of interest in the original image. At this stage, close neighbors and spots too close to the edge of the detected area were removed from further evaluation. The center of mass coordinates were used as starting parameters for the fit of a 16 × 16 pixel2 area around each particle to a two-dimensional Gaussian profile using a Levenberg-Marquart algorithm (36). Trajectories of individual particles were reconstructed by comparing the results of successive images of each series. Only trajectories with at least four time steps (five data points) were used in the presented analysis. Although this reduced the number of available traces, it improved the analysis and avoided artifacts from noise and photobleaching.
To calculate diffusion coefficients, we evaluated the mean-square displacements (MSDs)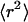 of the first three time steps from all trajectories as well as the cumulative distribution for the smallest time lag.
of the first three time steps from all trajectories as well as the cumulative distribution for the smallest time lag.
The MSDs were calculated according to
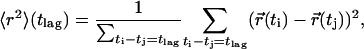 |
(5) |
(37,38) where 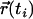 and
and 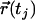 represent the positions of a fluorescent particle at times ti and tj = ti + tlag. The lateral diffusion coefficient D for free Brownian diffusion in the membrane plane is given by
represent the positions of a fluorescent particle at times ti and tj = ti + tlag. The lateral diffusion coefficient D for free Brownian diffusion in the membrane plane is given by
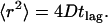 |
(6) |
Initial diffusion coefficients D were determined from linear fits to the first four steps of the MSD from all trajectories using the standard deviation of each MSD for each time lag as a weighting factor (39).
The cumulative distribution for the smallest time lag (32 ms for fast and 1 s for slow diffusing molecules) were analyzed by pooling the squared displacements 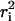 from all trajectories. To calculate the diffusion coefficients as well as to determine the mode of diffusion, we fitted three different cumulative distribution functions (CDF) to the data. First we followed the procedure described by Schütz et al. (40) to fit the CDF for one and two free diffusing fractions according to
from all trajectories. To calculate the diffusion coefficients as well as to determine the mode of diffusion, we fitted three different cumulative distribution functions (CDF) to the data. First we followed the procedure described by Schütz et al. (40) to fit the CDF for one and two free diffusing fractions according to
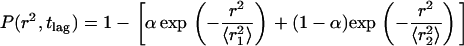 |
(7) |
with
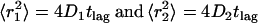 |
(8) |
and the size of the larger fractions α (α = 1 in case of one fraction).
To determine whether the observed cumulative distribution could be described better by a model for anomalous diffusion, we followed the approach by Deverall et al. (41) and used their equations for diffusion below the percolation threshold
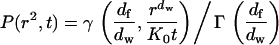 |
(9) |
with the complete and incomplete gamma functions Γ and γ, the fractal dimension of the substrate df, the fractal dimension of the walk dw, and 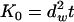 . In this approach, lipid diffusion in polymer-supported bilayers is analyzed with a model for particle diffusion in a Sierpinski gasket (42,43).
. In this approach, lipid diffusion in polymer-supported bilayers is analyzed with a model for particle diffusion in a Sierpinski gasket (42,43).
RESULTS
Constructing asymmetric bilayers with raft-like domain-forming lipids in one leaflet and single liquid-disordered phase-forming lipids in the other leaflet
Unlike mixtures composed of the outer leaflet lipids bPC, bSM, and cholesterol that produced micrometer-sized domains that increased in size with cholesterol content (12), mixtures of the inner leaflet lipids bPC, bPE, bPS, and cholesterol in various proportions showed no lo phase domains (Fig. 1). All attempts to create visible lo phase domains in planar bilayers made with these lipid mixtures failed. Bilayers of bPC, bPS, and cholesterol also showed no visible domains, whereas bilayers of bPE, bPS, and cholesterol (i.e., without bPC) contained large areas of defects (not shown).
Because inner leaflet lipids could not form lo phase domains on their own, we constructed an asymmetric planar bilayer system to examine whether lo phase domains in one leaflet could induce liquid-liquid phase separation in otherwise homogeneous opposing lipid monolayers. We chose a system consisting of a DPS-supported monolayer composed of bPC/bSM/cholesterol (2:2:1) that had been shown previously to produce large Rh-DPPE-excluding lo phase domains (12) and that was supported on a quartz slide by a tethered polymer cushion (44). Although the cholesterol concentration of 20 mol % is slightly below the cholesterol content of a typical PM (>∼30%), we wanted to keep it below the percolation threshold for PC/SM bilayers, which occurs at 20–30% cholesterol (12), to correctly assign the observed phases. The bilayer was completed by VF with a layer of bPC, bPE, and bPS (1:1:1) plus 20% cholesterol (Fig. 2, B and C) or POPC plus 20% cholesterol (Fig. 2, E and F). The resulting planar bilayers, which should have configurations as depicted either in Fig. 2 A (if domains are induced in the distal monolayer) or Fig. 2 B (if no domains are induced in the distal monolayer directly opposed to proximal monolayer lo phase domains), were observed by epifluorescence microscopy. When 20% cholesterol was included in the vesicles, the proximal lo phase domains in the resulting bilayer appeared stable, and the dye appeared to remain asymmetrically distributed. When only the LB monolayer that formed the bottom leaflet was stained with 0.02% Rh-DPPE (which is excluded from lo phase domains (12)), the lo phase domains in this leaflet were clearly defined (Fig. 2, B and E). In contrast, when 0.5% NBD-DMPE (which labels lo phase domains (12)) was added only to the vesicles that formed the top leaflet, the lo phase domains were clearly visible in the top leaflet when it was composed of bPC, bPE, bPS, and cholesterol (Fig. 2 C) but essentially nonexistent when the top leaflet was composed of POPC and cholesterol. When cholesterol was not included in the top-layer-forming POPC vesicles, the difference in appearance between the vesicle-stained and monolayer-stained samples was less clear, indicating a less stable asymmetric system (data not shown). Since cholesterol is known to rapidly flip-flop across membranes (45), we believe that equilibrating the cholesterol content between the two leaflets by including 20% cholesterol in both leaflets stabilizes the resulting bilayer.
FIGURE 2.

Asymmetric planar-supported bilayer systems with coexisting liquid phases in the proximal layer. Bilayers are supported on DPS polymers that are linked to quartz supports. The proximal monolayer contains stable lo phase domains in an ld phase lipid background. The transbilayer effect of the proximal domains is monitored by examining the distribution and diffusion of dyes in the distal layer. The presence of these domains may induce domains in the distal monolayer (A) or may have no effect on the distal monolayer (D). Bilayers were made by the LB/VF technique at room temperature. The LB monolayer was composed of bPC/bSM/cholesterol (2:2:1) + 3% DPS. The vesicles contained either bPC, bPE, bPS (1:1:1) + 20% cholesterol (B and C), or POPC + 20% cholesterol (E and F). 0.02% Rh-DPPE was added to the LB monolayer only (B and E) or 0.5% NBD-DMPE was added to the vesicles only (C and F). The same regions of double-labeled bilayers are shown in B and C, and E and F, respectively. The white bars represent 20 μm.
When the vesicles were stained, the formation of these complex bilayers could be monitored by TIRFM (Fig. 3, leftmost panels). The fluorescence intensity at the surface increased rapidly as vesicles fused to form the bilayer and reached a plateau once the surface was saturated. This process was completed in ∼30 min. When only the LB monolayer was stained, the stability of the lo phase domains upon vesicle injection could be monitored by imaging (Fig. 3, all other panels). The images shown in Fig. 3 A confirm that lo phase domains in tethered DPS-supported monolayers were stable during incubation with vesicles composed of POPC/cholesterol (4:1). When the unlabeled vesicles were composed of bPC, bPE, bPS, and cholesterol, the stability of the asymmetric system was further improved, with no observed reduction in contrast between the phases in the LB monolayer over time (Fig. 3 B). When cholesterol was omitted from the POPC vesicles, the tethered DPS-supported lo phase domains remained intact during incubation, but the contrast between the domains and the surrounding bilayer diminished over time, indicating that either the lo phase was being disrupted due to increased POPC concentrations or the Rh-DPPE was slowly flipping into the distal POPC layer and diffusing over the top of the domains (data not shown). In the absence of the tethering polymer lipid DPS, the lo phase domains in the directly supported bPC/bSM/cholesterol monolayers were washed away upon injection of POPC vesicles and the resulting holes in the remaining ld phase monolayer were quickly filled in by ld phase lipid from the fusing vesicles to form a uniform bilayer (Fig. 3 C). This instability of the directly supported phase-separated bilayers prompted us to use only tethered polymer-supported asymmetric bilayers in our studies of transbilayer coupling.
FIGURE 3.

Formation of planar-supported bilayers containing asymmetric lo phase domains. Bilayers were made by the LB/VF technique at room temperature. The LB monolayer was composed of bPC/bSM/cholesterol (2:2:1) + 3% DPS (A and B) or without DPS (C). The vesicles contained either POPC + 20% cholesterol (A), bPC, bPE, bPS, and cholesterol (B), or POPC only (C). Formation of the bilayer was monitored by TIRFM with 0.5% Rh-DPPE added to the vesicles only (panels on the left). The stability of the LB monolayer during incubation with vesicles was examined by imaging with 0.5% Rh-DPPE added to the LB monolayer only (micrographs on the right). The white bar represents 20 μm.
Liquid-ordered domains in supported monolayers are unstable when overflowed with vesicle suspensions, but can be stabilized by tethering to the solid support
We confirmed the surprising result of Fig. 3 C in a few single-phase systems (Fig. 4). When vesicles were injected on top of a directly supported POPC monolayer (uniform ld phase), most of the monolayer remained intact, with the exception of small, ∼4-μm diameter holes that quickly filled in as the vesicles fused to complete the bilayer (Fig. 4 A). However, when vesicles were injected over a directly supported monolayer of DPPC/cholesterol (1:1), which forms a uniform lo phase, almost the entire monolayer was washed away, leaving only streaks in the direction of the vesicle flow (Fig. 4 C). Over time, the POPC vesicles fused with the quartz, resulting in a uniform ld phase bilayer that was most likely composed of mostly vesicle-derived POPC. The addition of a tethered DPS support in the LB monolayer reduced the amount of lipid washed away upon injection of vesicles when the starting monolayer was a uniform ld monolayer (Fig. 4 B) or a uniform lo monolayer (Fig. 4 D). Although the amount of the DPPC/cholesterol LB monolayer that was washed away by the vesicles was much reduced by the presence of the tethered polymer, there were initially still streaks and holes in this lo phase monolayer that, however, healed over time.
FIGURE 4.

Effects of lipid phase and DPS polymers on the stability of LB monolayers during incubation with vesicles to form asymmetric planar-supported bilayers. Bilayers were made by the LB/VF technique at room temperature. The LB monolayer was stained with 0.5% Rh-DPPE and composed of POPC (A), POPC + 3% DPS (B), DPPC/cholesterol (C), or DPPC/cholesterol + 3% DPS (D). The vesicles contained only POPC in all cases. The white bar represents 20 μm.
The experiments shown in Figs. 3 and 4 illustrate a utility of tethered DPS supports that has not been previously shown, namely that they may serve to stabilize otherwise unstable LB monolayers of pure lo phase, or coexisting ld and lo phases, during and after the injection of vesicles in the process of forming LB/VF bilayers. In our experience, the combination of monolayer tethering and LB/VF for bilayer formation is the only technique that allows us to form stable asymmetric bilayers with coexisting lo and ld phase domains. The method therefore holds great promise for the engineering of asymmetric planar bilayers and the study of transbilayer lipid couplings.
Measuring lipid flip-flop in supported bilayers
In biological and model membranes, lipids can flip-flop at various rates across the bilayer from one leaflet to the other. Therefore, we wanted to know for how long the engineered lipid asymmetry could be maintained in planar-supported bilayers. We have recently shown that FLIC microscopy can be used to measure lipid asymmetry in planar-supported bilayers of various compositions and phases (26). Here, we show that this method also works to measure the time-dependent decay of lipid asymmetry. Fig. 5 A shows a FLIC chip covered with a lipid bilayer of POPC that was asymmetrically labeled with 0.5% Rh-DPPE in the distal monolayer. The different intensities correspond to squares of terraces of oxide layers with different heights on the silicon chip. The fraction of Rh-DPPE in the distal (as opposed to the proximal) leaflet of the bilayer, fD, was nearly 100% immediately after bilayer formation (Fig. 5 B). However, the asymmetry decayed gradually over a time course of more than 24 h (Fig. 5 C), supporting the notion that lipid asymmetry in these bilayers can be maintained for at least as long as in biological or other model membranes. The observed decay of asymmetry must be due to lipid flip-flop because the FLIC method measures the distribution of the labeled lipid between the two layers rather than just its absolute concentration in the distal leaflet.
FIGURE 5.

Degradation of lipid asymmetry in planar-supported bilayers monitored by FLIC microscopy. Bilayers were made by the LB/VF technique on 4-oxide FLIC chips at room temperature and stained with 0.5% Rh-DPPE in the vesicles only. (A) Fluorescence micrograph of an asymmetrically stained POPC bilayer on a 4-oxide FLIC chip. Each square on the chip is 5 × 5 μm2. (B) Histogram of measured average distances of dyes from the proximal face of the supported POPC bilayer immediately after its completion. The fraction fD of dye remaining in the distal layer is high at this time. A completely randomized bilayer would have an average dye distance 〈d〉 of 2 nm as indicated by the dashed line. (C) Time course of fraction of Rh-DPPE remaining in the distal monolayer of an asymmetrically stained supported POPC bilayer.
Lipid flip-flop can be considered a reversible first-order process, such that
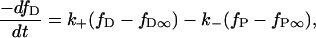 |
(10) |
where fP is the fraction of dyes in the proximal monolayer, fD∞ and fP∞ are the equilibrium fractions of dyes in the distal and proximal monolayers, respectively, k+ is the rate constant for distal-to-proximal flip, k− is the rate constant for proximal-to-distal flop, and t is time. Because fP + fD = 1, and assuming k+ = k− = k, Eq. 10 can be simplified and solved to give
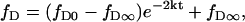 |
(11) |
where fD0 is fD at t = 0. A fit of the experimental data to Eq. 11 shows that the Rh-DPPE asymmetry in POPC bilayers decayed with a lifetime of t1/2 = ln(2)/2k of 15.2 h and became completely randomized (fD∞ = 0.43) over time (Fig. 5 B).
Lipid dynamics in heterogeneous asymmetric lipid bilayers by single particle tracking
To assess the effect that lo phase domains in the proximal leaflet of a planar-supported bilayer have on an otherwise homogeneous distal monolayer, we compared the lateral mobility of lipids in various phases of asymmetric bilayers by SPT. Tethered DPS-supported monolayers of bPC/bSM/cholesterol (2:2:1) were incubated with vesicles of POPC/cholesterol (4:1), i.e., the system depicted in Fig. 2 A and used to record the image in Fig. 2 C. The LB monolayer was stained with <10−2 mol % Rh-DPPE to allow for the identification of lo phase domains (Fig. 6 C), whereas the vesicles were stained with 10−2 (Fig. 6 A) or 10−5 (Fig. 6 B) mol % A647-DMPE. For comparison, diffusion was also measured by FRAP in the same system, in which case the vesicles were stained with 0.5 mol % NBD-DMPE and the LB monolayer was left unstained to prevent energy transfer between NBD and Rh. In separate control experiments, the diffusing dye (NBD-DMPE or A647-DMPE) was added to the LB monolayer instead of the vesicles. This allowed us to compare diffusion opposite lo phase domains to diffusion within these domains. Trajectories within or opposite lo phase domains were separated from those opposite or within the surrounding ld phase by overlaying them onto images of Rh-DPPE fluorescence (Fig. 6 D). An expanded view of two trajectories is shown in Fig. 6 E, and examples of the signal/noise that can be achieved when detecting single molecules in these bilayers are shown in the intensity profiles of Fig. 6 F. Spots such as those identified in Fig. 6 F decayed in single steps, proving that these intensities arose from single molecules (not shown).
FIGURE 6.

Single molecule tracking in asymmetric planar-supported bilayers. Bilayers were made by the LB/VF technique at room temperature. The LB monolayer was composed of bPC/bSM/cholesterol (2:2:1) + 3% DPS, and the vesicles were composed of POPC/cholesterol (4:1). In A, the vesicles were stained with 0.01% A647-DMPE to examine the overall distribution of the dye in the distal monolayer. In B, only 10−5% A647-DMPE was added to the vesicles for resolving and measuring the diffusion of single molecules. In C, 0.01% Rh-DPPE was added to the LB monolayer to confirm the position of lo phase domains in the proximal monolayer. B and C show the same region of a bilayer viewed through the A647-DMPE and Rh-DPPE channels, respectively. (D) Expanded view of boxed area in C. Trajectories from diffusing A647-DMPE molecules are overlaid onto the image of the proximal monolayer to distinguish between lipids diffusing in opposite different phases. (E) Twelvefold magnified view of the two trajectories shown in (D). (F) Two spatial fluorescence intensity distributions originating from one position each of the traces in D and E. Pixel size, 0.25 μm. Scale bars are 10 μm in A–C, 2 μm in D, and 0.25 μm in E.
Diffusion of A647-DMPE was characterized by analyzing two different statistical data sets. First we computed the MSD according to Eq. 5 and performed a linear fit of Eq. 6 to the first four data points to get the diffusion coefficient Dmsd for time lags up to 100 ms. MSD data and best fits are shown in the left column of Fig. 7. Best-fit results for Dmsd are summarized in the lower part of Table 1. When the distal monolayer was composed of POPC and cholesterol, diffusion was not significantly changed by the presence of adjacent lo phase domains in the proximal layer. Lipids directly opposite ld phase domains diffused with an initial diffusion coefficient D of 0.50 ± 0.07 μm2/s (Fig. 7 A). Diffusion of lipids diffusing opposite the lo phase was very similar, with D = 0.40 ± 0.04 μm2/s (Fig. 7 B). Diffusion measured by SPT in the proximal monolayer that contained lo phase domains was also measured and showed diffusion coefficients D of 0.21 ± 0.02 μm2/s in the ld phase domains and 0.15 ± 0.01 μm2/s in the lo phase domains (Fig. 7, C and D). Diffusion in the proximal layer was slower than in the distal layer. The general trends of these results were also confirmed with FRAP, which however only provided average diffusion coefficients integrated over larger areas (Table 1). When the distal layer was composed of bPC, bPE, bPS, and cholesterol, diffusion in the induced domains opposite the lo phase domains was 0.14 ± 0.01 μm2/s and diffusion in the induced domains opposite the ld phase domains was 0.09 ± 0.005 μm2/s (Table 1).
FIGURE 7.

Fits of SPT data using MSD (left) and CDF (right) methods. Bilayers were made by the LB/VF technique at room temperature. The LB monolayer was composed of bPC/bSM/cholesterol (2:2:1) + 3% DPS, and the vesicles were composed of POPC/cholesterol (4:1). Four sets of trajectories were analyzed: molecules diffusing in the distal monolayer opposite the ld phase of the proximal monolayer (A), molecules diffusing in the distal monolayer opposite lo phase domains in the proximal monolayer (B), molecules diffusing in the proximal monolayer within the ld phase (C), and molecules diffusing in the proximal monolayer within lo phase domains (D). The panels on the left show the MSD (Eq. 5) of the pooled trajectories up to a time lag of 150 ms. A weighted least-squares fit (solid line) was applied to the first four steps of the MSD to give Dmsd (Eq. 6). The panels on the right show the same data sets evaluated using the CDF of the square displacements (Eq. 7) with a time lag of 32 ms. Small dots show 1-P of the experimental data. Curves are fits to the CDF with α fixed at 1 (single diffusion coefficient D0, solid line) or the faster fraction α as a free variable (two diffusion coefficients D1 and D2, dashed line). The dotted lines are fits to the anomalous diffusion model (Eq. 9). Results from the first three fitting techniques are reported in Table 1.
TABLE 1.
Lipid diffusion measured by SPT and FRAP in single-phase symmetric and two-phase asymmetric planar-supported bilayers
| Bilayer composition | FRAP (NBD-DMPE)
|
SPT (A647-DMPE)
|
||||||
|---|---|---|---|---|---|---|---|---|
| mf, % | D, μm2/s | Dmsd, μm2/s* | D0, μm2/s† | D1, μm2/s‡ | D2, μm2/s‡ | α‡ | N§ | |
| Single-phase systems | ||||||||
| LB: POPC + DPS | 83 ± 4 | 0.93 ± 0.08 | 1.2 ± 0.1 | 0.90 ± 0.11 | 1.3 ± 0.2 | 0.27 ± 0.05 | 0.65 ± 0.03 | 31 (5) |
| Vesicles: POPC + dye | ||||||||
| LB: POPC + DPS + dye | 68 ± 9 | 0.8 ± 0.3 | 1.10 ± 0.07 | 1.00 ± 0.06 | 2.8 ± 1.0 | 0.5 ± 0.1 | 0.55 ± 0.03 | 69 (6) |
| Vesicles: POPC | ||||||||
| LB: POPC/chol (4:1) + DPS | 81 ± 4 | 0.61 ± 0.13 | 0.63 ± 0.05 | 0.61 ± 0.04 | 0.82 ± 0.06 | 0.16 ± 0.04 | 0.65 ± 0.02 | 69 (4) |
| Vesicles: POPC/chol (4:1) + dye | ||||||||
| LB: POPC/chol (4:1) + DPS + dye | 59 ± 11 | 0.47 ± 0.11 | 0.48 ± 0.06 | 0.46 ± 0.03 | 0.65 ± 0.05 | 0.09 ± 0.02 | 0.68 ± 0.02 | 57 (2) |
| Vesicles: POPC/chol (4:1) | ||||||||
| LB: bSM/chol (1:1) + DPS | 33 ± 14 | 0.02 ± 0.01 | 0.012 ± 0.004 | 0.0042 ± 0.0003 | 0.0026 ± 0.0003 | 0.023 ± 0.004 | 0.91 ± 0.01 | 61 (2) |
| Vesicles: bSM/chol (1:1) + dye | ||||||||
| Two-phase systems | ||||||||
| LB: bPC/bSM/chol (2:2:1) + DPS | 74 ± 7 | 0.53 ± 0.11 | 0.50 ± 0.07 (ld)¶ | 0.41 ± 0.04 (ld)¶ | 0.20 ± 0.4 (ld)¶ | 0.8 ± 0.1 (ld)¶ | 0.60 ± 0.05 (ld)¶ | 26 (4) |
| Vesicles: POPC/chol (4:1) + dye | 0.40 ± 0.04 (lo)¶ | 0.40 ± 0.03 (lo)¶ | 0.55 ± 0.06 (lo)¶ | 0.11 ± 0.04 (lo)¶ | 0.69 ± 0.04 (lo)¶ | 40 (4) | ||
| LB: bPC/bSM/chol (2:2:1) + DPS + dye | 74 ± 3 | 0.36 ± 0.06 | 0.21 ± 0.02 (ld)‖ | 0.23 ± 0.01 (ld)‖ | 0.17 ± 0.02 (ld)‖ | 0.41 ± 0.05(ld)‖ | 0.82 ± 0.05 (ld)‖ | 208 (11) |
| Vesicles: POPC/chol (4:1) | 0.15 ± 0.01 (lo)‖ | 0.20 ± 0.01 (lo)‖ | 0.15 ± 0.01 (lo)‖ | 0.48 ± 0.06 (lo)‖ | 0.92 ± 0.02 (lo)‖ | 312 (11) | ||
| LB: bPC/bSM/chol (2:2:1) + DPS | 20 ± 2 | 0.4 ± 0.2 | 0.090 ± 0.005 (ld)¶ | 0.16 ± 0.01 (ld)¶ | 0.08 ± 0.2 (ld)¶ | 0.22 ± 0.03 (ld)¶ | 0.61 ± 0.10 (ld)¶ | 183 (3) |
| Vesicles: bPC/bPE/bPS/chol (1.3:1.3:1.3:1) + dye | 0.14 ± 0.01 (lo)¶ | 0.18 ± 0.01 (lo)¶ | 0.09 ± 0.03 (lo)¶ | 0.26 ± 0.03 (lo)¶ | 0.56 ± 0.10 (lo)¶ | 109 (3) | ||
Analysis by mean-squared displacement.
Analysis by CDF with a single component.
Analysis by CDF with two components with component 1 comprising fraction α.
Total number of trajectories measured and number of bilayers in parentheses.
Molecules diffusing in the monolayer opposite to the phases indicated in parentheses.
Molecules diffusing within the phase indicated in parentheses.
The single particle diffusion data were also analyzed with CDFs, which although perhaps less intuitive yield significantly better statistics than analysis by MSD (40). The better statistics of this method permitted an analysis of the data according to the models for normal diffusion of one and two fractions (Eqs. 7 and 8) and anomalous diffusion (Eq. 9). The normalized distributions 1-P together with the best fit curves are shown in the logarithmic plots in the right-hand column of Fig. 7. The best fit parameters (diffusion coefficients D and fractions α) are also listed in Table 1. The results for the diffusion coefficient D from fits to the probability function for one fraction are very close to the results from the fits to the MSD function. In the distal leaflet opposite to the ld phase domain, D becomes 0.41 ± 0.04 μm2/s and opposite to the lo phase domain D becomes 0.40 ± 0.03 μm2/s when the distal layer is composed of POPC and cholesterol and 0.16 ± 0.01 μm2/s and 0.18 ± 0.01 μm2/s when the distal layer is composed of bPC, bPE, bPS, and cholesterol. The models for diffusion of two components and anomalous diffusion improve the fit, but not dramatically. These fits are represented in Fig. 7 as dashed and dotted lines, respectively, and numerical values for the two-component CDF are included in Table 1. The reduced χ2 values decrease by a factor of 2–5 when going to these more complex models, and they are practically the same for the two-component and anomalous diffusion models. Since the improvement by these more complex models is quite marginal, we regard the observed diffusion as sufficiently well described by the free Brownian motion of a single species.
When the results in the proximal leaflet were analyzed with the single-species CDF model, diffusion coefficients of 0.23 ± 0.01 μm2/s and 0.20 ± 0.01 μm2/s were obtained in the ld phase and lo phases, respectively. Again, fits to two-component diffusion or anomalous diffusion were improved, but not to a degree that was significant enough to justify their use.
To confirm the dynamical behavior of lipids observed in the complex lipid systems, we also measured lipid diffusion by SPT and FRAP in symmetric bilayers that contained only a single uniformly fluorescent lipid phase. Unlabeled tethered DPS-supported monolayers of POPC, bSM/cholesterol (1:1), or POPC/cholesterol (4:1) were incubated with either 0.5 mol % NBD-DMPE- or 10−5 mol % A647-DMPE-labeled vesicles of the matching lipid composition. The diffusion coefficients (and mobile fractions) obtained from these measurements are listed in the upper part of Table 1 and are graphically summarized in Fig. 8. The agreement between the SPT and FRAP measurements in all single-phase systems was very good. Lipids in pure POPC (ld) phases diffused with diffusion coefficients of 0.8–1.2 μm2/s, in POPC/cholesterol phases with diffusion coefficients of ∼0.5 μm2/s, and in bSM/cholesterol and phases with diffusion coefficients of 0.004–0.02 μm2/s.
FIGURE 8.

Diffusion coefficients of NBD-DMPE and A647-DMPE in the proximal and distal layers of single-phase symmetric bilayers of different fluidity (first three data groups) and two-phase asymmetric bilayers composed of indicated lipids in the proximal (lower labels) and distal (upper labels) layers (last two data groups). All proximal monolayers contain 3 mol % DPS. The single molecule diffusion coefficients shown are derived from fits to the MSD and to the CDF for normal diffusion of one fraction.
The complex mixtures in bilayers with asymmetric domain-forming lipid compositions had intermediate diffusion coefficients in the range 0.14–0.23 μm2/s that did not vary dramatically on whether the tracers were in the darker or brighter phases of either the inner or outer leaflet lipid mixtures (panels on the right in Fig. 8). The agreement between the SPT and FRAP measurements in the two-phase systems is also quite good because in the only case where there is an apparent discrepancy (last case on Table 1), the mobile fraction of the FRAP experiment that was carried out with a large stripe pattern was low and the slow diffusion steps measured by SPT were counted as “immobile” by FRAP in this particular experiment.
DISCUSSION
The primary goal of this study was to examine whether lo phase domains in one leaflet of a lipid bilayer could induce such domains epitaxially in the adjacent other leaflet even if the other leaflet was composed of lipids that normally do not form lo phase domains. This question was inspired by numerous cell biological observations of signal transduction through so-called “lipid rafts” and the fact that PMs of eukaryotic cells have asymmetric lipid distributions lacking typical “raft”-forming lipid mixtures in the inner leaflet. Our approach to this problem was to make asymmetric lipid bilayers on solid supports by assembling them from two individual monolayers with different lipid compositions and to examine both the microscopic appearance and lateral diffusion in each of the coexisting lipid compartments of these complex supported bilayers. In the course of these studies we learned that supported bilayers with this kind of complexity are not as stable as originally thought, which prompted us to first find conditions under which such bilayers can be prepared and under which their asymmetry can be maintained for the time needed to complete the experiments.
There are two principal methods to construct asymmetric supported bilayers: the LB/LS method (29) and the LB/LV fusion method (27). We knew from earlier experiments that stable asymmetric LB/LV bilayers can be made but that LB/LS bilayers do not retain much asymmetry, probably because the lipids mix between the two leaflets of the bilayer during or right after the LS touch-down (26). In this work, we found that lipid asymmetry is not always preserved even when the bilayers were prepared by the gentler LB/VF method. Fig. 4 shows that lipid monolayers in the lo phase get easily washed away by the incoming vesicle solution and that the bare areas get filled in with lipids from the vesicle solution. The removal of the first lo phase monolayer can be prevented to a large extent, but not completely, by stabilizing it with the polymer-lipid DPS, which tethers the bilayer covalently to the solid quartz substrate. Monolayers in the ld phase remain attached to the substrate in the absence or presence of DPS. We do not know why lo phase monolayers are less stable and adhere less to a hydrophilic quartz substrate than ld phase monolayers. It is possible that lateral lipid interactions and lipid cohesion are stronger in lo phase monolayers than in ld phase monolayers. In the absence of a tethering lipid, lo phase monolayers may thus be pealed off in sheets when the vesicle solution is flowed in, whereas lipids in the ld phase might interact less strongly laterally so that the interaction with the substrate dominates.
When examined in the system of coexisting lo and ld phases, we found that asymmetric domains were retained when the first monolayers were stabilized with the tethering polymer lipid DPS, but not in its absence (Fig. 3). Best results were obtained when the second monolayer-forming vesicles contained equal amounts of cholesterol as present in the substrate-exposed first monolayer (Figs. 2 and 3). In conclusion and based on these results and our experiences with these systems, we propose the following recipe for constructing asymmetric bilayers with complex compositions and phases: 1), use the LB/VF technique, 2), tether the LB monolayer to the substrate with DPS or possibly another tethering polymer, and 3), use equal amounts of cholesterol in the proximal and distal monolayer.
A second interesting aspect of our studies on the stability and maintenance of lipid asymmetry in supported bilayers was that we were able to measure by FLIC microscopy the time dependence of lipid asymmetry in these systems (Fig. 5). In a fluid lipid bilayer the rate of lipid flip-flop was 0.0228 h−1, which corresponds to a half-life time of 15.2 h at room temperature (∼22°C). This is not too dissimilar from the t1/2 of lipid flip-flop of 6.5 h that Kornberg and McConnell (46) reported for spin-labeled PC molecules in egg PC vesicles at 30°C. However, John et al. (47) measured the flip-flop of fluorescent PCs in DPPC vesicles to have a t1/2 of <1 h at 50°C. This higher rate may have been due to the highly curved nature of the small unilamellar vesicles, the higher temperature, or both that were used in that study. Finally, Liu and Conboy used sum-frequency vibrational spectroscopy on planar-supported bilayers made by the LB/LS method to measure a remarkably fast lipid flip-flop (t1/2 < 2 min) for DMPC at 20°C and rates that were too fast to be detected at higher temperatures (48). These extremely fast rates could be the result of the LB/LS deposition method, which we now know randomizes lipids very quickly (26). Liu and Conboy also found much lower flip-flop rates in bilayers made from lipids with longer saturated acyl chains. It appears that these bilayers were stable enough to be successfully produced with asymmetric lipid distributions by the LB/LS method. We did not examine such systems in this nor in our previous study because these lipids produce gel phase bilayers, which are not known to occur in cellular membranes. The asymmetry of bilayers with mixed gel phase and fluid phase lipid domains was also studied by Lin et al. (49) using atomic force microscopy.
The main purpose of our examination of lipid flip-flop in supported liquid phase bilayers was to determine the length of time available to study asymmetric fluid planar bilayers. As Fig. 5 C illustrates, a fluid lipid bilayer prepared by the LB/VF method should remain >95% asymmetric for 1 h, and >90% asymmetric for 2 h. Experiments taking longer than 2 h, or performed at elevated temperatures, may be compromised by lipid flip-flop.
Having established conditions to produce asymmetric lipid bilayers of complex coexisting fluid phases with high fidelity, we were able to examine our main question, namely whether SM- and cholesterol-rich domains in one leaflet can induce lipid domains in an adjacent leaflet that would normally not form domains on its own. The outcome of this experiment was quite interesting since the answer depends on the lipid composition of the normally unstructured leaflet. When this leaflet contained a very fluid synthetic phosphatidylcholine (POPC) and cholesterol, no domains were induced (Fig. 2 F). However, when this leaflet contained a natural mixture of phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, and cholesterol, domains were induced (Fig. 2 C). This experiment clearly shows that the transbilayer coupling of fluid lipid phases strongly depends on the lipid composition not only of the leaflet that mimics the outer leaflet, but also on the leaflet that mimics the inner leaflet of cellular membranes. Future experiments are needed to explore which components of the bPC, bPE, bPS, and cholesterol mixture are primarily responsible for the observed domain coupling in these bilayers. The coupling may require certain higher melting lipid chain compositions than the one found in POPC. Alternatively, PEs also have higher melting temperatures than the corresponding PC and PS counterparts because they can hydrogen-bond their headgroups and because they induce frustrated curvature strain into lipid bilayers. The system that we have developed here should be very useful to address many of these pending questions that are important to answer in our quest to better understand lipid mediated transbilayer signaling.
Our single molecule tracking studies show that the lipid diffusion coefficients in coexisting lo and ld phases made from natural brain lipids are not much different from each other. This similarity raises the question of whether the coexisting fluid phases that we observe in these complex lipid mixtures are indeed classical lo and ld phases as defined in simpler model systems. Although the differences between the observed “dark” and “bright” lipid domains may be more subtle, for a lack of better terms we still use the lo and ld phase terminology with the connotation that each of these observed phases are more “lo- and ld-like”, respectively. The diffusion coefficients in the complex proximal SM-containing leaflets are similar to those in the complex distal PE-containing leaflets (0.1–0.2 μm2/s). A similar diffusion coefficient of 0.28 ± 0.08 μm2/s was obtained by FRAP with uniform symmetric bilayers of bPC/bPE/bPS with 20% cholesterol (Fig. 1), indicating that the slow diffusion coefficient of the PE-containing layer is primarily intrinsic and at best only marginally reduced further by coupling to the SM-containing layer. Similar diffusion coefficients were also observed by fluorescence correlation spectroscopy and NMR in lo phases prepared from the synthetic lipids DLPC, DPPC, and cholesterol (50) or SM, DOPC, and cholesterol (51,52). The diffusion coefficients in the proximal layers are not slowed down by the polymer support in POPC bilayers but may be marginally reduced in POPC/cholesterol bilayers. The uniform control bilayers with POPC or POPC/cholesterol only, i.e., in the absence of DPS, exhibit very similar diffusion in both leaflets (data not shown). Twenty percent cholesterol slows the diffusion in POPC from ∼1.1 to ∼0.55 μm2/s. Interestingly, almost the same rate of diffusion is observed in the POPC/cholesterol leaflet adjacent to the complex SM-containing leaflet. This result provides further proof that there is no excess coupling between the two leaflets in this particular system.
The diffusion data of all asymmetric systems reported in this work should be considered limiting values because they assume perfect (100%) asymmetry of lipids between the two layers. Based on the data of Fig. 5, we think that this is a good approximation because all data were measured within typically 30 min to maximally an hour after preparation of the bilayers. We estimate that a maximum of (but typically much less than) 10–15% of lipids may have exchanged between the two leaflets when the measurements were taken. These lipids may contribute to the measured signal and thus slightly decrease observed differences from the actual differences of the diffusion coefficients between the two leaflets in cases where such differences exist. Therefore, the real differences between the proximal and distal layers and those between the different phases are at least those reported.
We were concerned that our failure to observe different diffusive properties in the lo- and ld-adjacent membrane compartments could be due to an overly simplistic analysis of our data with a simple mean-squared displacement function. However, several more sophisticated methods of data analysis that a), average over a much larger data set and therefore become statistically much more significant; b), analyze the data for two fractions of diffusion steps; or c), analyze the data in terms of anomalous, i.e., fractal, diffusion did not reveal significant differences between the modes of diffusion in these two compartments either. We are therefore confident that the diffusion properties of lipid probes are almost the same in lo- and ld-adjacent membrane compartments.
In conclusion, our results show that lipids can induce fluid-fluid phase separation in the opposing leaflet but only if certain yet to be explored compositional requirements are met. Since raft-like domains could not be induced by adjacent SM/cholesterol-rich domains in PC/cholesterol mixtures—but in more complex inner leaflet mixtures containing PEs and PSs—it is possible that these lipids can propagate raft-like domains to the inner leaflet; but they cannot nucleate them in the inner leaflet of cell membranes (53). This purely lipidic mechanism of signal transduction does not exclude that integral bilayer-spanning proteins could also act as transducers of lipid assembly perhaps by attracting “shells” of certain lipids (54). The mechanism does not exclude either that membrane proteins transiently interact with elements of the cytoskeleton and then drag some lipids and other proteins with them as depicted, for example, in the picket model of the PM (55). It should also be noted that on the molecular microscopic scale the macroscopically observed lipid domains could consist of lipid-cholesterol complexes (56). The techniques employed in this work cannot distinguish between models that postulate the existence of such complexes and those that do not. It is clear from these and many other studies that biological membranes are very complex systems. This work opens new possibilities to study these complexities from a new perspective. The results and techniques presented here may finally shed more light on the long-standing question of why cells use so many different lipid species in their membranes.
Acknowledgments
This work was supported by National Institutes of Health grant GM072694.
Volker Kiessling and Jonathan M. Crane contributed equally to this work.
Jonathan M. Crane's present address is Cardiovascular Research Center, University of California, San Francisco, CA 94118.
References
- 1.Mescher, M. F., and J. R. Apgar. 1985. The plasma membrane ‘skeleton’ of tumor and lymphoid cells: a role in cell lysis? Adv. Exp. Med. Biol. 184:387–400. [DOI] [PubMed] [Google Scholar]
- 2.Brown, D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- 3.Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 4.Rozelle, A. L., L. M. Machesky, M. Yamamoto, M. H. Driessens, R. H. Insall, M. G. Roth, K. Luby-Phelps, G. Marriott, A. Hall, and H. L. Yin. 2000. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 10:311–320. [DOI] [PubMed] [Google Scholar]
- 5.Vincent, S., D. Gerlier, and S. N. Manie. 2000. Measles virus assembly within membrane rafts. J. Virol. 74:9911–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindwasser, O. W., and M. D. Resh. 2001. Multimerization of human immunodeficiency virus type 1 Gag promotes its localization to barges, raft-like membrane microdomains. J. Virol. 75:7913–7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ono, A., and E. O. Freed. 2001. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA. 98:13925–13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bavari, S., C. M. Bosio, E. Wiegand, G. Ruthel, A. B. Will, T. W. Geisbert, M. Hevey, C. Schmaljohn, A. Schmaljohn, and M. J. Aman. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 195:593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freed, E. O. 2002. Virology. Rafting with Ebola. Science. 296:279. [DOI] [PubMed] [Google Scholar]
- 10.Scheiffele, P., A. Rietveld, T. Wilk, and K. Simons. 1999. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 274:2038–2044. [DOI] [PubMed] [Google Scholar]
- 11.Fridriksson, E. K., P. A. Shipkova, E. D. Sheets, D. Holowka, B. Baird, and F. W. McLafferty. 1999. Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry. 38:8056–8063. [DOI] [PubMed] [Google Scholar]
- 12.Crane, J. M., and L. K. Tamm. 2004. Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys. J. 86:2965–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veatch, S. L., and S. L. Keller. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bretscher, M. 1972. Asymmetrical lipid bilayer structure of biological membranes. Nat. New Biol. 61:11–12. [DOI] [PubMed] [Google Scholar]
- 15.Devaux, P. F. 1991. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 30:1163–1173. [DOI] [PubMed] [Google Scholar]
- 16.Quinn, P. J. 2002. Plasma membrane phospholipid asymmetry. Subcell. Biochem. 36:39–60. [DOI] [PubMed] [Google Scholar]
- 17.Boon, J. M., and B. D. Smith. 2002. Chemical control of phospholipid distribution across bilayer membranes. Med. Res. Rev. 22:251–281. [DOI] [PubMed] [Google Scholar]
- 18.Wang, T. Y., and J. R. Silvius. 2001. Cholesterol does not induce segregation of liquid-ordered domains in bilayers modeling the inner leaflet of the plasma membrane. Biophys. J. 81:2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodgers, W., B. Crise, and J. K. Rose. 1994. Signals determining protein tyrosine kinase and glycosyl-phosphatidylinositol-anchored protein targeting to a glycolipid-enriched membrane fraction. Mol. Cell. Biol. 14:5384–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baird, B., E. D. Sheets, and D. Holowka. 1999. How does the plasma membrane participate in cellular signaling by receptors for immunoglobulin E? Biophys. Chem. 82:109–119. [DOI] [PubMed] [Google Scholar]
- 21.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- 22.Feigenson, G. W., and J. T. Buboltz. 2001. Ternary phase diagram of dipalmitoyl-PC/dilauroyl-PC/cholesterol: nanoscopic domain formation driven by cholesterol. Biophys. J. 80:2775–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- 24.Veatch, S. L., and S. L. Keller. 2005. Miscibility phase diagrams of giant vesicles containing sphingomyelin. Phys. Rev. Lett. 94:148101. [DOI] [PubMed] [Google Scholar]
- 25.Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crane, J. M., V. Kiessling, and L. K. Tamm. 2005. Measuring lipid asymmetry in planar supported bilayers by fluorescence interference contrast microscopy. Langmuir. 21:1377–1388. [DOI] [PubMed] [Google Scholar]
- 27.Kalb, E., S. Frey, and L. K. Tamm. 1992. Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta. 1103:307–316. [DOI] [PubMed] [Google Scholar]
- 28.Kates, M. 1972. Techniques of Lipidology. Isolation, Analysis and Identification of Lipids. T. S. Work and E. Work, editors. Elsevier, Amsterdam.
- 29.Tamm, L. K., and H. M. McConnell. 1985. Supported phospholipid bilayers. Biophys. J. 47:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, B. A., and H. M. McConnell. 1978. Determination of molecular motion in membranes using periodic pattern photobleaching. Proc. Natl. Acad. Sci. USA. 75:2759–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambacher, A., and P. Fromherz. 1996. Fluorescence interference-contrast microscopy on oxidized silicon using a monomolecular dye layer. Appl. Phys. A. 63:207–216. [Google Scholar]
- 32.Braun, D., and P. Fromherz. 1997. Fluorescence interference-contrast microscopy of cell adhesion on oxidized silicon. Appl. Phys. A. 65:341–348. [Google Scholar]
- 33.Lambacher, A., and P. Fromherz. 2002. Luminescence of dye molecules on oxidized silicon and fluorescence interference contrast microscopy of biomembranes. J. Opt. Soc. Am. B. 19:1435–1453. [Google Scholar]
- 34.Kiessling, V., and L. K. Tamm. 2003. Measuring distances in supported bilayers by fluorescence interference-contrast microscopy: polymer supports and SNARE proteins. Biophys. J. 84:408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelles, J., B. J. Schnapp, and M. P. Sheetz. 1988. Tracking kinesin-driven movements with nanometre-scale precision. Nature. 331:450–453. [DOI] [PubMed] [Google Scholar]
- 36.Press, W. H., S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery. 1992. Numerical Recipes in C. Cambridge University Press, New York.
- 37.Kusumi, A., Y. Sako, and M. Yamamoto. 1993. Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys. J. 65:2021–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt, T., G. J. Schütz, W. Baumgartner, H. J. Gruber, and H. Schindler. 1995. Characterization of photophysics and mobility of single molecules in a fluid lipid membrane. J. Phys. Chem. 99:17662–17668. [Google Scholar]
- 39.Saxton, M. J. 1997. Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 72:1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schütz, G. J., H. Schindler, and T. Schmidt. 1997. Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J. 73:1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deverall, M. A., E. Gindl, H. Besir, J. Ruehe, M. J. Saxton, and C. A. Naumann. 2005. Membrane lateral mobility obstructed by polymer-tethered lipids studied at the single molecule level. Biophys. J. 88:1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Shaughnessy, B., and I. Procaccia. 1985. Analytical solutions for diffusion on fractal objects. Phys. Rev. Lett. 54:455–458. [DOI] [PubMed] [Google Scholar]
- 43.O'Shaughnessy, B., and I. Procaccia. 1985. Diffusion on fractals. Phys. Rev. A. 32:3073–3083. [DOI] [PubMed] [Google Scholar]
- 44.Wagner, M. L., and L. K. Tamm. 2000. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J. 79:1400–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith, R. J., and C. Green. 1974. The rate of cholesterol ‘flip-flop’ in lipid bilayers and its relation to membrane sterol pools. FEBS Lett. 42:108–111. [DOI] [PubMed] [Google Scholar]
- 46.Kornberg, R. D., and H. M. McConnell. 1971. Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry. 10:1111–1120. [DOI] [PubMed] [Google Scholar]
- 47.John, K., S. Schreiber, J. Kubelt, A. Herrmann, and P. Müller. 2002. Transbilayer movement of phospholipids at the main phase transition of lipid membranes: implications for rapid flip-flop in biological membranes. Biophys. J. 83:3315–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu, J., and J. C. Conboy. 2005. 1,2-diacyl-phosphatidylcholine flip-flop measured directly by sum-frequency vibrational spectroscopy. Biophys. J. 89:2522–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin, W. C., C. D. Blanchette, T. V. Ratto, and M. L. Longo. 2006. Lipid asymmetry in DLPC/DSPC-supported lipid bilayers: a combined AFM and fluorescence microscopy study. Biophys. J. 90:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kahya, N., D. K. Scherfeld, K. Bacia, B. Poolman, and P. Schwille. 2003. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 278:28109–28115. [DOI] [PubMed] [Google Scholar]
- 52.Filippov, A., G. Orädd, and G. Lindblom. 2004. Lipid lateral diffusion in ordered and disordered phases in raft mixtures. Biophys. J. 86:891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Meer, G. 2002. The different hues of lipid rafts. Science. 296:855–857. [DOI] [PubMed] [Google Scholar]
- 54.Anderson, R. G., and K. Jacobson. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 96:1821–1825. [DOI] [PubMed] [Google Scholar]
- 55.Kusumi, A., K. Suzuki, J. Kondo, N. Morone, and Y. Umemura. 2005. Protein-lipid interactions in the formation of raft microdomains in biological membranes. In Protein-Lipid Interactions. L. K. Tamm, editor. Wiley-VCH, Weinheim, Germany. 307–336.
- 56.McConnell, H. M., and M. Vrljic. 2003. Liquid-liquid immiscibility in membranes. Annu. Rev. Biophys. Biomol. Struct. 32:469–492. [DOI] [PubMed] [Google Scholar]