

Abstract
Our study examines the functional and structural effects of amino acid substitution in the distal side of β-chains of human Hb Duarte (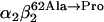 ). We have compared the functional properties of the purified Hb Duarte with those of HbA, and through proton NMR and molecular dynamics simulations we have investigated their tertiary and quaternary structures. The variant exhibits an increased oxygen affinity with a normal Hill coefficient and Bohr effect. The abnormal function of Hb Duarte is attributed to the presence of a proline residue at the β62 position, since the functional properties of another Hb variant in the same position, Hb J-Europa (β62Ala→Asp), have been described as normal. Thereafter 1H-NMR studies have shown that the β62 Ala→Pro substitution causes structural modifications of the tertiary structure of the β globins, leaving the quaternary structure unaltered. These results have been confirmed by extensive all-atom molecular dynamics simulations. All these findings lead to the conclusion that the β62 Ala→Pro substitution produces a destabilization of the E-helix extending downward to the CD corner. Particularly, a cavity near the distal histidine of the β-chains, connecting the heme pocket to the solvent, is affected, altering the functional properties of the protein molecule.
). We have compared the functional properties of the purified Hb Duarte with those of HbA, and through proton NMR and molecular dynamics simulations we have investigated their tertiary and quaternary structures. The variant exhibits an increased oxygen affinity with a normal Hill coefficient and Bohr effect. The abnormal function of Hb Duarte is attributed to the presence of a proline residue at the β62 position, since the functional properties of another Hb variant in the same position, Hb J-Europa (β62Ala→Asp), have been described as normal. Thereafter 1H-NMR studies have shown that the β62 Ala→Pro substitution causes structural modifications of the tertiary structure of the β globins, leaving the quaternary structure unaltered. These results have been confirmed by extensive all-atom molecular dynamics simulations. All these findings lead to the conclusion that the β62 Ala→Pro substitution produces a destabilization of the E-helix extending downward to the CD corner. Particularly, a cavity near the distal histidine of the β-chains, connecting the heme pocket to the solvent, is affected, altering the functional properties of the protein molecule.
INTRODUCTION
The crucial structure-function connection is classically represented by the role of hemoglobin (Hb) in the respiratory process (see the recent review of Lukin and Ho (1)). The allosteric behavior of the hemoglobin in favoring oxygen binding and releasing is a textbook example. However, despite the impressive number of investigations and amount of available data starting from the x-ray structures of Perutz (2,3), the open questions related to the structure-function paradigm in the respiratory proteins are far from being reduced and need an enlightening study. The complexity of the globular molecules at the level of number of degrees of freedom as well as of involved interactions calls for a continuous deepening process in the research. Hence, the effects of a point mutation in the amino acid structure of the protein are hardly traced back to an intuitive and immediate change in the protein function. However, this information is important at a clinical level for identifying possible connections between mutations and pathological aspects but can also open new scenarios for achieving results in the realization of blood substitutes (4,5). Thus, a multi-disciplinary study is strongly required to enlighten such issues.
The use of such an investigating strategy allows the comparison and the connection between different time and size scales, aiming at a strong interplay and a continuous feedback between techniques and investigation ranges. From the experimental side, proton NMR spectroscopy has been widely used to deepen the tertiary and quaternary structure of the HbA in the ligated (R) and unligated (T) status (6–10). Functional characterization can yield specific data of biochemical and biological interest, enabling the identification of mutants and their oxygen-binding properties. Especially for this latter, the timescales are meso- to macroscopic, but the extracted data take the burden of identifying specific biological aspects. Apparently, computer simulations cannot handle these temporal ranges, as nanosecond time intervals are already a severe challenge for classical molecular dynamics (MD) simulations. Thus, the goal of all-atom molecular modeling is not to directly tackle questions related to allosteric motions. However, since the biological activity of hemoglobins depends also on other factors (i.e., ligand partitioning between the solvent phase and the protein matrix, transport across the polypeptide matrix to the coordination position, and the binding process), there are several aspects that can be clarified by MD simulations and can be discussed in connection to experimental data.
All-atom MD simulations are a very powerful tool for investigating dynamical and structural features at a high accuracy level. Impressive results have been achieved on different biological systems: myoglobins, enzymes, and ion channels are only a few examples (11–14). However, due to the complexity of the interactions, effects, and mechanisms involved in hemoglobin problems, a very limited number of MD simulations are present in the literature and are always based on severe approximations concerning the simulation times and/or the description of the solvent. In the past, such approximations have led to an incorrect structure in the study of another mutation (15), as shown after a few years by experimental methods (16). Our MD study relies on an all-atom description of the system (hemoglobin and solvent) to possibly catch essential keynotes characterizing structure, function, and solvent interactions. Our state-of-the art microscopic modeling is the only way to capture such features.
In this work we investigate in detail the structure of a naturally occurring mutant with the aim to correlate functionality and structural properties at different level (mesoscopic and microscopic), possibly identifying the hallmarks of specific mechanisms. In tune with this spirit, analysis of the substitution-induced structural changes can provide a basis for understanding the functional consequences of the substitution: altered cooperativity, an oxygen affinity that can be moderately higher or moderately lower than that of HbA, and possible instability of the tetramers (17–20). We have focused our attention on the Hb Duarte (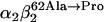 ) (21). The residue β62(E6) is external and lies close to distal heme-linked histidine (E7) that, although not directly involved in the heme contacts (22), helps maintain the stability of the E-helix. Since proline can only be tolerated as one of the first three residues of the regular E-helix, the substitution β62 Ala→Pro must be expected to disrupt the helix, consequently changing the functionality of the protein. In Fig. 1 the substituted alanines are shown in yellow.
) (21). The residue β62(E6) is external and lies close to distal heme-linked histidine (E7) that, although not directly involved in the heme contacts (22), helps maintain the stability of the E-helix. Since proline can only be tolerated as one of the first three residues of the regular E-helix, the substitution β62 Ala→Pro must be expected to disrupt the helix, consequently changing the functionality of the protein. In Fig. 1 the substituted alanines are shown in yellow.
FIGURE 1.

Deoxy hemoglobin with the α1 (red), α2 (orange), β1 (blue), and β2 (gray) subunits. Heme groups are evidenced via licorice representation, and the substituted ALA residues are in yellow. This figure as well as all the structural figures were obtained with the visualization program VMD (59).
Previous work (21) demonstrated that Hb Duarte has a high oxygen affinity. The presence of this hemoglobin variant offers the opportunity to deepen the role played by an amino acid in determining structural modification, which reflects on functional behavior. A normal oxygen affinity has been reported for Hb J-Europa, a variant affecting the same residue as Hb Duarte (23). In this case the Ala residue at E6 is replaced by Asp. The resulting variant, despite the introduction of a negatively charged residue near the distal histidine, shows normal oxygen-binding properties. However, a molecular picture of these yields is missing at the structural as well as the dynamical level; and to acquire such a picture, an NMR investigation and MD simulations at an all-atom level and in explicit solvent have been carried out on Hb Duarte for several nanoseconds with the aim to capture the local rearrangements produced by the mutation. Note that the affinity issue has raised interest in connection with the screening of possible blood substitutes. Recent work (4,5) has stressed the importance of having a high O2 affinity for cell-free polymerized hemoglobins to successfully ensure a reliable functionality.
An important question concerns the validity of the simulations on the hemoglobin variant starting from the HbA structure, that is, to extract the starting mutated structure not from x-ray data but from the HbA via a by-hand docking. Kim et al. (15) pointed out the importance of reliable NMR data for both hemoglobins to infer the difference on the quaternary structures. Only small departures between these latter validate the set-up procedure and the validity of the simulations. As described below, this is the case for this study, as shown by our NMR structural analysis. Simulations have been carried out for both deoxy- and oxyhemoglobins (HbA and Hb Duarte). Since the extracted trends associated with the mutation are similar in oxy and deoxy structures, we have essentially focused our discussion on the deoxy form.
MATERIALS AND METHODS
Functional techniques
Blood samples
After informal consent, venous samples from heterozygote carriers were collected using heparin as an anticoagulant. Cells were washed in an isoosmotic NaCl solution.
DNA analysis
DNA was extracted from peripheral blood samples with the saline method. To obtain the definition of mutations, DNAs of α and β globin genes were amplified by polymerase chain reaction (24) and directly sequenced with the Sanger procedure.
Hemoglobin analyses
The red cells were lysed by adding distilled water in a 1:1 ratio and then centrifuged for 30 min at 12,000 × g to remove the ghosts. The hemolysate was analyzed by isoelectrofocusing (IEF) in 5% thin layer polyacrilamide gels (Pharmalyte pH range 6.7–7.7; Amersham Pharmacia Biotec AB, Buckinghamshire, UK) (25).
Dissociated globin chains were analyzed by reverse-phase high performance liquid chromatography (RP-HPLC; Agilent 1100 series, Agilent Technologies, Milan, Italy) on a Zorbax 300SB-C18 column (Agilent Technologies). Each chromatogram was developed at room temperature with a linear gradient from 60% to 80% of the solvent A (50% acetonitrile, 20% methanol, 30% NaCl, 155 mM) in solvent B (25% acetonitrile, 40% methanol, 30% NaCl, 155 mM). The solvent program was a 90-min gradient with a flow rate of 1.4 ml/min. Absorbance was monitored at 215 nm. The components present in the hemolysate were purified by ion-exchange chromatography (IEC) using a HiLoad Q column (Amersham Pharmacia Biotec AB). The column was first equilibrated with 20 mM Tris-HCl buffer, pH 8.0; then the pH was decreased to 7.0 with a linear gradient. Absorbance was monitored at 280 nm. The purity of the two Hb variants' was checked by IEF and RP-HPLC. The 2,3-diphosphoglycerate (2,3-DPG) bound to abnormal Hb was removed during the process of isolation.
Functional studies
Oxygen-equilibrium curves were obtained at 25°C and 37°C, in the pH range 6.5–7.8, at an Hb concentration of 5 mg/ml by the tonometric method as described by Giardina and Amiconi (26). The values of p50 show an average standard deviation of ±3%. Experiments were carried out in 100 mM Tris/HCl or bisTris/HCl buffers, containing 100 mM NaCl, in the presence and absence of 5 mM 2,3-DPG. The amount of met-Hb contained in the samples, after oxygen-binding experiments, was determined spectrophotometrically and ranged from 2% to 7% of total Hb.
NMR
The 1H-NMR spectra were recorded on a Varian (Palo Alto, CA) Unity-Inova spectrometer at a resonance frequency of 399.948 MHz. All Hb samples (∼3%) were dissolved in 0.1 M sodium phosphate buffer (10% D2O) at pH 7.0. The experiments were performed at 29.0°C ± 0.1°C for Hb in the CO form, whereas 29.0°C, 36.0°C, and 41.0°C were the experimental temperatures used for Hb in the deoxy form. The samples treated at higher temperature underwent an additional check, collecting the spectrum at 29.0°C to verify that the observed chemical shift variation could not be ascribed to denaturation effects. All the 1H-NMR experiments were carried out on 5-mm Wilmad high-pressure NMR tubes (outside diameter 5 mm and inside diameter 4.2 mm) using 3.7-μs pulse (90°), 1-s repetition time, and spectral width of 12 kHz for CO form and 80 KHz for deoxy form. Fifty-hertz exponential line broadening was applied to increase the signal/noise ratio for the very low signals in the deoxy form, and 5 Hz was used for all the other signals in the deoxy and CO form.
Suppression of the intense water signal was achieved by direct saturation during the relaxation delay. The accuracy of chemical shift measurements in our samples was determined by repeating the experiments with three different samples and were found as follows: ±0.08 ppm for the resonances in the CO form and in the range 10–25 ppm in the deoxy form; ±0.3 ppm for the hyperfine shifted proximal resonances NδH exchangeable resonances in the region 60 to 80 ppm in the deoxy form. Chemical shifts in all spectra are referenced to DSS (2,2-dimethyl-2-silapentane-5-sulfonate) through the water signal set at 4.80 ± 0.05 ppm.
Molecular dynamics simulations
We performed state-of-the-art MD simulations at an all-atom level on the T deoxy form (Protein Data Bank (PDB) code: 1hbb; resolution 1.9 Å) and on the recent RR2 oxy structure (PDB code: 1MKO, 2.18 Å). The RR2 structure we used as starting point is more recent with respect to the R structure and with similar resolution provides the whole tetramer, contrary to the R structure where we have just two monomers and the tetramer is obtained by symmetry. All simulations have been performed using the program ORAC (27) with the Cornell et al. 1994 force field (28) and TIP3P water (29).
Given the spherical shape of hemoglobins, to reduce the number of water molecules without changing the dimension of the solvation shell, we inserted the protein in a truncated octahedron cell with an initial size of 80 Å, resulting in a system of ∼37,000 atoms for both oxy and deoxy forms (9500 water molecules including the crystallographic waters). It should be pointed out that previous MD simulations (15) on hemoglobins were based on severe approximations concerning the limited number of water molecules explicitly treated in the simulations. The smoothed particle mesh Ewald (SPME) scheme (30) has been used to treat long-range electrostatic interactions, assuming a 10-Å cutoff in the direct space (equal to the one assumed for the Lennard-Jones potential), an 80 × 80 × 80 grid (this corresponds to a grid space <1 Å in the relaxed structure) with a 5 B-spline order interpolation, and a 0.40-Å−1 cutoff in the reciprocal space. Constraints on the bonds involving H atoms were imposed via the SHAKE-RATTLE algorithm associated with a 5 shell rRespa algorithm (31) with time steps of 12, 4, 2, 1, and 0.5 fs, respectively. Starting from x-ray structures hydrogens are not assigned. This task is straightforward for all amino acids except the charged groups and histidines. Whereas for the former we used standard protonation at neutral pH, as done in other investigations, for the latter we followed the NMR assignments (32), without distinguishing between oxi and deoxy forms.
Another important point is the parametrization of the heme-histidine covalent environment that differs for the T and R structures. Although bonded and nonbonded parameters for heme are present in the Amber force field parm94 (28), we make use of the distances and bending angles involving the iron and nitrogens of the heme and the proximal histidine as evaluated by Rovira and co-workers in their ab-initio-based investigation (33,34): d(Fe,His-Nɛ) = 2.08 Å (bound) and 2.14 Å (unbound), Θ(NHeme,Fe,His-Nɛ) = 90° (bound) and 110° (unbound).
The simulation protocol for both deoxy and oxy forms consisted of a preliminary 3-ps minimization at 50 K with the protein's atoms kept fixed and free solvent molecules and of a subsequent slow-stepped-heating procedure: 100 K (6 ps), 200 K (18 ps), 250 K (36 ps), and finally 300 K (36 ps). Since the density of our system was unknown, the Parrinello-Rahman (35) scheme was followed at 250 K to carry out simulations under constant pressure, in our case 105 Pa, varying the box isotropically. This first relaxation produced a decrease of the size of the simulation cell from 80 to 77.6 Å. After the volume equilibration we simulated hemoglobins for 3 ns in the isothermal-isobaric (NPT) ensemble to obtain a good relaxation of the solvent. At the end of this latter cycle we introduced by hand the mutation substituting the alanine amino acids in prolines at the positions E6 of the β-chains. Both the HbA and mutated systems were simulated for an additional 6 ns (deoxy) and 3 ns (oxy) saving data every 1000 fs. This procedure allows us to follow relaxation of both systems and compare them to possibly point out the structural and dynamical perturbations induced by the mutation.
Cavities calculations
The cavity volumes of HbA and Hb Duarte hemoglobins were calculated with VOIDOO (36,37), a computational tool for evaluating molecular volumes and studying cavities in macromolecules such as proteins. In the program, the cavity volume is defined as the volume swept out by a probe sphere rolling over the molecule surfaces. The contacts between the probe sphere and the van der Waals protein surface delimit the probe-occupied cavity, which is similar to the Connely-type surface (38).
For the systems considered in this work volumes were calculated on 600 structures extracted from the MD simulations, corresponding to a structure each 10 ps of the simulation. The evaluated volumes appeared very sensitive to the input values of the grid size and the probe radius. Different probes, with radius ranging from 1.0 to 1.4 Å, and grids from 0.3 to 0.5 Å were tested. A probe radius of 1.4 Å (corresponding to a volume of 11 Å3) and an initial grid of 0.35 Å, with a volume refinement <1% (corresponding to a final grid of 0.15 Å), gave the most reliable results since the volumes were found consistent with those reported in the analysis of cavities in the sperm whale myoglobins (39).
RESULTS
Functional analysis
DNA analysis
Analysis of amplified DNA showed in the propositus the presence at codon 39 of the β-globin gene of a nonsense mutation in the heterozygous state. Direct DNA sequencing on amplified α globin genes revealed a single nucleotide mutation C→A at codon 68 that results in the Asn→Lys amino acid substitution, whereas sequencing of β-globin genes revealed a mutation at codon 62 G→C with the Ala→Pro substitution.
Hb analyses
The IEF procedure on hemolysate revealed the presence of two hemoglobins that have been separated by IEC. Fig. 2 a shows the hemoglobins purified. The Hb component that moves like HbA (20) corresponds to Hb Duarte (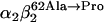 ) as confirmed by RP-HPLC analysis (Fig. 2 b).
) as confirmed by RP-HPLC analysis (Fig. 2 b).
FIGURE 2.

(a) IEF of total hemolysate (line 1), HbA (line 2), and Hb components purified by IEC (lines 3 and 4); (b) RP-HPLC of the globin chains of HbA and purified Hb Duarte.
Functional studies
The experimental data reported previously (21) have described only partially the functional properties of Hb Duarte. In this work, we have analyzed the oxygen ligand properties at 25°C and 37°C on purified Hb Duarte compared to HbA, both in the absence and presence of 2,3-DPG. From Fig. 3 it is clear that this variant possesses an oxygen affinity higher than that of HbA at all experimental conditions examined. Its cooperativity, 2,3-DPG response, and Bohr effect were not appreciably different from those of HbA. At 37°C the isopropanol test indicated that the hemolysate is slightly unstable compared with that of HbA.
FIGURE 3.

Effect of pH on the oxygen affinity (log p50) and cooperativity (n50) at (a) 25°C, and (b) 37°C of HbA (circles) and purified Hb Duarte (triangles) in the absence (open symbols) and in the presence (solid symbols) of 5 mM 2,3-DPG. Experimental conditions: 100 mM Bis-Tris/Tris + 100 mM NaCl. O2 pressure is expressed in torr.
NMR
NMR spectra of Hb Duarte β62Ala→Pro in the deoxy and carbonmonoxy form were compared to those of native HbA to define effects of amino acid change on the heme environment and subunit interface interactions.
Heme environment
The 1H-NMR spectra resonances of HbA and Hb Duarte in the CO form from 0.5 to −2.5 ppm are reported in Fig. 4. These 1H resonances arise from protons of the amino acid residues close to the heme distal and proximal cavities of the Hb molecules and are known to be sensitive to the heme orientation and their environment (6,40). The two resonances at −1.72 and −1.82 ppm, situated in the distal cavity, have been assigned to the γ2-methyl group of the E11Val of the α- (62Val) and β-(67Val) chains, respectively.
FIGURE 4.

Downfield ring current-shifted resonances of HbA and Hb Duarte in the CO form.
In the 1H-NMR spectrum from Hb Duarte, the resonance assigned to α62Val results shifted downfield to −1.71 ppm and the resonance assigned to β67Val shifts upfield to −1.84 ppm. The chemical shifts of the γ2-methyl group result essentially unaffected, in the limit of the experimental errors, by the 62Ala→Pro substitution in the β-chain compared to the corresponding signal in the HbA. Small differences in the intensity and frequencies of the unassigned resonances are observed in the Hb Duarte except in the HbA in the region from 0.5 to −1.5 ppm. These minor changes have been attributed to slight adjustments of the conformation of the heme and/or the amino acid residues in the heme proximity as a result of the mutation (8,9,15,40,41).
The 1H-NMR spectrum of Fig. 5 shows the large downfield hyperfine shifted signals assigned to NδH exchangeable protons of the proximal histidine residues (42,43) of the deoxy HbA and Hb Duarte collected at 29.0°C. In the HbA 1H-NMR spectrum the signal at 63.9 ppm is attributed to 87His in the α-chain, whereas the signal at 75.9 ppm is attributed to 92His in the β-chain. The chemical shift of the resonance assigned to the NδH in the Hb Duarte are downfield shifted to 76.5 ppm for the β92His, whereas the chemical shift of the α87His resonance, 63.9 ppm, does not change.
FIGURE 5.

Downfield hyperfined-shifted NδH of proximal histidines resonances of HbA and Hb Duarte in the deoxy form.
In the range 10–25 ppm of HbA and Hb Duarte in the deoxy form (Figs. 6 and 7), several resonances are present which arise from protons on the heme groups and nearby amino acid residues hyperfinely interacting with the unpaired electrons of iron in the heme (6).
FIGURE 6.

Hyperfine shifted proton resonances of HbA and Hb Duarte in the deoxy form collected at several temperatures.
FIGURE 7.

Hyperfine shifted and exchangeable proton resonances of HbA and Hb Duarte in the deoxy form collected at several temperatures.
The hyperfine shifted resonances of the Hb Duarte spectrum collected at 29°C show chemical shift changes at ∼11.9 and ∼14.6 ppm and at ∼22.8 and ∼21.8 ppm; these last two resonances are associated with protons in the β-chains of deoxy HbA (42). No changes are observed in the other range, 16–21 ppm, the resonance at ∼18.7 ppm is associated with protons in the β chains, and the resonance at ∼17.1 ppm is assigned to the protons in the α-chains of deoxy HbA (42). It is clear from these spectra that substitution at the β62Ala→Pro position causes adjustment of the configuration of the amino acid residues and/or the heme groups in the β-chains, leaving unperturbed the corresponding groups in the α-chains.
In the range 11.0–16.0 ppm new resonances appear at ∼12.1 and ∼15.1 ppm, this last resonance being very large. Contemporaneously, the disappearance of the signals at 14.6 and 11.9 ppm in the deoxy Hb Duarte spectrum collected at 29.0°C is observed. When the spectrum is collected at higher temperatures, 36.0° and 41.0°C, the two peats at 12.1 and 15.1 ppm are shifted. The temperature-sensitive chemical shift position indicates that these new resonances are likely to be hyperfine shifted resonances rather than exchangeable resonances (44,45). For HbA and Hb Duarte the differences in the ring current-shifted and hyperfine-shifted proton resonances clearly suggest that the substitution of β62Ala→Pro causes significant changes in the conformation of the heme pockets in the Hb Duarte.
Exchangeable proton resonances
Several exchangeable proton resonances in the range 10–15 ppm are present in the 1H-NMR spectrum (Fig. 7) of the deoxy HbA (T). They are considered sensitive indicators of the T quaternary structure (46,47). The resonances at 14.1 ppm and at 11.2 ppm have been identified as the intersubunit H-bond between α42Tyr and β99Asp and between α94Asp and β37Trp in the α1β2 interface. The analysis of these proton resonances gives indications on the quaternary conformational modifications, as a consequence of residue substitutions, in the T state (10,32,46–48). The resonance at 13.2 ppm has been assigned to the H-bond between α103His→β131Gln in the α1β1 subunits interfaces in the deoxy state of HbA (48). The chemical shift position of the resonance attributed to the H-bond between α103His→β131Gln in the spectrum of Hb Duarte is exactly the same as this of HbA, indicating no perturbation in the α1β1 subunit interfaces in the deoxy Hb Duarte. However, the resonances at 14.1 ppm and 11.2 ppm in the HbA are slightly downfield shifted, 0.11 ppm and 0.13 ppm, respectively, indicating that the perturbations are not just confined to the substitution site and that the β62 Ala→Pro mutation can affect the α1β2 subunits' interfaces in the deoxy state of Hb Duarte.
Fig. 8 shows the exchangeable proton resonances of HbA and Hb Duarte in the CO form. These resonances belong to the exchangeable proton resonances in the subunit interfaces. The 1H resonance at 12.8 ppm has been assigned to α122His, making a water-mediated hydrogen bond with β35Tyr, whereas the resonance at 12.1 ppm has been assigned to α103His hydrogen bonded to β131Gln (32,49–51). Both H-bonds are located in the α1β1 subunit interface of the HbA. These exchangeable 1H resonances are considered excellent markers for the legated quaternary structure (R). Their chemical shift positions in Hb Duarte are exactly the same as those of HbA, indicating no perturbation on the α1β1 subunit in the CO form of Hb Duarte. The resonances at 10.2 ppm, 10.4 ppm, and 10.7 ppm have been assigned to the 1Hɛ of β15Trp, α14Trp, and β37Trp in the spectrum of HbA in the CO form, respectively (10,32). These three resonances as observed for the resonances at 12.8 ppm and 12.1 ppm do not change in the spectrum Hb Duarte.
FIGURE 8.

Exchangeable proton resonances of HbA and Hb Duarte in the CO form.
MD results
The structural analysis was performed monitoring the following features of the deoxy tetramer:
Total Xrms. Xrms is the instantaneous mean-square deviation of Cα carbons from their x-ray structure. This deviation is calculated after performing a rigid body fit of the two structure (rigid translation and rotation) that minimize the deviation itself;
Chain Xrms (total and relative);
Secondary Xrms;
Hydrogen bonds involved in the α-β interfaces and in the mutation region.
The presentation of the results starts with those related to the HbA. Then, the mutated system will be reviewed. We have performed MD simulations for both deoxy and oxy hemoglobins (HbA and Hb Duarte). Since the extracted trends associated to the mutation are similar in oxy and deoxy structures, we have essentially focused our discussion on the deoxy form.
HbA
A first issue we addressed concerned the stability of our simulated system. According to the total Xrms shown in Fig. 9 a the structure is very stable: the Xrms is practically constant for the entire production run (9 ns) and does not exceed a value of 1.2 Å.
FIGURE 9.

Xrms of deoxy HbA: (a) tetramer, (b)–(e) β2, α2, β1, and α1 chains, respectively. The black curves refer to the deviations, the gray curves to the absolute deviations.
The result is remarkable because we are dealing with a protein made by four subunits held together by H-bonds. Hence, the predictive power of the simulations is potentially increased, relying on the validity of the interaction parametrization. The relative stability of the four subunits is inferred from the analysis of the deviations from the x-ray counterparts represented by the black curves in Fig. 9, b–e, for the β2, α2, β1, and α1 chains, respectively. The α-chains deviate less from the corresponding x-ray structures than the β ones, 0.8 Å vs. 1.2. Å. However, to discriminate between an internal motion of the subunit and a motion of the whole subunit, we plot in the same figure the absolute deviations (gray curves) that account also for the rotations and translations of the subunit within the box. Apart from the β2 chain at the beginning of the simulation (Fig. 9 b), the gray and black curves do not differ remarkably for each subunit. This means that the deviation from the x-ray structure during the simulation should be traced back to internal motion of the monomers, with possible larger fluctuations in the β-chains than in the α-subunits. It should be pointed out that the Xrms are evaluated considering the contributions of the backbone and not those of the lateral chains. The inclusion of these latter would slightly increase the deviations but not affect the previous conclusions on the stability of the simulations.
The simulations also yielded information on the secondary structure, i.e., on the proximal and distal (E- and F-) helices of the subunits (data not shown). For all the subunits the deviations from the x-ray structure are small for both kinds of helices (0.4 Å) with larger fluctuations for the β-helices. The calculated absolute deviations for these latter are of the order of 1.2. Å, practically the same value obtained for the entire β-chains. This accordance is a further fingerprint of the internal motion origin of the departure from the x-ray structure during the simulation. In conclusion, the analysis of the data for the HbA system provides evidences of a very stable simulated structure, enhancing the confidence in the simulation scheme as well as in the used force field.
Mutation and function-structure relation
The same kind of analysis used for the HbA is applied to the mutated structure. Fig. 10 shows the deviations from the x-ray structure of the HbA (black curves) and of the mutated tetramers (gray curves). The tetramer structure of the mutated hemoglobin is characterized by a slight increase of the Xrms with respect to that of the HbA (Fig. 10 a). Despite the increased deviation, the simulations can be considered quite stable. The tertiary structure exhibits a small drift for the β-chains (Fig. 10, b and d), whereas practically no changes are noticed in the α-chains (Fig. 10, c and e), this pointing out the localization of the induced rearrangement on the mutated chains. The inspection of the secondary structures reveals increased deviations from the x-ray structure for the two β-distal helices with respect to the HbA: 0.4 Å for this latter, 0.8 Å for the mutated system. The proximal helices appear to be unaffected by the mutation. The augmented deviation from the x-ray structure for the distal helices arises from the break of the helices exactly at the mutated site (E6). The breaking is accompanied by a displacement of the E1–E6 portion toward the heme. Additional insights into these changes can be gained by monitoring the contacts of the involved residues, as illustrated in the Discussion.
FIGURE 10.

Xrms of deoxy HbA (black curves) and mutated (gray curves) hemoglobins: (a) tetramer, (b)–(e) β2, α2, β1, and α1 chains, respectively.
DISCUSSION
The discussion is organized following an analysis of the structural behavior of the hemoglobin regions mostly affected by the mutation and/or crucial in possibly determining the affinity of the molecular system.
Quaternary and tertiary structures
The simulation findings and the NMR data point out the absence of mutation effects on the quaternary structure. According to the NMR data, the T state and the R state intersubunit contacts are unaffected by the β62Ala→Pro mutation. The exchangeable 1H resonances arising from α42Tyr–β99Asp and α94Asp–β37Trp, characteristics of the integrity of the T state, and the α122His–β35Tyr and α103His–β131Gln in the R state are at the same position in both HbA and Hb Duarte. The slight shift to low field observed for α42Tyr–β99Asp and α94Asp–β37Trp seems to be limited to minor adjustments of the α1β2 interface, whereas the α1β1 interface is totally unaffected.
The very minor structural perturbations observed in the quaternary structure at the level of the α1β2 interface do not seem to modify the protein functionality associated to conformational changes: the essential features of the quaternary transition R ↔ T are preserved upon mutation as confirmed by the unaltered cooperativity, DPG effect, and Bohr effect pinpointed by experiments (see the section Functional analysis).
In tune with this issue is the comparison of the simulated Xrms for the tetramer of HbA and Hb Duarte (Fig. 10 a) that suggests the absence of mutation effects on the quaternary structure, at least for the time spanned by MD calculations. Note that the MD simulation times are of the order of tenths of nanoseconds, whereas quaternary structural changes occur on the microsecond scale. Nevertheless, the successful comparison with the NMR data confirms that the fingerprints of a stable quaternary structure extracted from MD results could be valid on the longer timescale.
These conclusions are also confirmed by the results of the simulations collected in Fig. 11. Here the probability distributions of the distances characterizing the switch region for the HbA (solid curves) and Hb Duarte (dotted curves) are shown. The solid and dotted curves practically coincide for the α103His–β131Gln distance, whereas a slight broadening in the α42Tyr–β99Asp and α94Asp–β37Trp distance distributions is observed, in agreement with the experimental data. Notice the small departure of the most probable MD distances from the x-ray data (dot-dash lines in Fig. 11). Thus, the 1H-NMR and MD results of deoxy Hb Duarte indicate that the effect of the mutation is limited mostly to changes in the environment of the proximal heme pocket of the β-chains.
FIGURE 11.

Probability distributions of the distances in the switch region of the deoxy Hbs: (upper panel) Asp-94–Trp-37 at the α1β2 interface; (central panel) His-103–Gln-131 at the α1β1 interface; (lower panel) Tyr-42–Asp-99 at the α1β2 interface. Solid and dotted curves are for HbA and Duarte variants, respectively. The dash-dot lines indicate the corresponding distances in the x-ray structures.
Heme region
According to the NMR data, the mutation does produce appreciable perturbation in the region around the proximal heme pocket of the β-chains. Following the time evolution of the region during the simulations we have extracted some fingerprints of a structural change involving the histidines F8 (α87His and β92His) and the leucines F4 (α83Leu and β88Leu). Probability distributions of the representative distances and angles of H-bonds between F8 and F4 are collected in Figs. 12 and 13, respectively. The geometrical arrangement of the two residues is characterized via the reciprocal positions of two N atoms of F8 (Nδ and Nbckbn in the inset of Fig. 12) and the CO group of F4. For the α-chains, the curves for HbA and Hb Duarte perfectly overlap (see lower panels of Figs. 12 and 13) whereas some differences are present for the β chains (see Fig. 12). The probability distribution of the CO-Nbckbn distance is shifted toward larger values in the Hb Duarte variant than in HbA. Such a shift is not observed in the distribution of the CO-Nδ distance. An increasing distance weakens the strength of the associated H-bond. The angular distribution does not exhibit any difference between HbA and Hb Duarte, and only a general enhanced planarity of the CO-Nbckbn bond compared to the CO-Nδ one is observable in Fig. 13.
FIGURE 12.

Probability distribution of the distances Nδ(His F8)-CO(Leu F4) (black curves) and Nbckbn(His F8)-CO(LEU F4) (gray curves) for the deoxy Hbs. The solid and dotted curves refer to data for HbA and Hb Duarte, respectively. In the inset, the N ions and the leucine residue involved in the formation of H-bonds are shown.
FIGURE 13.

Probability distribution of the angles Nδ(His F8)-CO(Leu F4) (black curves) and Nbckbn(His F8)-CO(Leu F4) (gray curves) for the deoxy Hbs. The solid and dotted curves refer to data for HbA and Hb Duarte, respectively.
The decreased strength of the H-bond connecting the F8 and F4 residues might be interpreted as a tendency of the deoxy variant toward the R state with a consequent change in the affinity of the system. Table 1 emphasizes the H-bond removing action associated with the mutation. The occurrence frequencies of H-bonds detected during the simulation on HbA and Hb Duarte: are there collected in this latter there is a noticeable decrease of H-bonds in the β-chain with respect to HbA (the occurrence probabilities are 68% and 82%, respectively). However, the tendency toward R can be ruled out, since the structural parameters involving the heme do not show any fingerprints of R-state-like configuration. For example, the probability distributions of the angles and distances involving the atoms of the residues F8 in HbA and Hb Duarte overlap perfectly.
TABLE 1.
Occurrence of H-bonds in the deoxy Hbs during the simulations
| αδ | αbckbn | βδ | βbckbn | |
|---|---|---|---|---|
| HbA | 75 | 82 | 78 | 82 |
| Hb Duarte | 77 | 82 | 78 | 68 |
Probability (in %) over 12,000 MD steps for the occurrence of H-bonds in the deoxy Hbs (HbA and Hb Duarte). αδ and αbckbn denote the H-bonds involving Nδ and Nbckbn in the α-chains, respectively, and βδ and βbckbn refer to the analogous H-bonds in the β-chains. The criteria for labeling a H-bond between A and B are rAB < 3.5 Å and θAHB < 30°.
The MD results are in agreement with the NMR data, since only a slight perturbation of the hyperfine-shifted NδH-exchangeable proton resonance of the proximal histidine residue in the mutated β-chain is observed. The same agreement is reached for the region around the distal histidine: both experiment and theory do not provide evidence of mutation-induced perturbations. The probability distributions for the distances between Val E11 (α62Val and β67Val) and the heme, collected in Fig. 14, are paradigmatic. Here the characterizing distance is between one of the two CG carbon atoms of E11 and the Fe atom of the heme group (see inset in Fig. 14). During the simulation the two CG atoms exchange their position, thereupon the probability distribution refers to the shortest distance between one of the CG atom and Fe. No remarkable differences between Hb Duarte (dotted curves) and HbA (solid curves) emerge from the comparison, in agreement with the results of the NMR analysis.
FIGURE 14.

Distribution of the distance CG(1-2)(ValE11)-Fe (see inset) in the β- (upper panel) and α- (lower panel) chains for deoxy HbA (solid lines) and deoxy Hb Duarte (dashed lines) during the simulation.
E-helix and CD corner
In the previous section the unaffected behavior of a characterizing residue of the E-helix, the E11 valine, under mutation has been discussed. The situation for the other sector of the E-helix in the β-chains is different. As shown in Fig. 15 the mutation induces distortion of the helix section E1–E6 in Hb Duarte (blue tube) with respect to HbA structure (orange tube). Superimposing the two structures obtained for Hb Duarte and HbA, respectively, after 6 and 9 ns of simulation we obtained a tilt angle of ∼26°. In particular, the mutation and the related effects modify the contact map of β57Asn (E1), β59Lys (E3), and β61Lys (E5). The hydrogen bonds to the CD loop of β57Asn and β59Lys via contact with β44Ser (CD3) and β45Phe (CD4) in the HbA are eliminated by the mutation. β59Lys is now closer to the heme group and forms a hydrogen bond with a propionate group. This latter was linked to β66Lys (E10) in the HbA. The mutation-induced rearrangement includes a displacement of β61Lys toward β21Asp (B3), already in contact with β65Lys (E9). In the HbA β61Lys was linked to β55Met (D6), but the mutation annihilates this linkage. A further aspect related to the mutation and with a possible influence on the affinity is the structural change of the residue β45Phe (CD4) caused by the displacement of β59Lys from the CD loop. In the HbA β45Phe points toward the imidazole ring of the distal histidine protecting, in some sense, the accessibility to the distal cavity. The latter, after mutation, is protected by the long tail of β59Lys whereas β45Phe is displaced.
FIGURE 15.

Helix section E1–E6 in Hb Duarte (blue tube) and HbA (orange tube), respectively, after 6 and 9 ns of MD simulation.
To quantitatively monitor this feature, we have calculated the probability distribution of the distance between the carbon atoms CZ of β45Phe and CG of β63His (E7) as obtained from the simulations. The results are collected in Fig. 16 for the β- (upper panel) and α- (lower panel) chains. Solid and dashed lines refer to the HbA and the Hb Duarte hemoglobins, respectively. The difference between the simulated distances and those extracted from the x-ray structures (the dotted lines in Fig. 16) is clear. This is the expected difference for a system in crystalline form and in solution. The increased mobility can be recognized by the different shapes of the distribution in the upper and lower panels of Fig. 16. The results for the β-chains in the deoxy Hb Duarte hemoglobin show two well-separated maxima, whereas the deoxy-HbA data are characterized by a single maximum. In the α-chain (lower panel), the distributions for HbA and Hb Duarte differ only slightly but both keep the single-maximum behavior. We do observe experimentally several changes in the hyperfine shifted resonances (Figs. 6 and 7) at ∼11.9 and ∼14.6 ppm and at ∼22.8 and ∼21.8 ppm. These changes, in agreement with the MD simulation results, clearly indicate that the mutation at the β62Ala→Pro position provokes adjustment of the configuration of the amino acid residues and/or of the heme groups in the β-chains.
FIGURE 16.

Distribution of the distance CZ(PheCD4)-CG(HisE7) (see inset) in the β- (upper panel) and α- (lower panel) chains for deoxy HbA (solid lines) and deoxy Hb Duarte (dashed lines) during the simulation. The vertical dotted lines refer to the values extracted from the x-ray structure.
Cavities
A further issue addressed in our study is the analysis of the cavities in the protein structures during the simulations. Among the various cavities, we have focused our attention on that closer to the heme region (see Fig. 17) connecting the heme pocket to the distal histidine; we expect that this pocket may be more involved in determining the ligand trajectory. This cavity is located between the E-helix and the CD corner, the two structures most affected by the mutation. In particular the residues lining the cavity in the β-chain are Leu-28 (B10), Leu-32 (B14), Phe-42 (CD1), Phe-45 (CD4), Val-60 (E4), His-63 (E7-distal), and their equivalent in the α-chain. Their average volumes and their occurrence frequency during the simulations are reported in Table 2 for the a), deoxy and b), oxy species (HbA and Hb Duarte). In our evaluation, a cavity is considered when its volume is >11 Å3. A comparison of the occurrence probabilities immediately points out a remarkable difference between α- and β-chains in both HbA and Hb Duarte: The cavities occur more often in the α-chains than in the β ones (the occurrence probabilities are 81% vs. 20% in HbA, 72% vs. 25% in Hb Duarte).
FIGURE 17.

Cavity position in hemoglobin (see Table 2). The white spheres refer to the position of the center of mass of the cavities.
TABLE 2.
Volume of the cavities in the region close to the distal histidine in α- and β-chains
| Deoxy | α | β |
|---|---|---|
| HbA | 37 ± 11 (83%) | 33 ± 13 (20%) |
| Hb Duarte | 35 ± 10 (72%) | 45 ± 18 (25%) |
| Oxy | α | β |
| HbA | 38 ± 14 (75%) | 30 ± 10 (7%) |
| Hb Duarte | 36 ± 12 (67%) | 46 ± 14 (19%) |
Volume in Å3 of the cavity found close to the distal histidine during the simulation on the deoxy and oxy Hbs (see Fig. 17 for identifying the cavity in the structure). The analyzed structures were 600 corresponding to a structure every 10 ps of simulation. In parentheses, the occurrence probabilities (in %) of the cavities are reported.
The greatly different behavior can account for the well-known larger ligand accessibility of the α- than that of the β-chains in the deoxy form (30), reinforcing the idea of a strong correlation between affinity and cavities. The comparison between the deoxy and oxy data confirms the similar mutation-induced trends in the cavity behaviors, supporting our focus on the deoxy form in the discussion of the results. Interestingly, the mutation has an influence on the size of the considered cavities since in the β-subunits the average volume is larger in the Hb Duarte than in HbA. It should be pointed out that the computational error is quite large, ∼10 Å3, blurring somehow a clear-cut quantitative comparison. As a matter of comparison, the difference between the average volumes of the β-subunit in Hb Duarte and HbA amounts to 11.8 Å3 whereas the same difference is practically zero for the α-chain. The large fluctuations in the volumes are fingerprints of a remarkable, not negligible dynamics of the regions around the heme. Thus, the general larger affinity of the α-subunit is maintained upon mutation as stated by the values of the occurrence frequency (see Table 2), whereas the measured increased affinity of the Hb Duarte variant with respect to HbA can be traced back to the enlarged cavities observed in the simulations.
A substantial pocket closed to the heme region may provide additional degrees of freedom for ligands that can spend more time in the binding region. The importance of the cavity structure and dynamics in determining the binding kinetics of ligands has been pointed out in several works on myoglobins (52–54), and more recently it has been stressed by laser flash photolysis experiments on HbCO and HbO2 (55,56). According to these latter the ligand pathways into and out of the hemoglobin dynamically involve cavities of the heme region in analogy with the myoglobin case (57,58). A further refinement can be achieved considering the outcomes of the photolysis experiments (55,56): the escaping process of ligand to solvent occurs mainly from the distal pocket. It is reasonable to also expect that also the reverse process, i.e., from solvent to protein, can find a favorable gate in the same region. Thus, the ligand affinity seems to be strongly correlated to the structure and fluctuations of cavties close to the heme region.
CONCLUSIONS
In conclusion, our account of the characterization of Hb Duarte shows that the substitution of alanine by proline at position β62 (E6) leads to an increased oxygen affinity of the variant that can be related to structural modifications just behind the distal histidine. 1H-NMR and MD results on deoxy Hb Duarte clearly indicate that the effect of the mutation is limited to changes in the environment of the heme pocket of the β-chains. MD simulations limit the range of the perturbation to the first five residues of the E-helix and to some residues of the CD corner: new hydrogen bonds between residues and with the propionic group of the pyrrole III are observed together with displacements of involved amino acids. In particular, β45Phe (CD4), which points toward the imidazole ring in Hb A and protects the accessibility to the distal cavity, is now displaced by the mutation, and an increased mobility of the residue chain has been recognized. It should be pointed out that the imidazole ring could be partially protected also by the long tail of β59Lys, which is shifted by the mutation toward the propionic group.
The 1H-NMR and MD calculations do not pick out evident fingerprints of mutation-induced stabilization or destabilization of the R and/or T structure of Hb Duarte to account for the increased oxygen affinity. As pointed out, the modifications are limited at the level of the tertiary structure. The observed structural change of the residue β45Phe (CD4) caused by the displacement of β59Lys from the CD loop together with mutation-induced rearrangements close to the distal cavity might be one of the determining factors of the ligand affinity of the Hb Duarte. This must be correlated to the influence on the volume size of the pockets around the β-distal cavity. Note that the very same mutation-induced trend in the volumes is observed in both oxy and deoxy Hb Duarte. Our results seem to indicate that the increased oxygen affinity of Hb Duarte with respect to Hb A must be searched in the structural modifications of the region close to the distal cavity.
Acknowledgments
Calculations were carried out at CASPUR (Rome, Italy) and at CINECA (Bologna, Italy).
CPU time at CINECA was obtained within the framework of an INFM grant project and of a CINECA-Democritos agreement. This work was supported in part by MIUR through the project PON-CyberSar.
References
- 1.Lukin, J. A., and C. Ho. 2004. The structure-function relationship of hemoglobin in solution at atomic resolution. Chem. Rev. 104:1219–1230. [DOI] [PubMed] [Google Scholar]
- 2.Perutz, M. F. 1968. Preparation of haemoglobin crystals. J. Cryst. Growth. 2:54–56. [Google Scholar]
- 3.Perutz, M. F., G. Fermi, B. Luisi, B. Shaanan, and R. C. Liddington. 1987. Stereochemistry of cooperativity mechanisms in hemoglobin. Acc. Chem. Res. 20:309–321. [DOI] [PubMed] [Google Scholar]
- 4.Vandegriff, K. D., A. Bellelli, M. Samaja, A. Malavalli, M. Brunori, and R. M. Winslow. 2004. Kinetics of NO and O2 binding to maleimide poly(ethylene glycol)-conjugated human haemoglobin. Biochem. J. 382:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandegriff, K. D., A. Malavalli, J. Wooldridge, J. Lohman, and R. M. Winslow. 2003. MP4, a new nonvasoactive PEG-Hb conjugate. Transfusion. 43:509–516. [DOI] [PubMed] [Google Scholar]
- 6.Ho, C. 1992. Proton nuclear magnetic resonance studies on hemoglobin cooperative interactions and partially ligated intermediates. Adv. Protein Chem. 43:153–312. [DOI] [PubMed] [Google Scholar]
- 7.Ho, C., and J. R. Perussi. 1994. Proton nuclear magnetic resonance studies of hemoglobin. Methods Enzymol. 232:97–139. [DOI] [PubMed] [Google Scholar]
- 8.Jeong, S. T., N. T. Ho, M. P. Hendrich, and C. Ho. 1999. Recombinant hemoglobin (αα29leucine→ phenylalanine, α96valine→tryptophan, β108asparagine→lysine) exhibits low oxygen affinity and high cooperativity combined with resistance to autoxidation. Biochemistry. 38:13433–13442. [DOI] [PubMed] [Google Scholar]
- 9.Tsai, C. H., V. Simplaceanu, N. T. Ho, T. J. Shen, D. Wang, T. G. Spiro, and C. Ho. 2003. Site mutations disrupt inter-helical H-bonds (α14W-α67T and β15W-β72s) involved in kinetic steps in the hemoglobin R→T transition without altering the free energies of oxygenation. Biophys. Chem. 100:131–142. [DOI] [PubMed] [Google Scholar]
- 10.Fang, T. Y., V. Simplaceanu, C. H. Tsai, N. T. Ho, and C. Ho. 2000. An additional H-Bond in the α1β2 interface as the structural basis for the low oxygen affinity and high cooperativity of a novel recombinant hemoglobin (βL105W). Biochemistry. 30:13708–13718. [DOI] [PubMed] [Google Scholar]
- 11.Grayson, P., E. Tajkhorshid, and K. Schulten. 2003. Mechanisms of selectivity in channels and enzymes studied with interactive molecular dynamics. Biophys. J. 85:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tajkhorshid, E., P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- 13.Aksimentiev, A., and K. Schulten. 2004. Extending the molecular modeling methodology to study insertion of membrane nanopores. Proc. Natl. Acad. Sci. USA. 101:4337–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ceccarelli, M., C. Danelon, A. Laio, and M. Parrinello. 2004. Microscopic mechanism of antibiotics translocation through a porin. Biophys. J. 87:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, H. W., T. J. Shen, D. P. Sun, N. T. Ho, M. Madrid, M. F. Tam, and C. Ho. 1995. A novel low oxygen affinity recombinant hemoglobin (α96Val→Trp): switching quaternary structure without changing the ligation state. J. Mol. Biol. 248:867–882. [DOI] [PubMed] [Google Scholar]
- 16.Puius, Y. A., M. Zou, N. T. Ho, C. Ho, and S. C. Almo. 1998. Novel water-mediated hydrogen bonds as the structural basis for the low oxygen affinity of the blood substitute candidate rHb (α96Val→Trp). Biochemistry. 37:9258–9265. [DOI] [PubMed] [Google Scholar]
- 17.Wajcman, H., and F. Galacteros. 2005. Hemoglobins with high oxygen affinity leading to erythrocytosis. New variants and new concepts. Hemoglobin. 29:91–106. [PubMed] [Google Scholar]
- 18.Corda, M., M. Pellegrini, A. Olianas, A. Fais, M. T. Sanna, L. Manca, B. Masala, and B. Giardina. 2000. Adult and fetal HbJ Sardegna [His α50 (CE8)→Asp]: functional and molecular studies. Biochem. J. 346:193–199. [PMC free article] [PubMed] [Google Scholar]
- 19.Corda, M., A. Fais, L. Perseu, L. Cipollina, S. Barella, and R. Galanello. 2002. Identification and functional characterization of a new hemoglobin variant in Sardinia: Hb Muravera [β47GAT→GTT,(CD6)Asp→Val]. Haematologica. 87:1111–1112. [PubMed] [Google Scholar]
- 20.Miele, A. E., F. Draghi, A. Arcovito, A. Bellelli, M. Brunori, C. Travaglini-Allocatelli, and B. Vallone. 2001. Control of heme reactivity by diffusion: structural basis and functional characterization in hemoglobin mutants. Biochemistry. 40:14449–14458. [DOI] [PubMed] [Google Scholar]
- 21.Beutler, E., A. Lang, and H. Lehmann. 1974. Hemoglobin Duarte: (α2β2 62(E6)Ala leads to Pro): a new unstable hemoglobin with increased oxygen affinity. Blood. 43:527–535. [PubMed] [Google Scholar]
- 22.Sack, J. S., L. C. Andrews, K. A. Magnus, J. C. Hanson, J. Rubin, and W. E. Love. 1978. Location of amino acid residues in human hemoglobin. Hemoglobin. 2:153–169. [DOI] [PubMed] [Google Scholar]
- 23.Kiger, L., J. Kister, P. Groff, G. Kalmes, D. Promé, F. Galactéros, and H. Wajcman. 1996. Hb J-Europa [β 62(E6)Ala→Asp]: normal oxygen binding properties in a new variant involving a residue located distal to the heme. Hemoglobin. 20:135–140. [DOI] [PubMed] [Google Scholar]
- 24.Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis, and H. A. Herlich. 1988. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 239:487–491. [DOI] [PubMed] [Google Scholar]
- 25.Masala, B., and L. Manca. 1991. Detection of the common Hb F Sardinia [A γ(E19)Ile—Thr] variant by isoelectric focusing in normal newborns and in adults affected by elevated fetal hemoglobin syndromes. Clin. Chim. Acta. 198:195–202. [DOI] [PubMed] [Google Scholar]
- 26.Giardina, B., and G. Amiconi. 1981. Measurement of binding of gaseous and nongaseous ligands to hemoglobins by conventional spectrophotometric procedures. Methods Enzymol. 76:417–427. [DOI] [PubMed] [Google Scholar]
- 27.Procacci, P., E. Paci, T. Darden, and M. Marchi. 1997. ORAC: a molecular dynamics program to simulate complex molecular systems with realistic electrostatic interactions. J. Comput. Chem. 18:1848–1862. [Google Scholar]
- 28.Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- 29.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 30.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 31.Procacci, P., and M. Marchi. 1996. Taming the Ewald sum in molecular dynamics simulations of solvated proteins via a multiple time step algorithm. J. Chem. Phys. 104:3003–3012. [Google Scholar]
- 32.Simplaceanu, V., J. A. Lukin, T.-Y. Fang, M. Zou, N. T. Ho, and C. Ho. 2000. Chain-selective isotopic labeling for NMR studies of large multimeric proteins: application to hemoglobin. Biophys. J. 79:1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rovira, C., and M. Parrinello. 2000. First-principles molecular dynamics simulations of models for the myoglobins active center. Int. J. Quantum. Chem. 80:1172–1180. [Google Scholar]
- 34.Rovira, C., B. Schulze, M. Eichinger, J. D. Evanseck, and M. Parrinello. 2001. Influence of the heme pocket conformation on the structure and vibrations of the Fe-CO bond in myoglobins: a QM/MM density functional study. Biophys. J. 81:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52:7182–7190. [Google Scholar]
- 36.Kleywegt, G. J., J. Y. Zou, M. Kjeldgaard, and T. A. Jones. 2001. In International Tables for Crystallography, Vol. F. M. G. Rossman and E. Arnold, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 353–356. Chapter 17.1.
- 37.Kleywegt, G. J., and T. A. Jones. 1994. Detection, delineation, measurement and display of cavities in macromolecular structures. Acta Crystallogr. D50:178–185. [DOI] [PubMed] [Google Scholar]
- 38.Connolly, M. L. 1993. The molecular surface package. J. Mol. Graph. 11:139–143. [DOI] [PubMed] [Google Scholar]
- 39.Tilton, R. F. Jr., U. C. Singh, S. J. Weiner, M. L. Connelly, I. D. Kuntz Jr., and P. A. Kollmann. 1984. Computational studies of the interaction of myoglobin and xenon. Biochemistry. 23:2849–2857.6466620 [Google Scholar]
- 40.Shen, T.-J., N. T. Ho, V. Simplaceanu, M. Zou, B. N. Green, M. F. Tam, and C. Ho. 1993. Production of unmodified human adult hemoglobin in Escherichia coli. Proc. Natl. Acad. Sci. USA. 90:8108–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun, D. P., M. Zou, N. T. Ho, and C. Ho. 1997. Contribution of surface histidyl residues in the alpha-chain to the Bohr effect of human normal adult hemoglobin: roles of global electrostatic effect. Biochemistry. 36:6663–6673. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi, S., A. K. C. Lin, and C. Ho. 1980. Proton nuclear magnetic resonance studies of hemoglobins M Boston (α58E7His→Tyr) and M Milwaukee (β67E11 Val→Glu): spectral assignments of hyperfine-shifted proton resonances and of proximal histidine (E7) NH resonances to the α and β chains of normal adult hemoglobin. Biochemistry. 19:5196–5202. [DOI] [PubMed] [Google Scholar]
- 43.La Mar, G. N., K. Nagai, T. Jue, D. L. Budd, K. Gersonde, H. Sick, T. Kagimoto, A. Hayashi, and F. Takeda. 1980. Assignment of proximal histidyl imidazole exchangeable proton NMR resonances to individual subunits in hemoglobins A, Boston, Iwate and Milwaukee. Biochem Biophys. Res. Commun. 96:1172–1177. [DOI] [PubMed] [Google Scholar]
- 44.Johnson, M. E., and C. Ho. 1974. Effects of ligands and organic phosphates on functional properties of human adult hemoglobin. Biochemistry. 13:3653–3661. [DOI] [PubMed] [Google Scholar]
- 45.Kurland, R. J., and B. R. McGarvey. 1970. Isotropic NMR shifts in transition metal complexes: the calculation of the Fermi contact and pseudocontact terms. J. Magn. Reson. 2:286–301. [Google Scholar]
- 46.Perutz, M. F. 1970. Stereochemistry of cooperative effects in haemoglobin. Nature. 228:226–233. [DOI] [PubMed] [Google Scholar]
- 47.Fung, L. W.-M., and C. Ho. 1975. A proton nuclear magnetic resonance study of the quaternary structure of human hemoglobins in water. Biochemistry. 14:2526–2535. [DOI] [PubMed] [Google Scholar]
- 48.Ishimori, K., K. Imai, G. Miyazaki, T. Kitagawa, Y. Wada, H. Morimoto, and I. Morishima. 1992. Site-directed mutagenesis in hemoglobin: functional and structural role of inter- and intrasubunit hydrogen bonds as studied with 37β and 145β mutations. Biochemistry. 31:3256–3264. [DOI] [PubMed] [Google Scholar]
- 49.Chang, C. K., V. Simplaceanu, and C. Ho. 2002. Effects of amino acid substitution at β131 on the structure and properties of hemoglobin: evidence for communication between α1β1 and α1β2 subunit interfaces. Biochemistry. 41:5644–5655. [DOI] [PubMed] [Google Scholar]
- 50.Russu, I. M., N. T. Ho, and C. Ho. 1987. A proton nuclear magnetic Overhauser effect investigation of the subunit interfaces in human normal adult hemoglobin. Biochim. Biophys. Acta. 914:40–48. [DOI] [PubMed] [Google Scholar]
- 51.Cheng, Y., T.-J. Shen, V. Simplaceanu, and C. Ho. 2002. Ligand binding properties and structural studies of recombinant and chemically modified hemoglobins altered at β93 cysteine. Biochemistry. 41:11901–11913. [DOI] [PubMed] [Google Scholar]
- 52.Scott, E. E., and Q. H. Gibson. 1997. Ligand migration in sperm whale myoglobin. Biochemistry. 36:11909–11917. [DOI] [PubMed] [Google Scholar]
- 53.Scott, E. E., Q. H. Gibson, and J. S. Olson. 2001. Mapping the pathways for O2 entry into and exit from myoglobins. J. Biol. Chem. 276:5177–5188. [DOI] [PubMed] [Google Scholar]
- 54.Lamb, D. C., K. Nienhaus, A. Arcovito, F. Draghi, A. E. Miele, M. Brunori, and G. U. Nienhaus. 2002. Structural dynamics of myoglobin. J. Biol. Chem. 277:11636–11644. [DOI] [PubMed] [Google Scholar]
- 55.Sottini, S., S. Abbruzzetti, C. Viappiani, S. Bettati, L. Ronda, and A. Mozzarelli. 2005. Evidence for two geminate rebinding states following laser photolysis of R state hemoglobin encapsulated in wet silica gels. J. Phys. Chem. B. 109:11411–11413. [DOI] [PubMed] [Google Scholar]
- 56.Lepeshkevich, S. V., J. Karpiuk, I. V. Sazanovich, and B. M. Dzhagarov. 2004. A kinetic description of dioxygen motion within α- and β-subunits of human hemoglobin in the R-state: geminate and bimolecular stages of the oxygenation reaction. Biochemistry. 43:1675–1684. [DOI] [PubMed] [Google Scholar]
- 57.Brunori, M., and Q. H. Gibson. 2001. Cavities and packing defects in the structural dynamics of myoglobins. EMBO Rep. 2:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agmon, N. 2004. Coupling of protein relaxation to ligand binding and migration in myoglobins. Biophys. J. 87:1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]