

Abstract
Relapsing polychondritis is an autoimmune disease that affects cartilage in the ear, nose, and respiratory tract. A pathogenic immune response has been proposed and antibodies to several cartilage proteins are detected in sera from these patients. To investigate the role of the humoral immune response in relapsing polychondritis, we used the matrilin-1-induced relapsing polychondritis model. Mice deficient of B cells (μMT) and mice congenic at the complement factor 5, were immunized with matrilin-1, a cartilage-specific protein mainly detected in the tracheal cartilage. To investigate the binding properties and tissue selection of matrilin-1-specific antibodies we produced matrilin-1-specific B-cell hybridomas. Although 83% of the μMT heterozygous mice developed respiratory distress and erosive chondritis in the respiratory tract, none of the B-cell-deficient mice were susceptible to disease. In addition, we show that complement factor 5 is important for the induction of matrilin-1-induced relapsing polychondritis. Monoclonal matrilin-1-specific antibodies injected into neonatal mice bound specifically to cartilage of the respiratory tract and adult B-cell-deficient mice injected with the same antibodies developed erosive chondritis in the respiratory tract. We conclude that relapsing polychondritis can be mediated by a pathway involving tissue-specific antibodies and complement activation.
Autoimmune diseases in which the patients present an inflammation in or around cartilage are widespread in the population. Relapsing polychondritis (RP) is a chronic relapsing disorder in which cartilage in the ear, nose, and respiratory tract are affected.1 The mechanisms that are involved in the initial triggering of RP, the maintenance of the pathogenic immune response, and subsequent destruction of the cartilage are poorly understood. Previous reports implicate a role for the humoral immune response, particularly in the effector phase. It has been demonstrated that antibodies to CII are present in the acute phase of RP and that the antibody titers seem to correlate with the severity of the symptoms.2–4 Antibodies to collagen type IX (CIX) and type XI (CXI) are detected in patient sera as well.5,6 Furthermore, antibodies to matrilin-1, an extracellular matrix protein predominantly expressed in tracheal cartilage,7 and antibodies to cartilage oligomeric matrix protein (COMP) are detected in these patients.8 A role for immune complexes and a subsequent activation of the complement system have also been suggested in RP because C3 depositions and Igs were detected in affected auricular cartilage and in renal mesangium.9,10
RP has been associated with HLA-DR4, which implicates a role for the MHC class II molecule and the involvement of T cells.11–13 However, the influence of T cells in RP pathogenesis has been poorly investigated and only a few reports have been published. In separate reports of two patients with severe tracheomalacia, T-cell responses to CIX and CXI,6 and to matrilin-114 were demonstrated. Our earlier findings from the matrilin-1-induced relapsing polychondritis (MIRP),15 an animal model mimicking respiratory distress and nasal chondritis that represent a subset of the severe symptoms in RP, indicate that both T cells and B cells specific for matrilin-1 are activated. Strikingly, the B-cell response consisted mainly of autoreactive antibodies of the IgG type suggesting that immune tolerance to matrilin-1 had been broken. The role of B cells and pathogenic antibodies in rheumatoid arthritis (RA) have recently been revived thanks to findings made in animal models such as the K/BxN mice in which antibodies reactive with glucose phosphoisomerase are highly arthritogenic16 and in the more classical collagen-induced arthritis (CIA) model.17–19
To investigate whether B cells could be involved also in the pathogenesis of RP we used the MIRP model in the mouse. We found that both B cells and complement-dependent pathways are critical based on mice deleted for B cells and complement factor (C5) and we also established pathogenic matrilin-1-specific monoclonal antibodies.
Materials and Methods
Mice
All animals were bred and kept at the animal department at Medical Inflammation Research, Lund University. Male mice were used at an age of 8 to 13 weeks (intradermal immunization) or 4 to 6 months (intravenous injection). They were kept in a climate-controlled environment with cycles of 12 hours light/dark and sound. μMT mice, produced by a deletion in the IgM μ-chain in a cross of C57BL/6×129,20 were backcrossed into a B10.Q (H-2q) background for eight generations and intercrossed for two generations to generate homozygous littermates lacking B cells (μMT-BQ). We also backcrossed the μMT into the DBA/1 (H-2q) strain (μMT-DQ) for five generations. Crossing mice from the B10.Q and NOD.Q strains according to the speed congenic protocol produced the C5 congenic strain. These mice were intercrossed to get a homozygous NOD.Q fragment for the C5 locus. The NOD.Q fragment in the C5 congenic strain covers ∼30 cM between the markers D2Mit116 and D2Mit91.
Induction and Evaluation of Disease
Mice were immunized intradermally at the back of the tail with 100 μg of matrilin-121 emulsified in complete Freund’s adjuvant (Difco, Detroit, MI). The mice were boosted at day 35 (μMT mice) or at day 40 (C5 congenic mice) with 50 μg of matrilin-1 in incomplete Freund’s adjuvant (Difco). Scoring was performed according to modified scoring grades previously developed for the rat model:15 score 1, suspicion of respiratory distress; score 2, discontinuous inspiratory stridor; 3, continuous inspiratory stridor; 4, continuous inspiratory stridor and abnormal breathing pattern; 5, cyanosis.
Tissue Preparation and Staining
Tissue was dissected and immediately either snap-frozen at −70°C or fixed in 4% paraformaldehyde solution for 24 hours and further embedded in paraffin. Joints were decalcified for 2 to 3 weeks in ethylenediaminetetraacetic acid solution. Sections of 5 to 6 μm were stained with hematoxylin and erythrosine.
Production of Monoclonal Antibodies and in Vivo Injections
Five matrilin-1-specific hybridomas (AM2, AM4, AM5, AM7, AM8) were produced according to established protocols by using interleukin-10-deficient22 B10.Q mice, immunized with matrilin-1 as described above and boosted a second time in the footpad at day 140 with 50 μg of matrilin-1 in incomplete Freund’s adjuvant. The interleukin-10-deficient B10.Q mice were used because of their sensitivity to develop severe and chronic chondritis of the respiratory tract (AS Hansson et al, unpublished data). Neonatal mice, ie, 2-day-old mice, were injected intraperitoneally with 100 μl of each biotinylated monoclonal antibody. Tissue was collected 24 hours later and snap-frozen. Immunostainings, detected by diaminobenzidine, were used to visualize the bound antibodies on the tissue. Mice, 4 to 6 months old, were injected intraperitoneally with combinations of two to three different matrilin-1-specific antibodies. The doses varied between total doses of 4 to 5 mg per mouse or 9 mg per mouse (the μMT-BR mice), with equal concentrations of the different monoclonal antibodies, volumes varied between ∼400 to 1000 μl, injections with larger volumes were divided into several occasions with a few hours in between. At day 5, 50 μg of lipopolysaccharide were injected intraperitoneally. Tissue was collected and fixed in 4% paraformaldehyde solution as described above.
Antibody Detection
Sera were collected and stored in −20°C until assayed. Enzyme-linked immunosorbent assay was performed according to standard protocols.8 Plates (Costar; Corning Inc., Corning, NY) were coated with 1 μg/ml of denatured bovine matrilin-1,21 10 μg/ml of CII, or 10 μg/ml of COMP. Enzyme-linked immunosorbent assay on μMT mice was performed with sera diluted 1/10 and titrated in steps of 10. In the C5-congenic and corresponding control mice sera were diluted 1/10,000 (sera at day 77, the isotypes IgG1 and IgG2a were diluted 1/1000). We used secondary antibodies detecting sheep anti-mouse IgG Fcγ labeled with alkaline phosphatase, (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) or a peroxidase-conjugated secondary antibody detecting Ig isotypes (goat anti-mouse IgG1 and goat anti-mouse IgG2a) of the matrilin-1 response. The plates were developed with p-nitrophenol or ABTS solution as the substrates and the amount of antibody was estimated as absorbency at 405 nm by using a Titertek Multiscan filterphotometer. All plates detecting the same antigen or isotype were analyzed at the same time point.
Statistical Analysis
Antibody levels were analyzed with Mann-Whitney U-test. If not indicated otherwise, P<0.05 was considered significant.
Results
B-Cell-Deficient Mice Do Not Develop MIRP
To investigate the role of B cells in MIRP, we used murine littermates on the B10.Q background that were either heterozygous (n = 11) or homozygous (n = 12) for the deletion of IgM (μMT). Approximately 7 weeks after immunization (day 46 ± 6) the heterozygous mice developed respiratory distress symptoms with high incidence (Figure 1a). A few mice reached score 5, that is cyanosis, in the acute phase and were taken off the experiment whereas the majority of the remaining symptomatic mice were seen with a subsequent relapsing disease (Figure 1; a to d). The homozygous mice, ie, B cell-deficient, did not present any respiratory distress or other pathological clinical sign. Macroscopic joint inflammation was not detected in any mouse.
Figure 1.

Disease course of mice immunized with matrilin-1. B-cell-deficient (homozygous) and B-cell-sufficient (heterozygous) μMT mice on a B10.Q background are scored for respiratory distress. A: Incidence. B: Mean clinical score of all mice. C and D: Clinical scores of two individual mice with a characteristic relapsing disease. For clinical score see Material and Methods.
Tissue sections were analyzed in the acute phase of the disease and at the end of the experiment, at day 140. An aggressive inflammatory infiltrate was found in the tracheal cartilage during the onset period of the respiratory symptoms. This infiltrate, that destroyed major parts of the cartilage tissue, consisted of mainly neutrophils (Figure 2a). Laryngeal and nasal cartilages were also affected by mainly a neutrophil infiltration, but with a milder destruction of the affected cartilage (Figure 2b). In the chronic phase of the disease, an active inflammation with a mix of neutrophils, macrophages, and lymphocytes could still be detected in a few mice, but new formation of cartilage and tissue reorganization were the most prominent features (Figure 2c). Joint cartilage was not affected in the acute phase and only mildly affected in the late phase of the disease, which could possibly be explained by induction of an immune response to cartilage proteins other than matrilin-1. No clinical or histological finding was detected in the B-cell-deficient mice.
Figure 2.
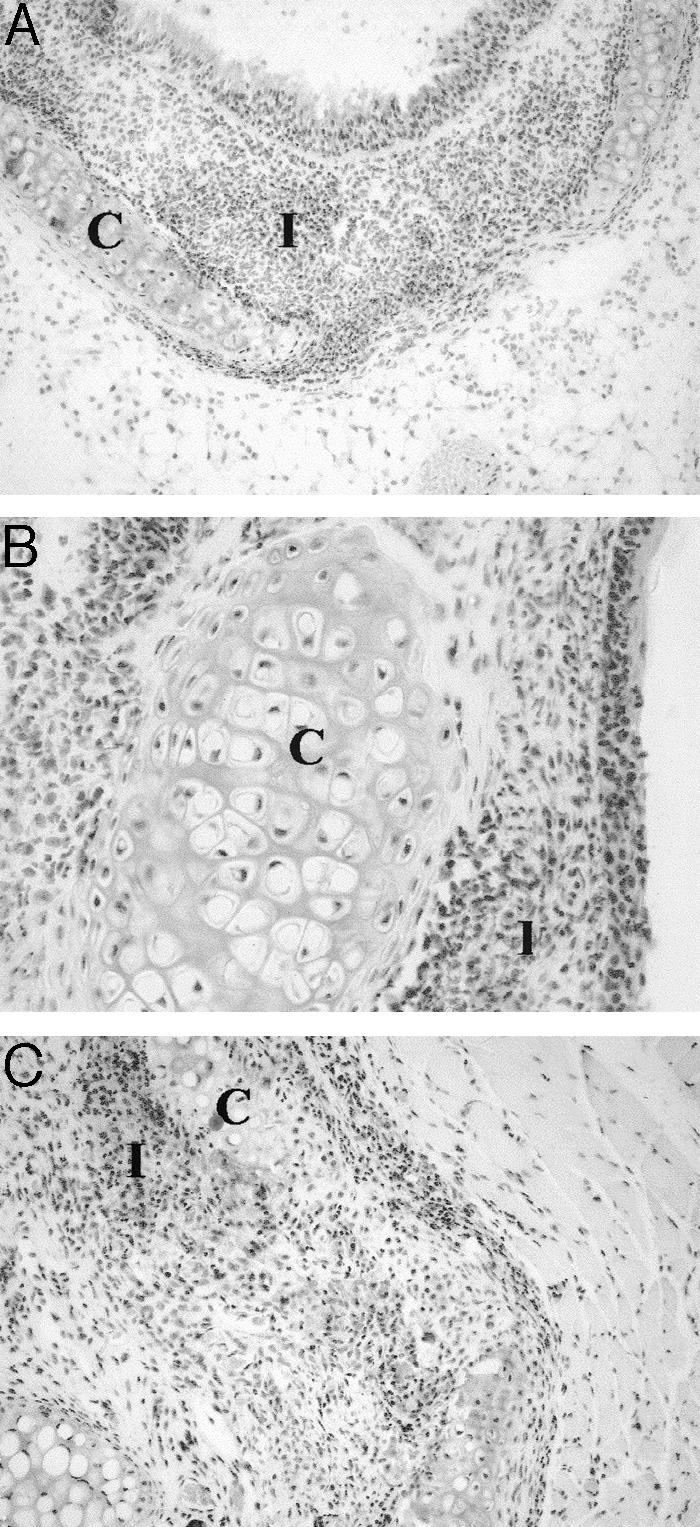
Tissue sections from a μMT-B10.Q-sufficient (heterozygous) mouse, immunized with matrilin-1. A and B: Tracheal tissue with a destructed tracheal ring (A) and inflammatory infiltrate in nasal tissue (B), both from the acute phase of the disease. C: Section from laryngeal cartilage 140 days after immunization. C, Cartilage tissue; I, inflammatory infiltrate. Staining with hematoxylin and erythrosine. Original magnifications: ×75 (A, C); ×200 (B).
Heterozygous Mice Produce Antibodies to Matrilin-1; B-Cell-Deficient Mice Do Not
All μMT mice immunized with matrilin-1 were investigated for an antibody response to matrilin-1. A significant increase in antibody titer was detected in the heterozygous mice, both at day 35 and day 140 (data not shown). The B-cell-deficient mice did not produce any antibodies, nor did the control mice. Four heterozygous mice mounted low antibody titers to COMP and three had low titers to CII (data not shown). No correlation was found between any of the investigated parameters and the antibody titers of CII or COMP, but importantly, all mice with disease had high levels of antibodies to matrilin-1.
Monoclonal Antibodies to Matrilin-1 Bind Neonatal Cartilage in Vivo
To further investigate the role of B cells and immunoglobulins, we established five B-cell hybridomas, which produced monoclonal antibodies specific to matrilin-1. Specificity of the clones (AM2, AM4, AM5, AM7, and AM8) was tested by enzyme-linked immunosorbent assay, Western blot, and tissue staining.8 Crossreactivities of various degrees were demonstrated between some of the clones, and accordingly they most likely shared some of the matrilin-1 epitopes (Figure 3). All clones were identified as IgG1 isotypes, except for AM4 that was indefinable, but most likely of the IgG2c isotype.
Figure 3.

Inhibition assay of five matrilin-1-specific monoclonal antibodies. Five clones (AM2, AM4, AM5, AM7, and AM8) were produced and tested for cross-reactivity. The results of the enzyme-linked immunosorbent assay are presented as percentage of inhibition when blocking a biotinylated antibody with a nonbiotinylated antibody.
To test the in vivo binding capacity of the biotinylated monoclonal antibodies, we injected the antibodies into neonatal mice and visualized the same by immunohistochemical staining 24 hours later. The five clones bound with different affinity to the tracheolaryngeal cartilage tissue; AM5 was defined as the strongest binder (Figure 4a), AM2 was a moderate one and AM4, AM7, and AM8 bound weakly (data not shown). The fact that matrilin-1 is purified under denaturing conditions raises the possibility that the weakly bound antibodies recognize epitopes on native matrilin-1 and which, because of several steps in tissue and staining preparation, are no longer present. The nasal cartilage tissue exhibited a similar staining pattern as the laryngeal one. No antibody bound to the articular cartilage surface in the hind paws (Figure 4b).
Figure 4.
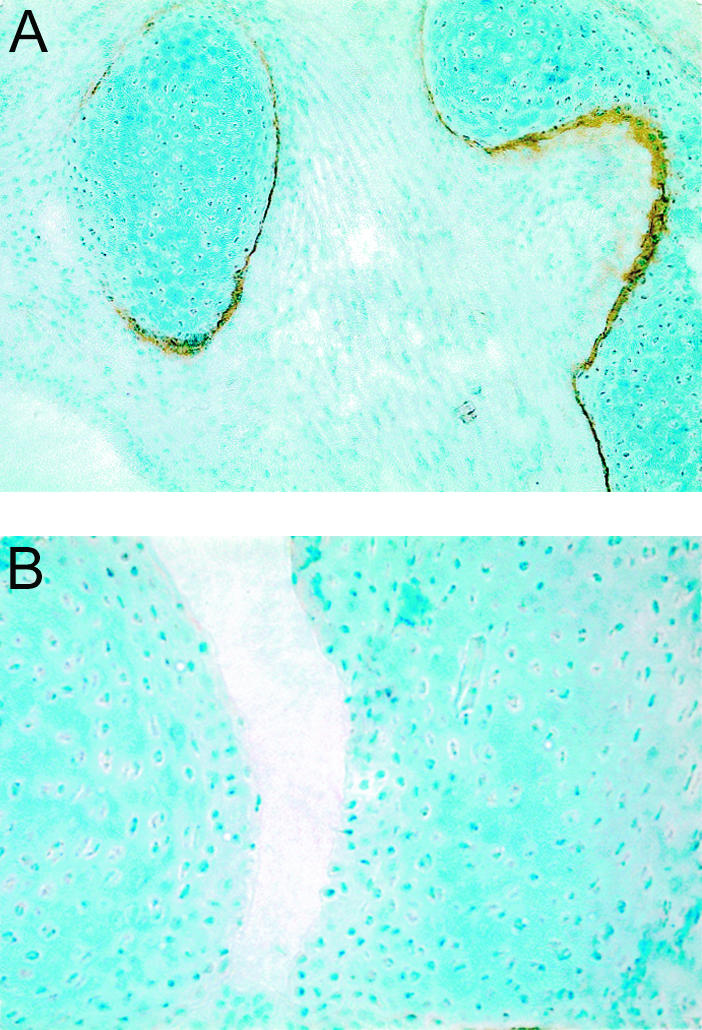
Tissue sections from neonatal mice that were injected with matrilin-1-specific monoclonal antibodies. A: Laryngeal cartilage from a neonate injected with AM5. B: Hind paw from a neonate injected with AM5. Background staining with methyl green. Original magnifications: ×65 (A); ×170 (B).
Monoclonal Antibodies to Matrilin-1 Induce Inflammation in the Cartilage of the Respiratory Tract
To investigate the effect of our matrilin-1-specific antibodies in adult B-cell-deficient mice, the mice were injected with different combinations of our monoclonal antibodies followed by an injection of lipopolysaccharide at day 5. When we used the combination of AM2 + AM5 at a concentration of 2 mg per antibody, two of three μMT-DQ mice presented mild clinical scores. In the tissue sections of these two mice, we could detect neutrophils and macrophages in an erosive active inflammation of the laryngeal cartilage (Figure 5a). In addition, an infiltration of mainly neutrophils was found in the mucosal part of the nasal septum (Figure 5b). Tissue samples were collected and stained from the ear and the joints as well, but no inflammation could be detected. Furthermore, we used μMT-BR mice, a mouse strain that we have recently shown is more sensitive to antibody-induced arthritis than the μMT-B10.Q strain (KS Nandakumar et al, unpublished data). Two of six of these mice died before the planned collection of tissue specimen. This was likely because of an acute respiratory distress caused by a severe chondritis of the respiratory tract, which is commonly seen in several other mouse strains, as for example the QD and the B10.Q strains (AS Hansson et al, unpublished data). Interestingly, one of these mice was injected with a cocktail of the same monoclonal antibodies that induced cartilage erosions in the μMT-DQ mice (AM2 + AM5), while the other one received AM2 + AM4. The fact that a total concentration of 9 mg of monoclonal antibodies (instead of 4 to 5 mg) was injected in the μMT-BR mice most probably also contributed to the lethal outcome. The remaining four μMT-BR mice were sacrificed on day 12 after the first injection and tissue was analyzed as described for the earlier experiment but no major inflammatory changes were detected. In addition we tried to induce disease in μMT-BQ, BALB/c, and QD mice, all of which are strains known as susceptible to antibody-induced arthritis and/or matrilin-1-induced chondritis, but with negative outcome. Nor were we able to transfer disease by serum from affected QD mice into μMT-BR mice. Control monoclonal antibodies of all isotypes were used but no signs of arthritis or chondritis were observed (data not shown).
Figure 5.
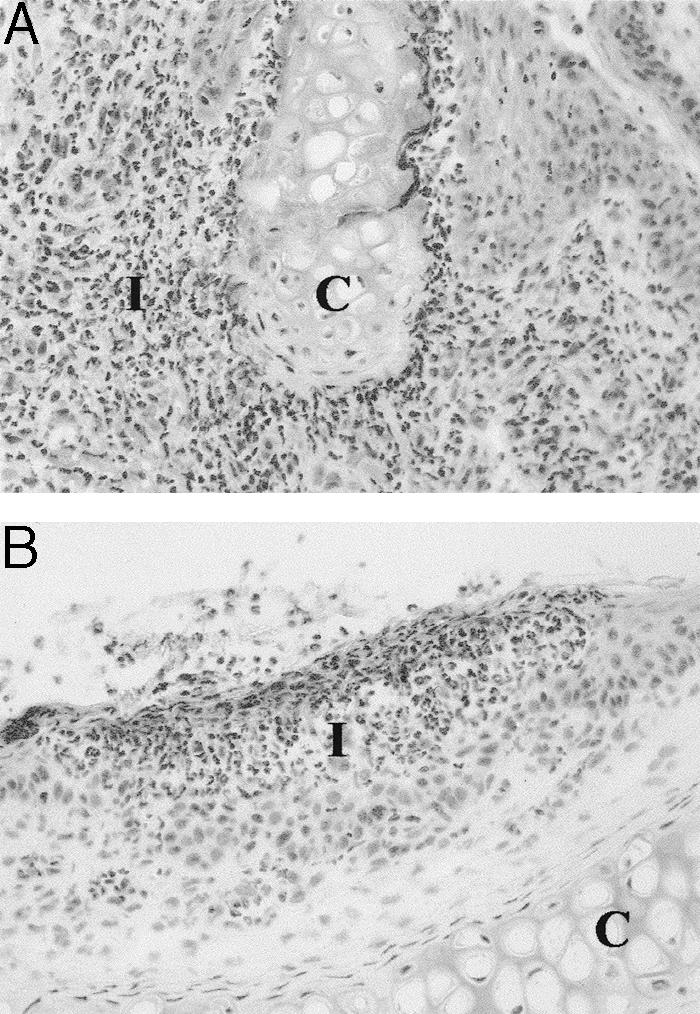
Tissue sections from a μMT-QD B-cell-deficient (homozygous) mouse that was injected with matrilin-1-specific monoclonal antibodies. A: Tissue section from laryngeal cartilage showing an erosive inflammation. B: Nasal tissue showing an inflammatory infiltrate in the mucosal part of the septum. C, Cartilage; I, inflammatory infiltrate. Staining with hematoxylin and erythrosine. Original magnifications, ×180.
Complement Factor 5 Plays a Major Role in the Pathogenesis of MIRP
We wanted to analyze the role of complement in MIRP. We used B10.Q mice, congenic for the C5 locus with genes from the NOD mouse, which carries a 2-bp deletion in the C5-encoding gene making it nonfunctional. B10.Q control mice as well as C5-congenic mice were immunized with matrilin-1 according to standard protocol. Respiratory distress was observed in 78% of the control mice but only in 25% of the C5-congenic mice. In addition, a significantly higher mean maximum score was found in the control mice (Table 1). No difference was found between the groups when analyzing only affected mice for severity, onset day, or weight loss during the disease course (data not shown). We could however demonstrate a significant difference in antibody response to matrilin-1 between the two groups (Figure 6). The level of antibodies of the IgG1 isotype was significantly lower in the C5-congenic mice compared to the control group whereas no difference was found for the IgG2a/c isotype.
Table 1.
Clinical Characteristics in Mice Immunized with Matrilin-1 with a Functional (B10.Q) or a Nonfunctional (B10.Q C5 Congenic) C5 Molecule
| Incidence | Mean maximum score (all animals included) | Onset day | |
|---|---|---|---|
| B10.Q | 7/9* | 2.9 ± 1.7† | 52 ± 8 |
| B10.Q C5 congenic | 2/8* | 0.6 ± 1.2† | 52 ± 10 |
P = 0.06, bilateral Fisher’s test.
P < 0.02, Mann-Whitney U-test.
Figure 6.

Serum levels of matrilin-1-specific antibodies in mice immunized with matrilin-1. Sera were diluted 1/10,000 in all assays except for sera from day 77 detecting IgG1 and IgG2a, which were diluted 1/1000. Titers are expressed as OD values detecting IgG Fcγ (A), IgG1 (B), and IgG2a/c (C).
Discussion
Few investigations have been reported on the pathogenic pathways in RP and its corresponding animal models. In this study we show that B cells are crucial for the induction of RP-like symptoms in the murine MIRP model and that C5 plays a prominent role in the induction phase of this disease. We also demonstrate that matrilin-1-specific monoclonal antibodies bind cartilage of the respiratory tract and that these antibodies are pathogenic. These findings confirm and extend the recent suggestion that matrilin-1 is a critical autoantigen eliciting respiratory symptoms in rodents as well as in human patients.8,14,15 Contrary to RP, RA and the arthritis models have been thoroughly explored throughout many years. RP and RA share several clinical manifestations as well as features on the molecular level, but large differences, such as for example the site of chondritis, appear as well which is also demonstrated in animal models.15,23–25 In this discussion, we deal with the immunological characteristics of models mimicking RP as well as models for RA because we hypothesize that some of the basic mechanisms are valid in both diseases and that the differences appearing are important to penetrate the understanding of the tissue-specific immune response.
A humoral immune response is required for the development of CIA.18,26,27 High levels of anti-CII IgG are usually detected in the sera from CII-immunized animals but does not automatically lead to arthritis although all arthritic mice have a minimal level of anti-CII IgG antibodies in serum.28,29 These observations indicate that antibodies are not the sole factor for CIA but clearly they are essential components. In the recently described T-cell receptor transgenic K/BxN model of arthritis it has been shown that antibodies to glucose phosphoisomerase accumulate on cartilage surface and induce severe arthritis.30 However, it needs to be emphasized that in some other models of arthritis, B cells are likely to play only a minor, or possibly a protective role, for development of arthritis.31,32 These differences in pathogenic pathways leading to the same end results, arthritis, may be caused by factors such as differences in the mode of induction, the genetic background, or environmental surrounding. It is also unclear whether the pathogenic pathways leading to chondritis are the same as those leading to arthritis and in this case both the immune specificity and the target tissue are different. We show that matrilin-1-specific antibodies induce cartilage inflammation and erosion in the respiratory tract, indicating that pathogenic antibodies operate in this model of RP. Although we only succeeded to induce mild respiratory symptoms at injection of matrilin-1-specific antibodies in B-cell-deficient mice it is possible that adjusting the dose, the use of combinations of antibodies and another selection of epitope-specificity would increase the pathogenicity. These possibilities have been described in more detail using arthritis models, the CIA model, and the spontaneous TCR transgenic model K/BxN, in which a cocktail of several monoclonal antibodies with selected epitope-specificity enhanced disease induction.16,19,33,34 In addition, serum transfer from diseased animals to B-cell-deficient mice has been reported to induce a mild and transient arthritis in the K/BxN model.35 We did not induce any clinical or histological chondritis by serum transfer from MIRP-diseased mice. However, considering that only a mild and transient articular inflammation was detected in the K/BxN mice, it will be difficult to catch these inflammatory changes in the MIRP model because the tracheal cartilage is not visible to the eye during active inflammation and hence quite strong inflammation is needed for clinical symptoms to arise. The fact that we induced inflammation in the μMT-DQ but not in the μMT-BQ strain fits with our recent results that the QD strain is very susceptible to MIRP induction compared to all other strains tested, including the B10.Q (AS Hansson et al, unpublished data). Furthermore, the mouse that we chose for isolation of matrilin-1-specific B-cell hybridomas was immunized 140 days before the fusion of the lymph nodes. Despite the fact that this mouse developed severe respiratory distress in a relapsing manner (AS Hansson et al, unpublished data) with no macro- or microscopical signs of joint involvement, the cartilage reorganization is likely to reveal new epitopes originating from tracheal cartilage proteins such as matrilin-1, COMP, CII, CIX, and CXI. This might result in hybridomas producing antibodies that are reactive to epitopes appearing only in the chronic phase of the disease, as a consequence of tissue destruction, and not to the epitopes that are important for the initiation of a matrilin-1 immune response.
In recent years the role of immunoglobulin receptors has been highlighted in experimental arthritis as important players in the antibody-mediated effector phase. Data suggest that FcγRI and FcγRIII are crucial for the development of arthritis in DBA/1 mice immunized with CII while the FcγRII was protective against disease.36 The complement factors have also been proposed as major contributors in this phase of disease and particularly C5 has been shown to play a critical role in several arthritis models.37–41 In this study we show that C5 is one of the major players for induction of autoimmune chondritis in the respiratory tract and nose. However, C5 is not an absolute requirement for the development of MIRP because a few mice devoid of a functional C5 were still affected by respiratory distress, which indicates that alternative routes not dependent on the C5 molecule were activated. Surprisingly, the C5 congenic mice produced less antibodies of the IgG1 isotype indicating that a Th2-like immune response plays a pathogenic role in the MIRP model. The observation that most of the hybridomas produced IgG1 antibodies could be coincidental but are also in line with data recently presented on the murine K/BxN serum-transferred arthritis model where IgG1 was determined as the main and only isotype required for arthritis induction.16 Consequently, several factors such as the Ig isotype, IgG1, the impact of neutrophil influx in the initial phase and the importance of the alternative pathway of the complement system indicate major similarities between the MIRP pathogenesis and the K/BxN model. Alternative pathogenic pathways are obviously involved in CIA where most IgG subclasses (1, 2a, and 2b) have been shown to induce arthritis19,33,34,42 (KS Nandakumar et al, unpublished observations).
In conclusion, our results emphasize the requirement of a humoral immune response in autoimmune chondritis. They further indicate that several pathogenic pathways most likely are involved in murine models that mimic human diseases such as RP and RA. We show that B cells, matrilin-1-specific antibodies, and C5 play major roles in the pathogenesis of cartilage inflammation of the respiratory tract. However, additional investigations on the fine specificity of the pathogenic B cells, immunoglobulins, and complement factors are needed to reveal the complexity of the autoimmune inflammation in cartilage tissue.
Acknowledgments
We thank Estelle Eriksson-Bajtner for advice when producing the hybridomas and Carlos Palestro for excellent animal care.
Footnotes
Address reprint requests to Ann-Sofie Hansson, Dept of Clinical Immunology, Göteborg University, Guldhedsgatan 10, 413 46 Gothenburg, Sweden. E-mail: ann-sofie.hansson@vgregion.se.
Supported by grants from the Anna Greta Crafoord Foundation for Rheumatological Research, King Gustaf V:s 80-Year Foundation, Greta and Johan Kock’s Foundations, Alfred Österlund’s Foundation, the Swedish Association against Rheumatism, and the Swedish Medical Research Council.
References
- McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore) 1976;55:193–215. [PubMed] [Google Scholar]
- Foidart JM, Abe S, Martin GR, Zizic TM, Barnett EV, Lawley TJ, Katz SI. Antibodies to type II collagen in relapsing polychondritis. N Engl J Med. 1978;299:1203–1207. doi: 10.1056/NEJM197811302992202. [DOI] [PubMed] [Google Scholar]
- Ebringer R, Rook G, Swana GT, Bottazzo GF, Doniach D. Autoantibodies to cartilage and type II collagen in relapsing polychondritis and other rheumatic diseases. Ann Rheum Dis. 1981;40:473–479. doi: 10.1136/ard.40.5.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer O, Cyna J, Dryll A, Cywiner-Golenzer C, Wassef M, Ryckewaert A. Relapsing polychondritis—pathogenic role of anti-native collagen type II antibodies. A case report with immunological and pathological studies. J Rheumatol. 1981;8:820–824. [PubMed] [Google Scholar]
- Yang CL, Brinckmann J, Rui HF, Vehring KH, Lehmann H, Kekow J, Wolff HH, Gross WL, Muller PK. Autoantibodies to cartilage collagens in relapsing polychondritis. Arch Dermatol Res. 1993;285:245–249. doi: 10.1007/BF00371591. [DOI] [PubMed] [Google Scholar]
- Alsalameh S, Mollenhauer J, Scheuplein F, Stoss H, Kalden JR, Burkhardt H, Burmester GR. Preferential cellular and humoral immune reactivities to native and denatured collagen types IX and XI in a patient with fatal relapsing polychondritis. J Rheumatol. 1993;20:1419–1424. [PubMed] [Google Scholar]
- Paulsson M, Heinegard D. Radioimmunoassay of the 148-kilodalton cartilage protein. Distribution of the protein among bovine tissues. Biochem J. 1982;207:207–213. doi: 10.1042/bj2070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson AS, Heinegard D, Piette JC, Burkhardt H, Holmdahl R. The occurrence of autoantibodies to matrilin 1 reflects a tissue-specific response to cartilage of the respiratory tract in patients with relapsing polychondritis. Arthritis Rheum. 2001;44:2402–2412. doi: 10.1002/1529-0131(200110)44:10<2402::aid-art405>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Bergfeld WF. Relapsing polychondritis with positive direct immunofluorescence. Arch Dermatol. 1978;114:127. doi: 10.1001/archderm.1978.01640130085034. [DOI] [PubMed] [Google Scholar]
- Chang-Miller A, Okamura M, Torres VE, Michet CJ, Wagoner RD, Donadio JV, Jr, Offord KP, Holley KE. Renal involvement in relapsing polychondritis. Medicine (Baltimore) 1987;66:202–217. doi: 10.1097/00005792-198705000-00004. [DOI] [PubMed] [Google Scholar]
- Zeuner M, Straub RH, Rauh G, Albert ED, Scholmerich J, Lang B. Relapsing polychondritis: clinical and immunogenetic analysis of 62 patients. J Rheumatol. 1997;24:96–101. [PubMed] [Google Scholar]
- Lang B, Rothenfusser A, Lanchbury JS, Rauh G, Breedveld FC, Urlacher A, Albert ED, Peter HH, Melchers I. Susceptibility to relapsing polychondritis is associated with HLA-DR4. Arthritis Rheum. 1993;36:660–664. doi: 10.1002/art.1780360513. [DOI] [PubMed] [Google Scholar]
- Stastny P. Association of the B-cell alloantigen DRw4 with rheumatoid arthritis. N Engl J Med. 1978;298:869–871. doi: 10.1056/NEJM197804202981602. [DOI] [PubMed] [Google Scholar]
- Buckner JH, Wu JJ, Reife RA, Terato K, Eyre DR. Autoreactivity against matrilin-1 in a patient with relapsing polychondritis. Arthritis Rheum. 2000;43:939–943. doi: 10.1002/1529-0131(200004)43:4<939::AID-ANR28>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Hansson AS, Heinegard D, Holmdahl R. A new animal model for relapsing polychondritis, induced by cartilage matrix protein (matrilin-1). J Clin Invest. 1999;104:589–598. doi: 10.1172/JCI5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccioni M, Zeder-Lutz G, Huang H, Ebel C, Gerber P, Hergueux J, Marchal P, Duchatelle V, Degott C, van Regenmortel M, Benoist C, Mathis D. Arthritogenic monoclonal antibodies from K/BxN mice. J Exp Med. 2002;195:1071–1077. doi: 10.1084/jem.20011941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Townes AS, Kang AH. Nature and specificity of the immune response to collagen in type II collagen-induced arthritis in mice. J Clin Invest. 1982;69:673–683. doi: 10.1172/JCI110495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J Exp Med. 1983;158:378–392. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson AC, Hansson AS, Nandakumar KS, Backlund J, Holmdahl R. IL-10-deficient B10. Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J Immunol. 2001;167:3505–3512. doi: 10.4049/jimmunol.167.6.3505. [DOI] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- Paulsson M, Heinegard D. Purification and structural characterization of a cartilage matrix protein. Biochem J. 1981;197:367–375. doi: 10.1042/bj1970367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Bradley DS, Das P, Griffiths MM, Luthra HS, David CS. HLA-DQ6/8 double transgenic mice develop auricular chondritis following type II collagen immunization: a model for human relapsing polychondritis. J Immunol. 1998;161:5046–5053. [PubMed] [Google Scholar]
- Cremer MA, Pitcock JA, Stuart JM, Kang AH, Townes AS. Auricular chondritis in rats. An experimental model of relapsing polychondritis induced with type II collagen. J Exp Med. 1981;154:535–540. doi: 10.1084/jem.154.2.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen an experimental model of arthritis. J Exp Med. 1977;146:857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Cremer MA, Townes AS, Kang AH. Type II collagen-induced arthritis in rats. Passive transfer with serum and evidence that IgG anticollagen antibodies can cause arthritis. J Exp Med. 1982;155:1–16. doi: 10.1084/jem.155.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin Exp Immunol. 1998;111:521–526. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson M, Tarkowski A. Origin of the autoreactive anti-type II collagen response. I. Frequency of specific and multispecific B cells in primed murine lymph nodes. Immunology. 1987;61:369–374. [PMC free article] [PubMed] [Google Scholar]
- Holmdahl R, Andersson M, Goldschmidt TJ, Gustafsson K, Jansson L, Mo JA. Type II collagen autoimmunity in animals and provocations leading to arthritis. Immunol Rev. 1990;118:193–232. doi: 10.1111/j.1600-065x.1990.tb00817.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3:360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- Plows D, Kontogeorgos G, Kollias G. Mice lacking mature T and B lymphocytes develop arthritic lesions after immunization with type II collagen. J Immunol. 1999;162:1018–1023. [PubMed] [Google Scholar]
- Holmdahl R, Lorentzen JC, Lu S, Olofsson P, Wester L, Holmberg J, Pettersson U. Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev. 2001;184:184–202. doi: 10.1034/j.1600-065x.2001.1840117.x. [DOI] [PubMed] [Google Scholar]
- Terato K, Hasty KA, Reife RA, Cremer MA, Kang AH, Stuart JM. Induction of arthritis with monoclonal antibodies to collagen. J Immunol. 1992;148:2103–2108. [PubMed] [Google Scholar]
- Burkhardt H, Koller T, Engstrom A, Nandakumar KS, Turnay J, Kraetsch HG, Kalden JR, Holmdahl R. Epitope-specific recognition of type II collagen by rheumatoid arthritis antibodies is shared with recognition by antibodies that are arthritogenic in collagen-induced arthritis in the mouse. Arthritis Rheum. 2002;46:2339–2348. doi: 10.1002/art.10472. [DOI] [PubMed] [Google Scholar]
- Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen-induced arthritis is dependent on distinct fcgamma receptors. J Exp Med. 2000;191:1611–1616. doi: 10.1084/jem.191.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose PJ, Moss IK, Maini RN, Williams TJ. Measurement of the chemotactic complement fragment C5a in rheumatoid synovial fluids by radioimmunoassay: role of C5a in the acute inflammatory phase. Ann Rheum Dis. 1990;49:747–752. doi: 10.1136/ard.49.10.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen-induced arthritis in mice. J Immunol. 2002;169:454–459. doi: 10.4049/jimmunol.169.1.454. [DOI] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol. 2000;164:4340–4347. doi: 10.4049/jimmunol.164.8.4340. [DOI] [PubMed] [Google Scholar]
- Johansson AC, Sundler M, Kjellen P, Johannesson M, Cook A, Lindqvist AK, Nakken B, Bolstad AI, Jonsson R, Alarcon-Riquelme M, Holmdahl R. Genetic control of collagen-induced arthritis in a cross with NOD and C57BL/10 mice is dependent on gene regions encoding complement factor 5 and FcgammaRIIb and is not associated with loci controlling diabetes. Eur J Immunol. 2001;31:1847–1856. doi: 10.1002/1521-4141(200106)31:6<1847::aid-immu1847>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Holmdahl R, Rubin K, Klareskog L, Larsson E, Wigzell H. Characterization of the antibody response in mice with type II collagen-induced arthritis, using monoclonal anti-type II collagen antibodies. Arthritis Rheum. 1986;29:400–410. doi: 10.1002/art.1780290314. [DOI] [PubMed] [Google Scholar]